
A Novel Chimeric Oncolytic Virus Mediates a Multifaceted Cellular Immune Response in a Syngeneic B16 Melanoma Model

 and
and 
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Tumor Cell Lines and Virus
2.2. Mouse Studies
2.3. RT-qPCR
2.4. In Vitro Co-Culture
2.5. Flow Cytometry
2.6. Statistical Analysis
3. Results
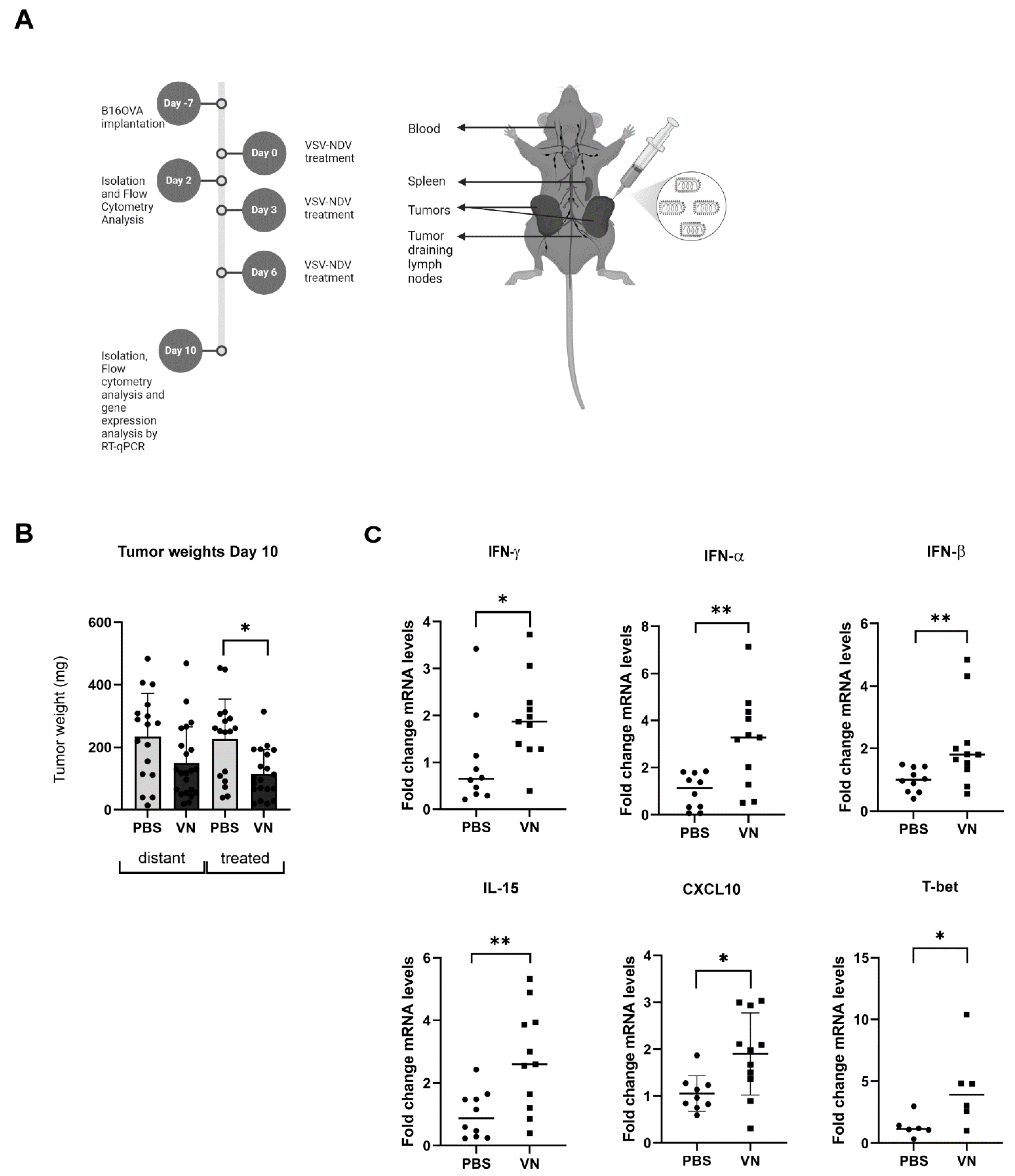
3.1. VSV-NDV Reduces the Tumor Mass and Induces the Expression of Inflammatory Markers in a Syngeneic B16 Melanoma Model
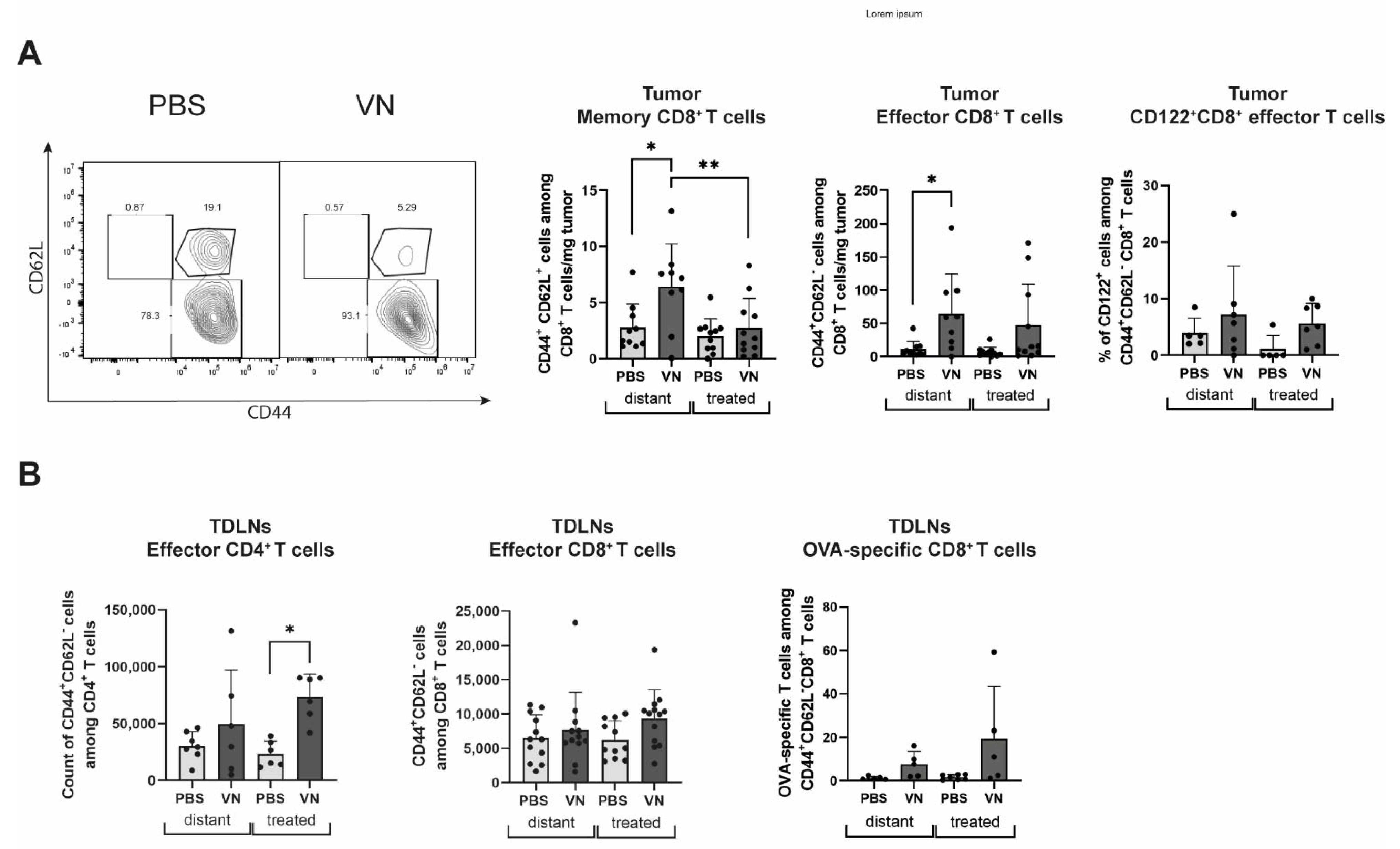
3.2. VSV-NDV Treatment Leads to an Increase in Activated Tumor-Specific CD8+ T Cells and Memory and Effector T Cells
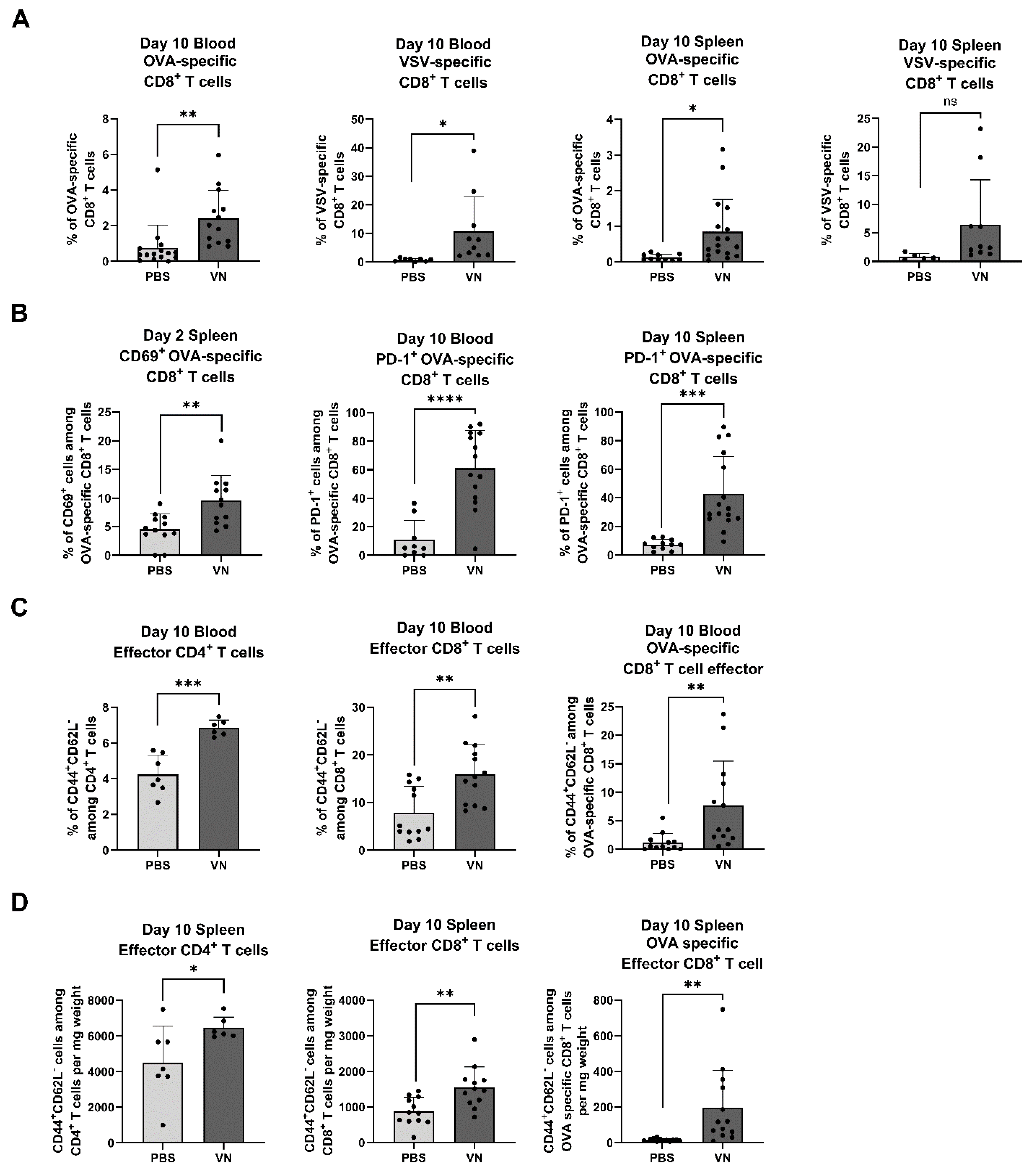
3.3. Intratumoral VSV-NDV Treatment Leads to an Enhanced Antigen-Specific T Cell Response in Peripheral Blood and Spleen
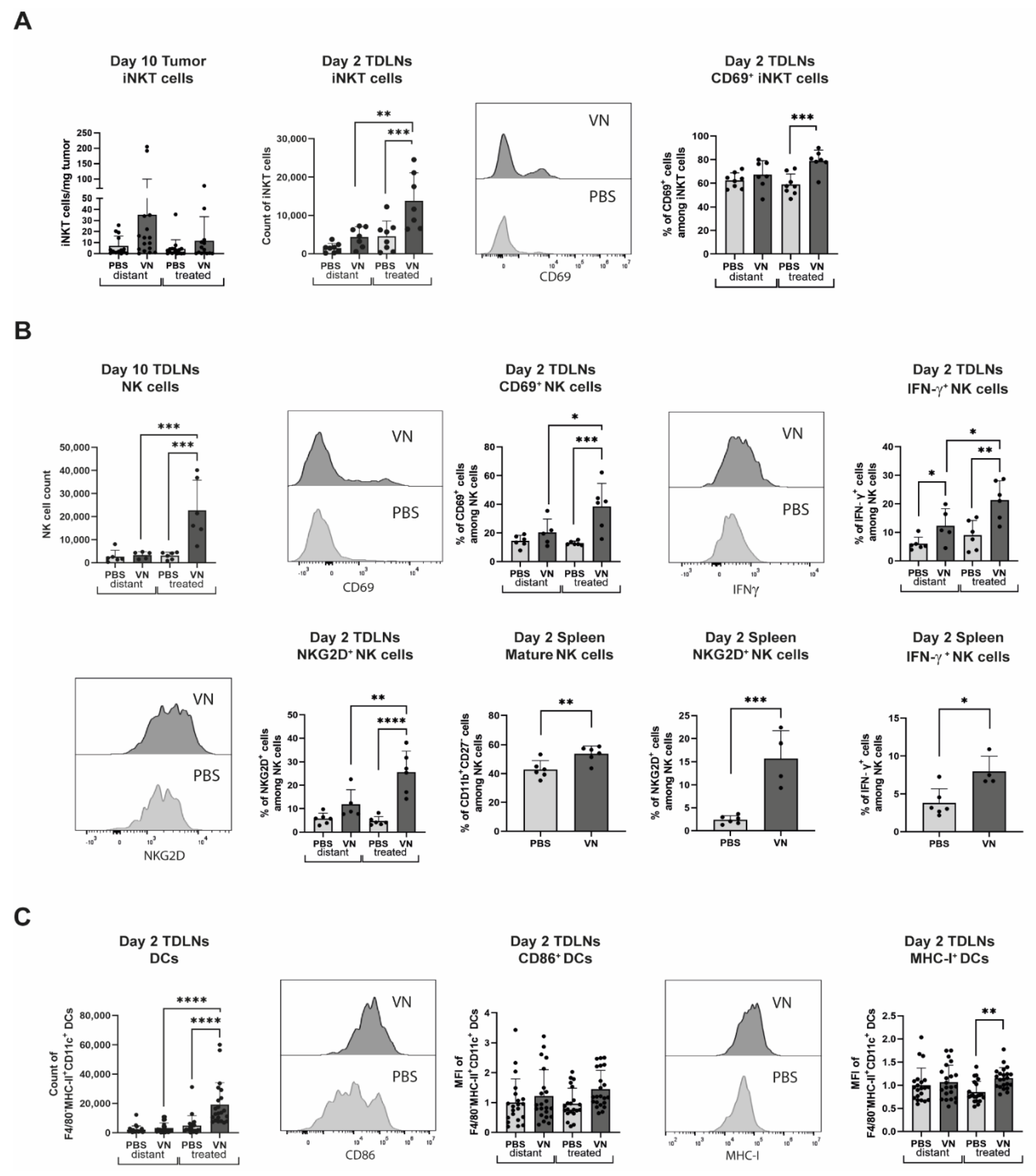
3.4. VSV-NDV Elicits an Innate Immune Response in the TDLNs, Spleen, and Tumors
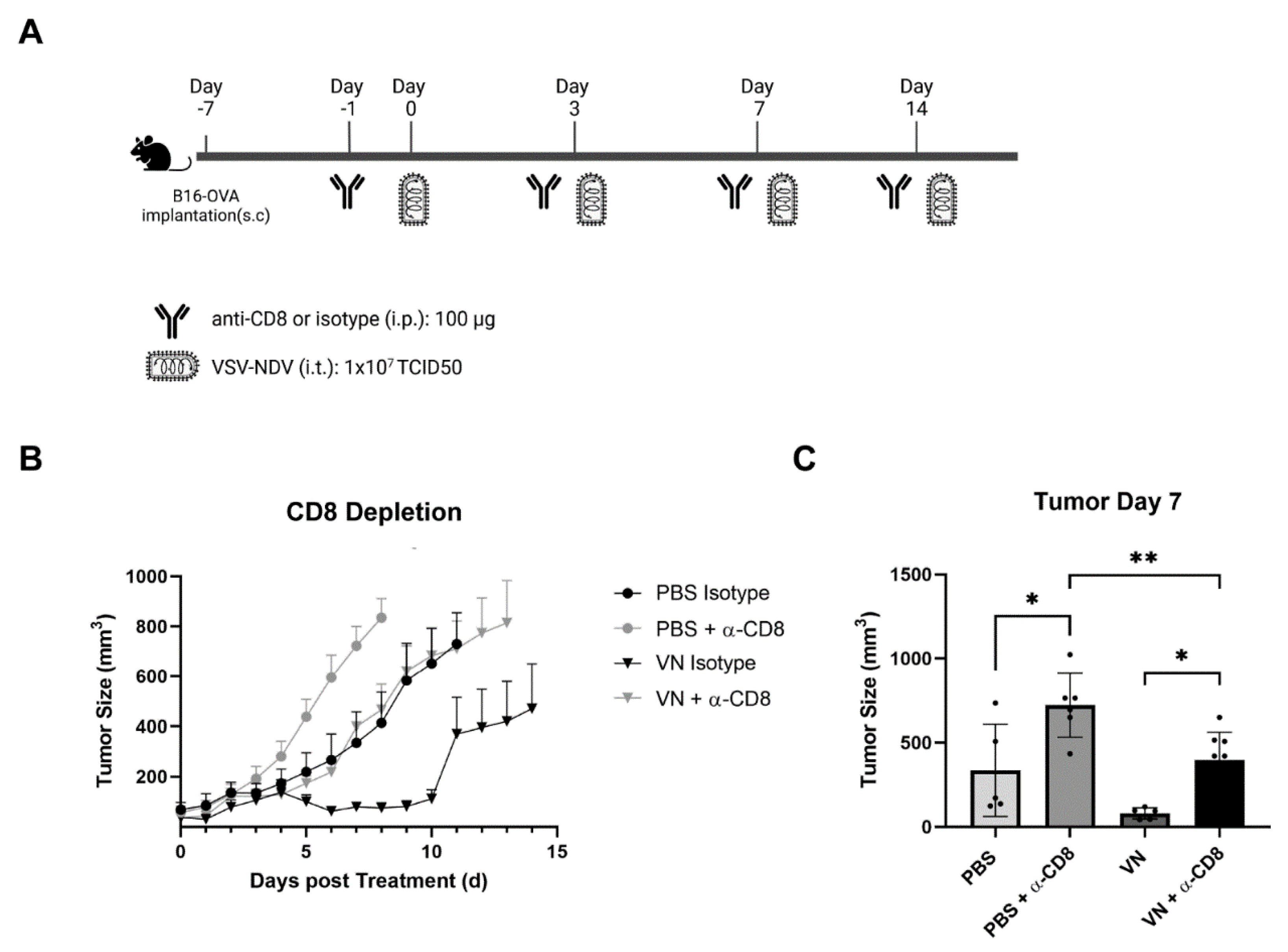
3.5. The In Vivo Depletion of CD8+ T Cells Reverses the Beneficial Effect of rVSV-NDV Treatment in the B16 OVA
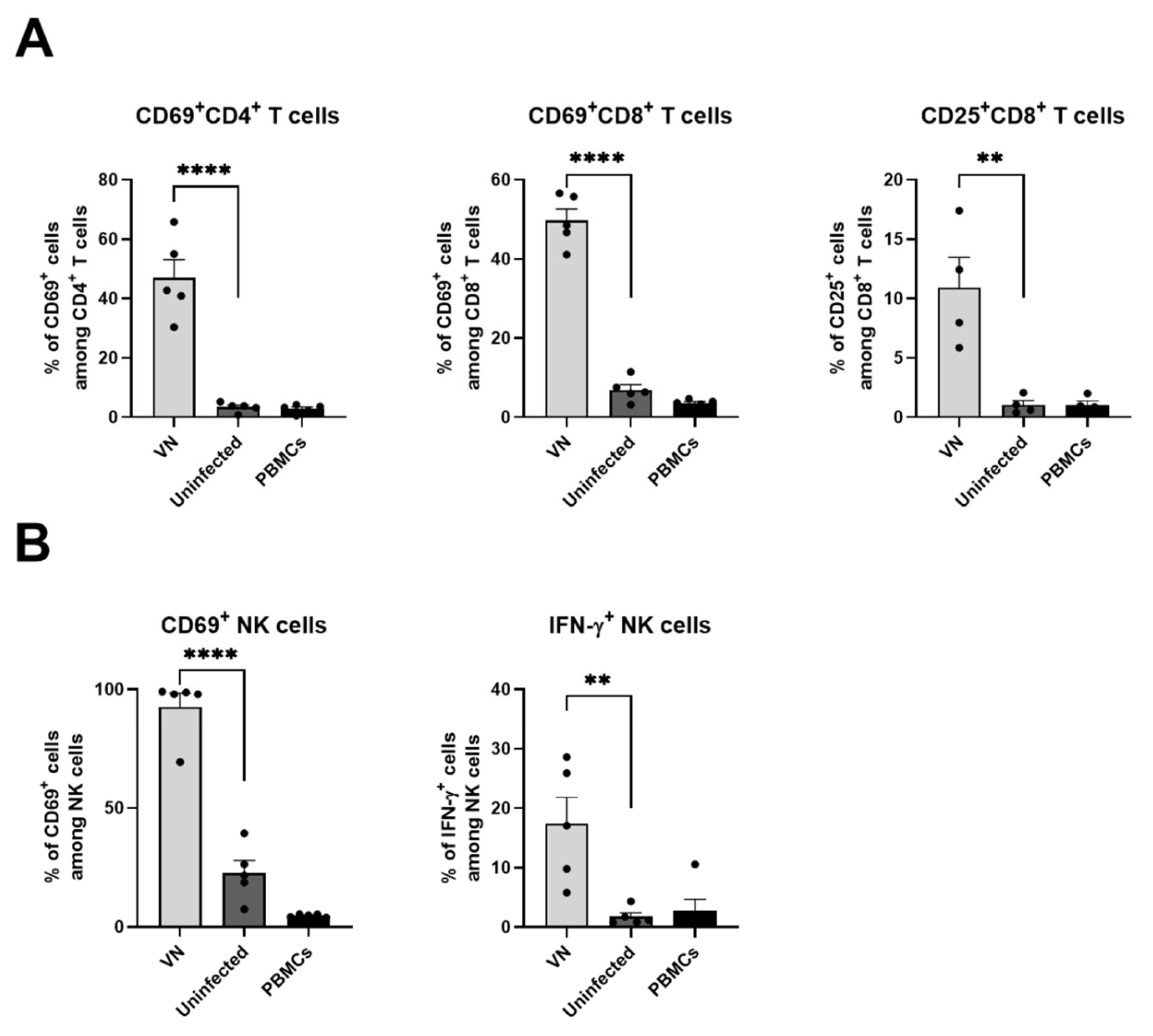
3.6. Virus Infection of Cancer Cells Leads to Activation of Human T Cells and NK Cells
4. Discussion
5. Conclusions
6. Patents
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Poh, A. First Oncolytic Viral Therapy for Melanoma. Cancer Discov. 2016, 6, 6. [Google Scholar] [CrossRef]
- Todo, T.; Ito, H.; Ino, Y.; Ohtsu, H.; Ota, Y.; Shibahara, J.; Tanaka, M. Intratumoral oncolytic herpes virus G47∆ for residual or recurrent glioblastoma: A phase 2 trial. Nat. Med. 2022, 28, 1630–1639. [Google Scholar] [CrossRef] [PubMed]
- Macedo, N.; Miller, D.M.; Haq, R.; Kaufman, H.L. Clinical landscape of oncolytic virus research in 2020. J. Immunother. Cancer 2020, 8, e001486. [Google Scholar] [CrossRef] [PubMed]
- Chaurasiya, S.; Fong, Y.; Warner, S.G. Oncolytic Virotherapy for Cancer: Clinical Experience. Biomedicines 2021, 9, 419. [Google Scholar] [CrossRef]
- Li, K.; Zhao, Y.; Hu, X.; Jiao, J.; Wang, W.; Yao, H. Advances in the clinical development of oncolytic viruses. Am. J. Transl. Res. 2022, 14, 4192–4206. [Google Scholar] [PubMed]
- Stojdl, D.F.; Lichty, B.; Knowles, S.; Marius, R.; Atkins, H.; Sonenberg, N.; Bell, J.C. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat. Med. 2000, 6, 821–825. [Google Scholar] [CrossRef]
- Xia, T.; Konno, H.; Barber, G.N. Recurrent Loss of STING Signaling in Melanoma Correlates with Susceptibility to Viral Oncolysis. Cancer Res. 2016, 76, 6747–6759. [Google Scholar] [CrossRef]
- Xia, T.; Konno, H.; Ahn, J.; Barber, G.N. Deregulation of STING Signaling in Colorectal Carcinoma Constrains DNA Damage Responses and Correlates With Tumorigenesis. Cell Rep. 2016, 14, 282–297. [Google Scholar] [CrossRef]
- Melcher, A.; Parato, K.; Rooney, C.M.; Bell, J.C. Thunder and lightning: Immunotherapy and oncolytic viruses collide. Mol. Ther. 2011, 19, 1008–1016. [Google Scholar] [CrossRef]
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef] [PubMed]
- Tough, D.F.; Borrow, P.; Sprent, J. Induction of bystander T cell proliferation by viruses and type I interferon in vivo. Science 1996, 272, 1947–1950. [Google Scholar] [CrossRef] [PubMed]
- Kolumam, G.A.; Thomas, S.; Thompson, L.J.; Sprent, J.; Murali-Krishna, K. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J. Exp. Med. 2005, 202, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Abdullahi, S.; Jakel, M.; Behrend, S.J.; Steiger, K.; Topping, G.; Krabbe, T.; Colombo, A.; Sandig, V.; Schiergens, T.S.; Thasler, W.E.; et al. A Novel Chimeric Oncolytic Virus Vector for Improved Safety and Efficacy as a Platform for the Treatment of Hepatocellular Carcinoma. J. Virol. 2018, 92, 10.1128. [Google Scholar] [CrossRef]
- Krabbe, T.; Marek, J.; Groll, T.; Steiger, K.; Schmid, R.M.; Krackhardt, A.M.; Altomonte, J. Adoptive T Cell Therapy Is Complemented by Oncolytic Virotherapy with Fusogenic VSV-NDV in Combination Treatment of Murine Melanoma. Cancers 2021, 13, 1044. [Google Scholar] [CrossRef]
- Marek, J.; Hanesch, L.; Krabbe, T.; El Khawanky, N.; Heidegger, S.; Altomonte, J. Oncolytic virotherapy with chimeric VSV-NDV synergistically supports RIG-I-dependent checkpoint inhibitor immunotherapy. Mol. Ther. Oncolytics 2023, 30, 117–131. [Google Scholar] [CrossRef]
- Manji, F.; Laister, R.C.; Kuruvilla, J. An evaluation of pembrolizumab for classical Hodgkin lymphoma. Expert Rev. Hematol. 2022, 15, 285–293. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Abu-Sbeih, H.; Ascierto, P.A.; Brufsky, J.; Cappelli, L.C.; Cortazar, F.B.; Gerber, D.E.; Hamad, L.; Hansen, E.; Johnson, D.B.; et al. Society for Immunotherapy of Cancer (SITC) clinical practice guideline on immune checkpoint inhibitor-related adverse events. J. Immunother. Cancer 2021, 9, e002435. [Google Scholar] [CrossRef]
- Okobi, T.J.; Uhomoibhi, T.O.; Akahara, D.E.; Odoma, V.A.; Sanusi, I.A.; Okobi, O.E.; Umana, I.; Okobi, E.; Okonkwo, C.C.; Harry, N.M. Immune Checkpoint Inhibitors as a Treatment Option for Bladder Cancer: Current Evidence. Cureus 2023, 15, e40031. [Google Scholar] [CrossRef]
- Wang, L.; Geng, H.; Liu, Y.; Liu, L.; Chen, Y.; Wu, F.; Liu, Z.; Ling, S.; Wang, Y.; Zhou, L. Hot and cold tumors: Immunological features and the therapeutic strategies. MedComm (2020) 2023, 4, e343. [Google Scholar] [CrossRef]
- Reschke, R.; Yu, J.; Flood, B.; Higgs, E.F.; Hatogai, K.; Gajewski, T.F. Immune cell and tumor cell-derived CXCL10 is indicative of immunotherapy response in metastatic melanoma. J. Immunother. Cancer 2021, 9, e003521. [Google Scholar] [CrossRef] [PubMed]
- Au, K.K.; Peterson, N.; Truesdell, P.; Reid-Schachter, G.; Khalaj, K.; Ren, R.; Francis, J.A.; Graham, C.H.; Craig, A.W.; Koti, M. CXCL10 alters the tumour immune microenvironment and disease progression in a syngeneic murine model of high-grade serous ovarian cancer. Gynecol. Oncol. 2017, 145, 436–445. [Google Scholar] [CrossRef] [PubMed]
- Engeland, C.E.; Grossardt, C.; Veinalde, R.; Bossow, S.; Lutz, D.; Kaufmann, J.K.; Shevchenko, I.; Umansky, V.; Nettelbeck, D.M.; Weichert, W.; et al. CTLA-4 and PD-L1 checkpoint blockade enhances oncolytic measles virus therapy. Mol. Ther. 2014, 22, 1949–1959. [Google Scholar] [CrossRef] [PubMed]
- Wongthida, P.; Diaz, R.M.; Galivo, F.; Kottke, T.; Thompson, J.; Melcher, A.; Vile, R. VSV oncolytic virotherapy in the B16 model depends upon intact MyD88 signaling. Mol. Ther. 2011, 19, 150–158. [Google Scholar] [CrossRef]
- Koske, I.; Rossler, A.; Pipperger, L.; Petersson, M.; Barnstorf, I.; Kimpel, J.; Tripp, C.H.; Stoitzner, P.; Banki, Z.; von Laer, D. Oncolytic virotherapy enhances the efficacy of a cancer vaccine by modulating the tumor microenvironment. Int. J. Cancer 2019, 145, 1958–1969. [Google Scholar] [CrossRef]
- Leveille, S.; Goulet, M.L.; Lichty, B.D.; Hiscott, J. Vesicular stomatitis virus oncolytic treatment interferes with tumor-associated dendritic cell functions and abrogates tumor antigen presentation. J. Virol. 2011, 85, 12160–12169. [Google Scholar] [CrossRef]
- Chen, L.; Zhou, C.; Chen, Q.; Shang, J.; Liu, Z.; Guo, Y.; Li, C.; Wang, H.; Ye, Q.; Li, X.; et al. Oncolytic Zika virus promotes intratumoral T cell infiltration and improves immunotherapy efficacy in glioblastoma. Mol. Ther. Oncolytics 2022, 24, 522–534. [Google Scholar] [CrossRef]
- Yamashita, M.; Tasaki, M.; Murakami, R.; Arai, Y.; Nakamura, T.; Nakao, S. Oncolytic vaccinia virus induces a novel phenotype of CD8(+) effector T cells characterized by high ICOS expression. Mol. Ther. Oncolytics 2021, 20, 422–432. [Google Scholar] [CrossRef]
- Liikanen, I.; Basnet, S.; Quixabeira, D.C.A.; Taipale, K.; Hemminki, O.; Oksanen, M.; Kankainen, M.; Juhila, J.; Kanerva, A.; Joensuu, T.; et al. Oncolytic adenovirus decreases the proportion of TIM-3(+) subset of tumor-infiltrating CD8(+) T cells with correlation to improved survival in patients with cancer. J. Immunother. Cancer 2022, 10, e003490. [Google Scholar] [CrossRef]
- Nelson, A.; McMullen, N.; Gebremeskel, S.; De Antueno, R.; Mackenzie, D.; Duncan, R.; Johnston, B. Fusogenic vesicular stomatitis virus combined with natural killer T cell immunotherapy controls metastatic breast cancer. Breast Cancer Res. 2024, 26, 78. [Google Scholar] [CrossRef]
- Krabbe, T.; Altomonte, J. Fusogenic Viruses in Oncolytic Immunotherapy. Cancers 2018, 10, 216. [Google Scholar] [CrossRef] [PubMed]
- Webb, M.J.; Sangsuwannukul, T.; van Vloten, J.; Evgin, L.; Kendall, B.; Tonne, J.; Thompson, J.; Metko, M.; Moore, M.; Chiriboga Yerovi, M.P.; et al. Expression of tumor antigens within an oncolytic virus enhances the anti-tumor T cell response. Nat. Commun. 2024, 15, 5442. [Google Scholar] [CrossRef] [PubMed]
- Bhat, R.; Dempe, S.; Dinsart, C.; Rommelaere, J. Enhancement of NK cell antitumor responses using an oncolytic parvovirus. Int. J. Cancer 2011, 128, 908–919. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Tai, L.H.; Ilkow, C.S.; Alkayyal, A.A.; Ananth, A.A.; de Souza, C.T.; Wang, J.; Sahi, S.; Ly, L.; Lefebvre, C.; et al. Maraba MG1 virus enhances natural killer cell function via conventional dendritic cells to reduce postoperative metastatic disease. Mol. Ther. 2014, 22, 1320–1332. [Google Scholar] [CrossRef]
- Tai, L.H.; de Souza, C.T.; Belanger, S.; Ly, L.; Alkayyal, A.A.; Zhang, J.; Rintoul, J.L.; Ananth, A.A.; Lam, T.; Breitbach, C.J.; et al. Preventing postoperative metastatic disease by inhibiting surgery-induced dysfunction in natural killer cells. Cancer Res. 2013, 73, 97–107. [Google Scholar] [CrossRef]
- Ogbomo, H.; Zemp, F.J.; Lun, X.; Zhang, J.; Stack, D.; Rahman, M.M.; McFadden, G.; Mody, C.H.; Forsyth, P.A. Myxoma virus infection promotes NK lysis of malignant gliomas in vitro and in vivo. PLoS ONE 2013, 8, e66825. [Google Scholar] [CrossRef]
- Gujar, S.A.; Pan, D.A.; Marcato, P.; Garant, K.A.; Lee, P.W. Oncolytic virus-initiated protective immunity against prostate cancer. Mol. Ther. 2011, 19, 797–804. [Google Scholar] [CrossRef]
- Rintoul, J.L.; Lemay, C.G.; Tai, L.H.; Stanford, M.M.; Falls, T.J.; de Souza, C.T.; Bridle, B.W.; Daneshmand, M.; Ohashi, P.S.; Wan, Y.; et al. ORFV: A novel oncolytic and immune stimulating parapoxvirus therapeutic. Mol. Ther. 2012, 20, 1148–1157. [Google Scholar] [CrossRef]
- Bottcher, J.P.; Bonavita, E.; Chakravarty, P.; Blees, H.; Cabeza-Cabrerizo, M.; Sammicheli, S.; Rogers, N.C.; Sahai, E.; Zelenay, S.; Reis e Sousa, C. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 2018, 172, 1022–1037.e14. [Google Scholar] [CrossRef]
- Le Bon, A.; Etchart, N.; Rossmann, C.; Ashton, M.; Hou, S.; Gewert, D.; Borrow, P.; Tough, D.F. Cross-priming of CD8+ T cells stimulated by virus-induced type I interferon. Nat. Immunol. 2003, 4, 1009–1015. [Google Scholar] [CrossRef]
- Gauvrit, A.; Brandler, S.; Sapede-Peroz, C.; Boisgerault, N.; Tangy, F.; Gregoire, M. Measles virus induces oncolysis of mesothelioma cells and allows dendritic cells to cross-prime tumor-specific CD8 response. Cancer Res. 2008, 68, 4882–4892. [Google Scholar] [CrossRef] [PubMed]
- Schulz, O.; Diebold, S.S.; Chen, M.; Naslund, T.I.; Nolte, M.A.; Alexopoulou, L.; Azuma, Y.T.; Flavell, R.A.; Liljestrom, P.; Reis e Sousa, C. Toll-like receptor 3 promotes cross-priming to virus-infected cells. Nature 2005, 433, 887–892. [Google Scholar] [CrossRef] [PubMed]
- Prestwich, R.J.; Errington, F.; Ilett, E.J.; Morgan, R.S.; Scott, K.J.; Kottke, T.; Thompson, J.; Morrison, E.E.; Harrington, K.J.; Pandha, H.S.; et al. Tumor infection by oncolytic reovirus primes adaptive antitumor immunity. Clin. Cancer Res. 2008, 14, 7358–7366. [Google Scholar] [CrossRef] [PubMed]
- Franks, M.L.; An, J.H.; Leavenworth, J.W. The Role of Natural Killer Cells in Oncolytic Virotherapy: Friends or Foes? Vaccines 2024, 12, 721. [Google Scholar] [CrossRef] [PubMed]
- Aranda, F.; Llopiz, D.; Diaz-Valdes, N.; Riezu-Boj, J.I.; Bezunartea, J.; Ruiz, M.; Martinez, M.; Durantez, M.; Mansilla, C.; Prieto, J.; et al. Adjuvant combination and antigen targeting as a strategy to induce polyfunctional and high-avidity T-cell responses against poorly immunogenic tumors. Cancer Res. 2011, 71, 3214–3224. [Google Scholar] [CrossRef]
- Uche, I.K.; Stanfield, B.; Rudd, J.; Kousoulas, K.; Rider, P.J. Abstract 3570: Prospects for personalized cancer vaccines: The oncolytic and immunotherapeutic HSV-1(VC2) virus expressing an ovalbumin (OVA) antigen elicits robust anti-OVA immune responses. Cancer Res. 2022, 82, 3570. [Google Scholar] [CrossRef]
- Hirano, N.; Butler, M.O.; Xia, Z.; Berezovskaya, A.; Murray, A.P.; Ansen, S.; Nadler, L.M. Efficient presentation of naturally processed HLA class I peptides by artificial antigen-presenting cells for the generation of effective antitumor responses. Clin. Cancer Res. 2006, 12, 2967–2975. [Google Scholar] [CrossRef]
- Henare, K.; Yung, R.; Dunbar, P.R.; Print, C.G.; Ching, L.-M. Abstract A19: Characterizing the tumor stroma of B16 melanoma at different sites. Cancer Res. 2015, 75, A19. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Forward (5′-3′) | Reverse (5′-3′) |
|---|---|---|
| GAPDH | GCCTTCTCCATGGTGGTGAA | GCACAGTCAAGGCCGAGAAT |
| IFN-γ | TCAAGTGGCATAGATGTGGAAGAA | TGGCTCTGCAGGATTTTCATG |
| IL-15 | CATCCATCTCGTGCTACTTGTGTT | TGGCTCTGCAGGATTTTCATG |
| T-bet | TAAGCAAGGACGGCGAATGTT | TGCCTTCTGCCTTTCCACAC |
| IFN-α | ATGGCTAGRCTCTGTGCTTTCCT | GTCTCGTCTTYAGACCTCTCGGGA |
| IFN-β | GGAGATGACGGAGAAGATGC | CCCAGTGCTGGAGAAATTGT |
| CXCL10 | CCA AGT GCT GCC GTC ATT TTC | GGC TCG CAG GGA TGA TTT CAA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Glauß, S.; Neumeyer, V.; Hanesch, L.; Marek, J.; Hartmann, N.; Wiedemann, G.M.; Altomonte, J. A Novel Chimeric Oncolytic Virus Mediates a Multifaceted Cellular Immune Response in a Syngeneic B16 Melanoma Model. Cancers 2024, 16, 3405. https://doi.org/10.3390/cancers16193405
Glauß S, Neumeyer V, Hanesch L, Marek J, Hartmann N, Wiedemann GM, Altomonte J. A Novel Chimeric Oncolytic Virus Mediates a Multifaceted Cellular Immune Response in a Syngeneic B16 Melanoma Model. Cancers. 2024; 16(19):3405. https://doi.org/10.3390/cancers16193405
Chicago/Turabian StyleGlauß, Sonja, Victoria Neumeyer, Lorenz Hanesch, Janina Marek, Nina Hartmann, Gabriela M. Wiedemann, and Jennifer Altomonte. 2024. "A Novel Chimeric Oncolytic Virus Mediates a Multifaceted Cellular Immune Response in a Syngeneic B16 Melanoma Model" Cancers 16, no. 19: 3405. https://doi.org/10.3390/cancers16193405
APA StyleGlauß, S., Neumeyer, V., Hanesch, L., Marek, J., Hartmann, N., Wiedemann, G. M., & Altomonte, J. (2024). A Novel Chimeric Oncolytic Virus Mediates a Multifaceted Cellular Immune Response in a Syngeneic B16 Melanoma Model. Cancers, 16(19), 3405. https://doi.org/10.3390/cancers16193405