
Preclinical Evaluation of Novel Folate Receptor 1-Directed CAR T Cells for Ovarian Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.1.1. Primary Human Blood and Tissues
2.1.2. Ethics Approval
2.2. Methods
2.2.1. Tissue Processing for Microscopy
2.2.2. Cyclic Immunofluorescence Staining with the MACSima Imaging Platform
2.2.3. MACSima Data Analysis
2.2.4. Generation of CAR Plasmids
2.2.5. Generation of CAR T Cells
2.2.6. Cell Lines and Culture Conditions
2.2.7. Cell Line Generation
2.2.8. Spheroid Generation
2.2.9. Manufacturing of CAR T Cells with the CliniMACS Prodigy
2.2.10. Cytotoxicity Assays
2.2.11. Flow Cytometry
2.2.12. qPCR and VCN Determination
2.2.13. gDNA Sequencing
2.2.14. In Vivo Experiments
2.2.15. Light Sheet Microscopy
2.2.16. Statistical Analysis
3. Results
3.1. FOLR1 Is Differentially Expressed in Ovarian Carcinoma and Healthy Tissues
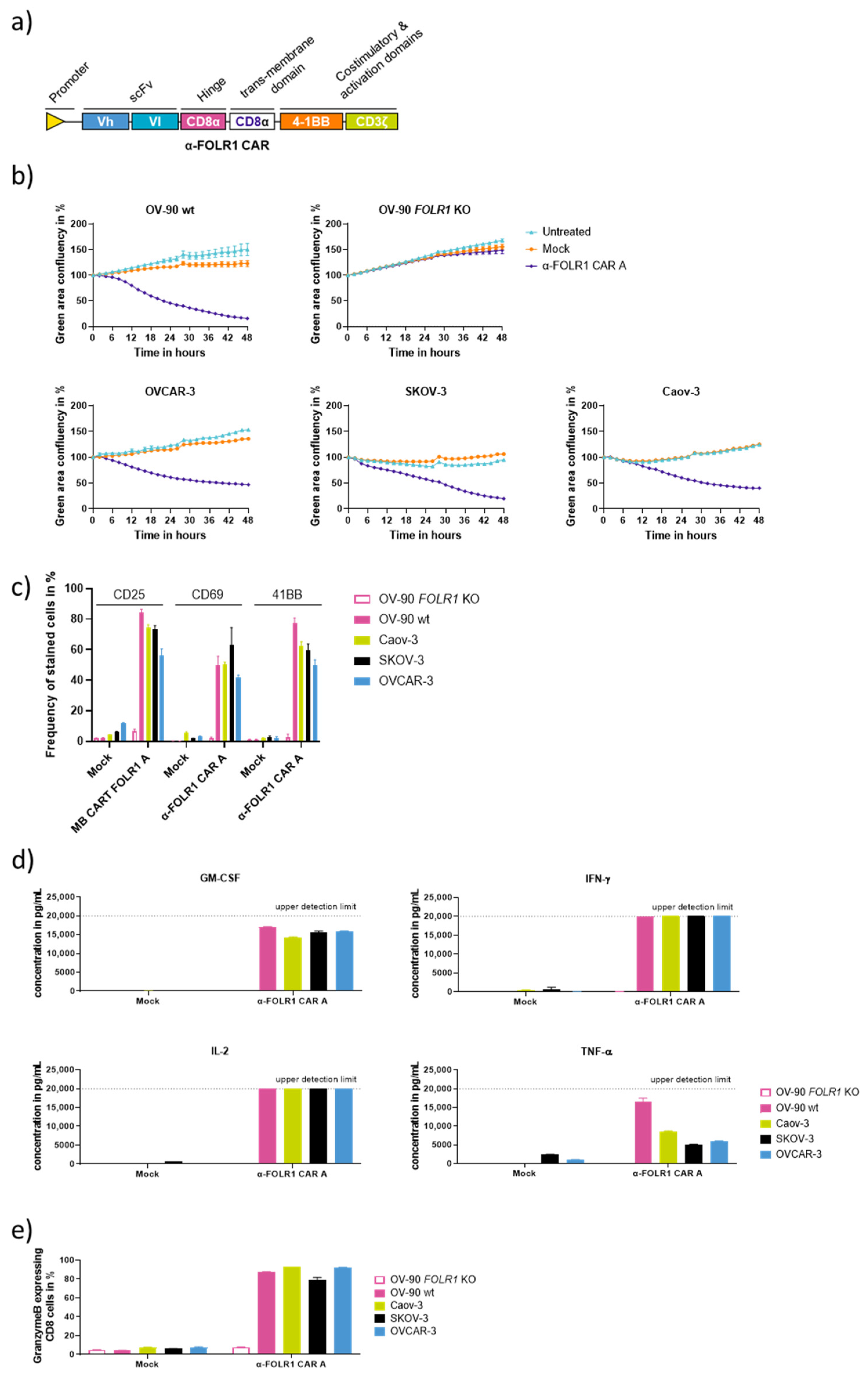
3.2. Characterization of Anti-FOLR1 CAR Designs Reveals Efficient and Specific CAR Candidates In Vitro
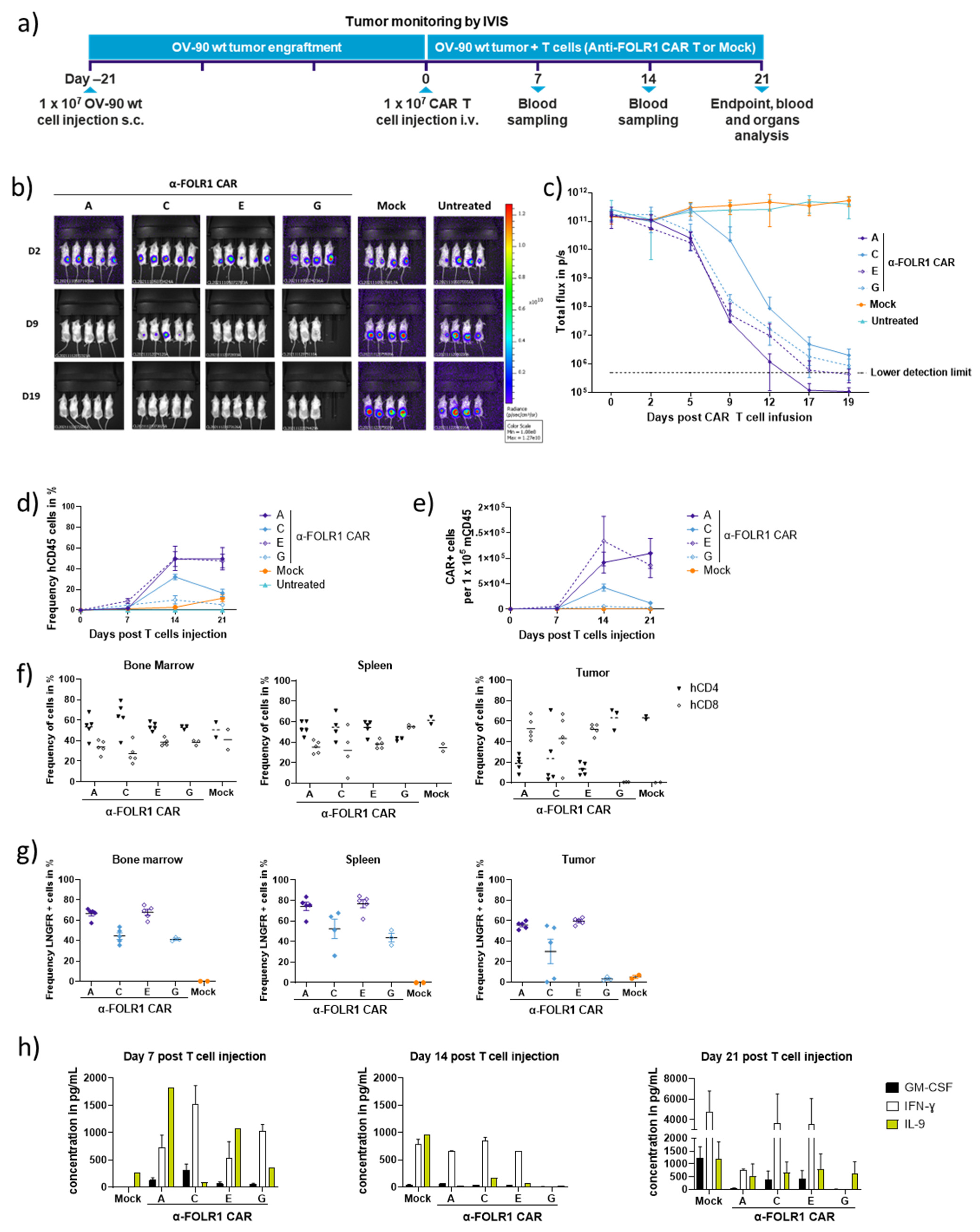
3.3. Rapid In Vivo Eradication of Advanced OV-90 Xenografts by Anti-FOLR1 CAR T Cells Depends on the CAR Design
3.4. Optimized Anti-FOLR1 CAR T Cells Are Functional In Vitro
3.5. Anti-FOLR1 CAR T Cells Infiltrate Ovarian Cancer Spheroids and Lyse Patient Tumor Cells In Vitro
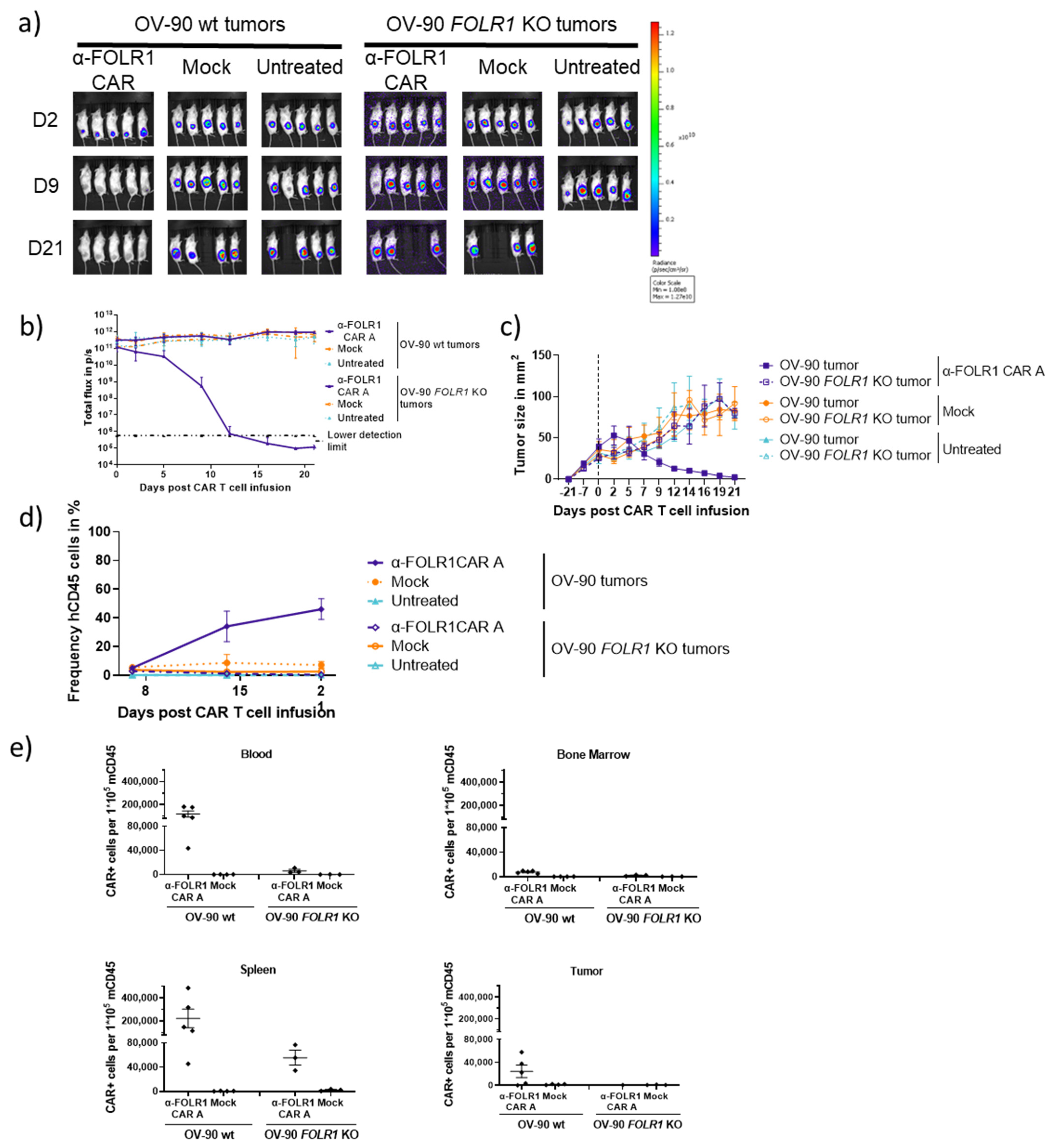
3.6. Optimized Anti-FOLR1 CAR T Cells Specifically Reduce Tumor Burden Antigen-Dependently in an Ovarian Cancer Model
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2022. CA A Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Gaona-Luviano, P.; Medina-Gaona, L.A.; Magaña-Pérez, K. Epidemiology of Ovarian Cancer. Chin. Clin. Oncol. 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Secord, A.A.; O’Malley, D.M.; Sood, A.K.; Westin, S.N.; Liu, J.F. Rationale for Combination PARP Inhibitor and Antiangiogenic Treatment in Advanced Epithelial Ovarian Cancer: A Review. Gynecol. Oncol. 2021, 162, 482–495. [Google Scholar] [CrossRef]
- Scaranti, M.; Cojocaru, E.; Banerjee, S.; Banerji, U. Exploiting the Folate Receptor α in Oncology. Nat. Rev. Clin. Oncol. 2020, 17, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Kalli, K.R.; Oberg, A.L.; Keeney, G.L.; Christianson, T.J.; Low, P.S.; Knutson, K.L.; Hartmann, L.C. Folate receptor alpha as a tumor target in epithelial ovarian cancer. Gynecol. Oncol. 2008, 108, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Parker, N.; Turk, M.J.; Westrick, E.; Lewis, J.D.; Low, P.S.; Leamon, C.P. Folate Receptor Expression in Carcinomas and Normal Tissues Determined by a Quantitative Radioligand Binding Assay. Anal. Biochem. 2005, 338, 284–293. [Google Scholar] [CrossRef]
- Kelemen, L.E. The Role of Folate Receptor α in Cancer Development, Progression and Treatment: Cause, Consequence or Innocent Bystander? Int. J. Cancer 2006, 119, 243–250. [Google Scholar] [CrossRef]
- Van Dam, G.M.; Themelis, G.; Crane, L.M.A.; Harlaar, N.J.; Pleijhuis, R.G.; Kelder, W.; Sarantopoulos, A.; de Jong, J.S.; Arts, H.J.G.; van der Zee, A.G.J.; et al. Intraoperative Tumor-Specific Fluorescence Imaging in Ovarian Cancer by Folate Receptor-α Targeting: First in-Human Results. Nat. Med. 2011, 17, 1315–1319. [Google Scholar] [CrossRef]
- Kuroki, L.; Guntupalli, S.R. Treatment of Epithelial Ovarian Cancer. BMJ 2020, 371, m3773. [Google Scholar] [CrossRef]
- Shi, H.; Guo, J.; Li, C.; Wang, Z. A Current Review of Folate Receptor Alpha as a Potential Tumor Target in Non-Small-Cell Lung Cancer. Drug Des. Dev. Ther. 2015, 9, 4989–4996. [Google Scholar] [CrossRef]
- Sato, S.; Itamochi, H. Profile of Farletuzumab and Its Potential in the Treatment of Solid Tumors. OncoTargets Ther. 2016, 9, 1181–1188. [Google Scholar] [CrossRef] [PubMed]
- Stewart, D.; Cristea, M. Antibody–Drug Conjugates for Ovarian Cancer. Curr. Opin. Obstet. Gynecol. 2019, 31, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Heo, Y.-A. Mirvetuximab Soravtansine: First Approval. Drugs 2023, 83, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Li, X.; Wang, X.; Cheng, L.; Li, Z.; Zhang, C.; Ye, Z.; Qian, Q. Current Progress in CAR-T Cell Therapy for Solid Tumors. Int. J. Biol. Sci. 2019, 15, 2548–2560. [Google Scholar] [CrossRef] [PubMed]
- Morand, S.; Devanaboyina, M.; Staats, H.; Stanbery, L.; Nemunaitis, J. Ovarian Cancer Immunotherapy and Personalized Medicine. Int. J. Mol. Sci. 2021, 22, 6532. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, M.H.; Westwood, J.A.; Parker, L.L.; Wang, G.; Eshhar, Z.; Mavroukakis, S.A.; White, D.E.; Wunderlich, J.R.; Canevari, S.; Rogers-Freezer, L.; et al. A Phase I Study on Adoptive Immunotherapy Using Gene-Modified T Cells for Ovarian Cancer. Clin. Cancer Res. 2006, 12, 6106–6115. [Google Scholar] [CrossRef] [PubMed]
- Blache, U.; Popp, G.; Dünkel, A.; Koehl, U.; Fricke, S. Potential Solutions for Manufacture of CAR T Cells in Cancer Immunotherapy. Nat. Commun. 2022, 13, 5225. [Google Scholar] [CrossRef] [PubMed]
- Ganeeva, I.; Zmievskaya, E.; Valiullina, A.; Kudriaeva, A.; Miftakhova, R.; Rybalov, A.; Bulatov, E. Recent Advances in the Development of Bioreactors for Manufacturing of Adoptive Cell Immunotherapies. Bioengineering 2022, 9, 808. [Google Scholar] [CrossRef]
- Kinkhabwala, A.; Herbel, C.; Pankratz, J.; Yushchenko, D.A.; Rüberg, S.; Praveen, P.; Reiß, S.; Rodriguez, F.C.; Schäfer, D.; Kollet, J.; et al. MACSima Imaging Cyclic Staining (MICS) Technology Reveals Combinatorial Target Pairs for CAR T Cell Treatment of Solid Tumors. Sci. Rep. 2022, 12, 1911. [Google Scholar] [CrossRef]
- Schäfer, D.; Tomiuk, S.; Küster, L.N.; Al Rawashdeh, W.; Henze, J.; Tischler-Höhle, G.; Agorku, D.J.; Brauner, J.; Linnartz, C.; Lock, D.; et al. Identification of CD318, TSPAN8 and CD66c as Target Candidates for CAR T Cell Based Immunotherapy of Pancreatic Adenocarcinoma. Nat. Commun. 2021, 12, 1453. [Google Scholar] [CrossRef]
- Patra, B.; Lateef, M.A.; Brodeur, M.N.; Fleury, H.; Carmona, E.; Péant, B.; Provencher, D.; Mes-Masson, A.-M.; Gervais, T. Carboplatin Sensitivity in Epithelial Ovarian Cancer Cell Lines: The Impact of Model Systems. PLoS ONE 2020, 15, e0244549. [Google Scholar] [CrossRef] [PubMed]
- Lock, D.; Mockel-Tenbrinck, N.; Drechsel, K.; Barth, C.; Mauer, D.; Schaser, T.; Kolbe, C.; Al Rawashdeh, W.; Brauner, J.; Hardt, O.; et al. Automated Manufacturing of Potent CD20-Directed Chimeric Antigen Receptor T Cells for Clinical Use. Hum Gene Ther. 2017, 28, 914–925. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Gires, O.; Pan, M.; Schinke, H.; Canis, M.; Baeuerle, P.A. Expression and function of epithelial cell adhesion molecule EpCAM: Where are we after 40 years? Cancer Metastasis Rev. 2020, 39, 969–987. [Google Scholar] [CrossRef]
- Fernández, M.; Faiza, J.; Vijay, C. Advances in targeting the folate receptor in the treatment/imaging of cancers. Chem. Sci. 2017, 9, 790–810. [Google Scholar] [CrossRef]
- Guedan, S.; Calderon, H.; Posey, A.D., Jr.; Maus, M.V. Engineering and Design of Chimeric Antigen Receptors. Mol. Ther. Methods Clin. Dev. 2018, 31, 145–156. [Google Scholar] [CrossRef]
- Stoiber, S.; Cadilha, B.L.; Benmebarek, M.-R.; Lesch, S.; Endres, S.; Kobold, S. Limitations in the Design of Chimeric Antigen Receptors for Cancer Therapy. Cells 2019, 8, 472. [Google Scholar] [CrossRef]
- Jamali, A.; Kapitza, L.; Schaser, T.; Johnston, I.C.D.; Buchholz, C.J.; Hartmann, J. Highly Efficient and Selective CAR-Gene Transfer Using CD4- and CD8-Targeted Lentiviral Vectors. Mol. Ther. Methods Clin. Dev. 2019, 16, 371–379. [Google Scholar] [CrossRef]
- Lynn, R.C.; Poussin, M.; Kalota, A.; Feng, Y.; Low, P.S.; Dimitrov, D.S.; Powell, D.J., Jr. Targeting of folate receptor β on acute myeloid leukemia blasts with chimeric antigen receptor-expressing T cells. Blood 2015, 125, 3466–3476. [Google Scholar] [CrossRef]
- Rojas-Zuleta, W.G.; Sanchez, E. IL-9: Function, Sources, and Detection. Methods Mol. Biol. 2017, 1585, 21–35. [Google Scholar] [CrossRef]
- Almeida-Nunes, D.L.; Mendes-Frias, A.; Silvestre, R.; Dinis-Oliveira, R.J.; Ricardo, S. Immune Tumor Microenvironment in Ovarian Cancer Ascites. Int. J. Mol. Sci. 2022, 23, 10692. [Google Scholar] [CrossRef]
- Brehm, M.A.; Kenney, L.L.; Wiles, M.V.; Low, B.E.; Tisch, R.M.; Burzenski, L.; Mueller, C.; Greiner, D.L.; Shultz, L.D. Lack of acute xenogeneic graft- versus-host disease, but retention of T-cell function following engraftment of human peripheral blood mononuclear cells in NSG mice deficient in MHC class I and II expression. FASEB J. 2019, 33, 3137–3151. [Google Scholar] [CrossRef]
- Tallent, A. FDA Gives Nod to Mirvetuximab Soravtansine. Cancer Discov. 2023, 13, 8. [Google Scholar] [CrossRef]
- Haslauer, T.; Greil, R.; Zaborsky, N.; Geisberger, R. CAR T-Cell Therapy in Hematological Malignancies. Int. J. Mol. Sci. 2021, 22, 8996. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Latchoumanin, O.; Wu, G.; Zhou, G.; Hebbard, L.; George, J.; Qiao, L. Recent Clinical Trials Utilizing Chimeric Antigen Receptor T Cells Therapies against Solid Tumors. Cancer Lett. 2017, 390, 188–200. [Google Scholar] [CrossRef] [PubMed]
- Hartnett, E.G.; Knight, J.; Radolec, M.; Buckanovich, R.J.; Edwards, R.P.; Vlad, A.M. Immunotherapy Advances for Epithelial Ovarian Cancer. Cancers 2020, 12, 3733. [Google Scholar] [CrossRef]
- Kandalaft, L.E.; Powell, D.J.; Coukos, G. A Phase I Clinical Trial of Adoptive Transfer of Folate Receptor-Alpha Redirected Autologous T Cells for Recurrent Ovarian Cancer. J. Transl. Med. 2012, 10, 157. [Google Scholar] [CrossRef] [PubMed]
- Brooks, A.R.; Harkins, R.N.; Wang, P.; Qian, H.S.; Liu, P.; Rubanyi, G.M. Transcriptional Silencing Is Associated with Extensive Methylation of the CMV Promoter Following Adenoviral Gene Delivery to Muscle. J. Gene Med. 2004, 6, 395–404. [Google Scholar] [CrossRef]
- Herbst, F.; Ball, C.R.; Tuorto, F.; Nowrouzi, A.; Wang, W.; Zavidij, O.; Dieter, S.M.; Fessler, S.; Van Der Hoeven, F.; Kloz, U.; et al. Extensive Methylation of Promoter Sequences Silences Lentiviral Transgene Expression During Stem Cell Differentiation In Vivo. Mol. Ther. 2012, 20, 1014–1021. [Google Scholar] [CrossRef]
- Huang, R.; Li, X.; He, Y.; Zhu, W.; Gao, L.; Liu, Y.; Gao, L.; Wen, Q.; Zhong, J.F.; Zhang, C.; et al. Recent Advances in CAR-T Cell Engineering. J. Hematol. Oncol. 2020, 13, 86. [Google Scholar] [CrossRef]
- Levin, A.G.; Rivière, I.; Eshhar, Z.; Sadelain, M. CAR T Cells: Building on the CD19 Paradigm. Eur. J. Immunol. 2021, 51, 2151–2163. [Google Scholar] [CrossRef]
- Zhang, H.; Snyder, K.M.; Suhoski, M.M.; Maus, M.V.; Kapoor, V.; June, C.H.; Mackall, C.L. 4-1BB Is Superior to CD28 Costimulation for Generating CD8+ Cytotoxic Lymphocytes for Adoptive Immunotherapy. J. Immunol. 2007, 179, 4910–4918. [Google Scholar] [CrossRef] [PubMed]
- Guedan, S.; Posey, A.D., Jr.; Shaw, C.; Wing, A.; Da, T.; Patel, P.R.; McGettigan, S.E.; Casado-Medrano, V.; Kawalekar, O.U.; Uribe-Herranz, M.; et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight. 2018, 3, e96976. [Google Scholar] [CrossRef] [PubMed]
- Bashour, K.T.; Gondarenko, A.; Chen, H.; Shen, K.; Liu, X.; Huse, M.; Hone, J.C.; Kam, L.C. CD28 and CD3 Have Complementary Roles in T-Cell Traction Forces. Proc. Natl. Acad. Sci. USA 2014, 111, 2241–2246. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, S.; Mastaglio, S.; Bondanza, A.; Ponzoni, M.; Sanvito, F.; Aldrighetti, L.; Radrizzani, M.; La Seta-Catamancio, S.; Provasi, E.; Mondino, A.; et al. IL-7 and IL-15 Allow the Generation of Suicide Gene–Modified Alloreactive Self-Renewing Central Memory Human T Lymphocytes. Blood 2009, 113, 1006–1015. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daigre, J.; Martinez-Osuna, M.; Bethke, M.; Steiner, L.; Dittmer, V.; Krischer, K.; Bleilevens, C.; Brauner, J.; Kopatz, J.; Grundmann, M.D.; et al. Preclinical Evaluation of Novel Folate Receptor 1-Directed CAR T Cells for Ovarian Cancer. Cancers 2024, 16, 333. https://doi.org/10.3390/cancers16020333
Daigre J, Martinez-Osuna M, Bethke M, Steiner L, Dittmer V, Krischer K, Bleilevens C, Brauner J, Kopatz J, Grundmann MD, et al. Preclinical Evaluation of Novel Folate Receptor 1-Directed CAR T Cells for Ovarian Cancer. Cancers. 2024; 16(2):333. https://doi.org/10.3390/cancers16020333
Chicago/Turabian StyleDaigre, Julie, Manuel Martinez-Osuna, Maria Bethke, Larissa Steiner, Vera Dittmer, Katrin Krischer, Cathrin Bleilevens, Janina Brauner, Jens Kopatz, Matthias David Grundmann, and et al. 2024. "Preclinical Evaluation of Novel Folate Receptor 1-Directed CAR T Cells for Ovarian Cancer" Cancers 16, no. 2: 333. https://doi.org/10.3390/cancers16020333
APA StyleDaigre, J., Martinez-Osuna, M., Bethke, M., Steiner, L., Dittmer, V., Krischer, K., Bleilevens, C., Brauner, J., Kopatz, J., Grundmann, M. D., Praveen, P., Eckardt, D., Bosio, A., & Herbel, C. (2024). Preclinical Evaluation of Novel Folate Receptor 1-Directed CAR T Cells for Ovarian Cancer. Cancers, 16(2), 333. https://doi.org/10.3390/cancers16020333