
FL118 Enhances Therapeutic Efficacy in Colorectal Cancer by Inhibiting the Homologous Recombination Repair Pathway through Survivin–RAD51 Downregulation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Compounds and Antibodies
2.2. Cell Culture and SN38-Resistant LOVO Cell Establishment
2.3. Cell Viability Assay and Normal Cell Toxicity
2.4. Western Blot
2.5. Flow Cytometry Analysis
2.6. Comet Assay
2.7. Immunofluorescence and Foci Analysis
2.8. qRT-PCR
2.9. Drug Combination Analysis
2.10. Expression and RNA-Seq Data Analysis
2.11. Transfection
2.12. Xenografts
2.13. Immunohistochemistry
2.14. Statistical Analysis
3. Results
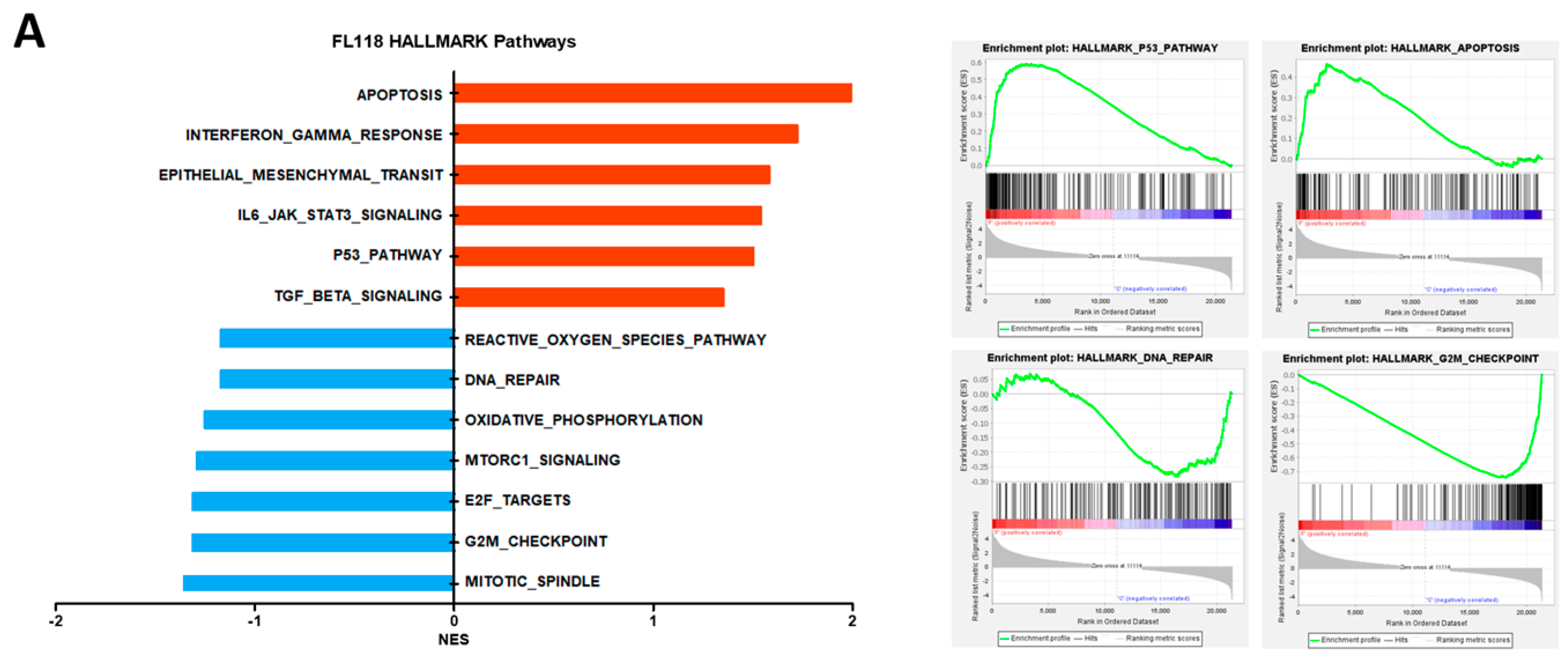
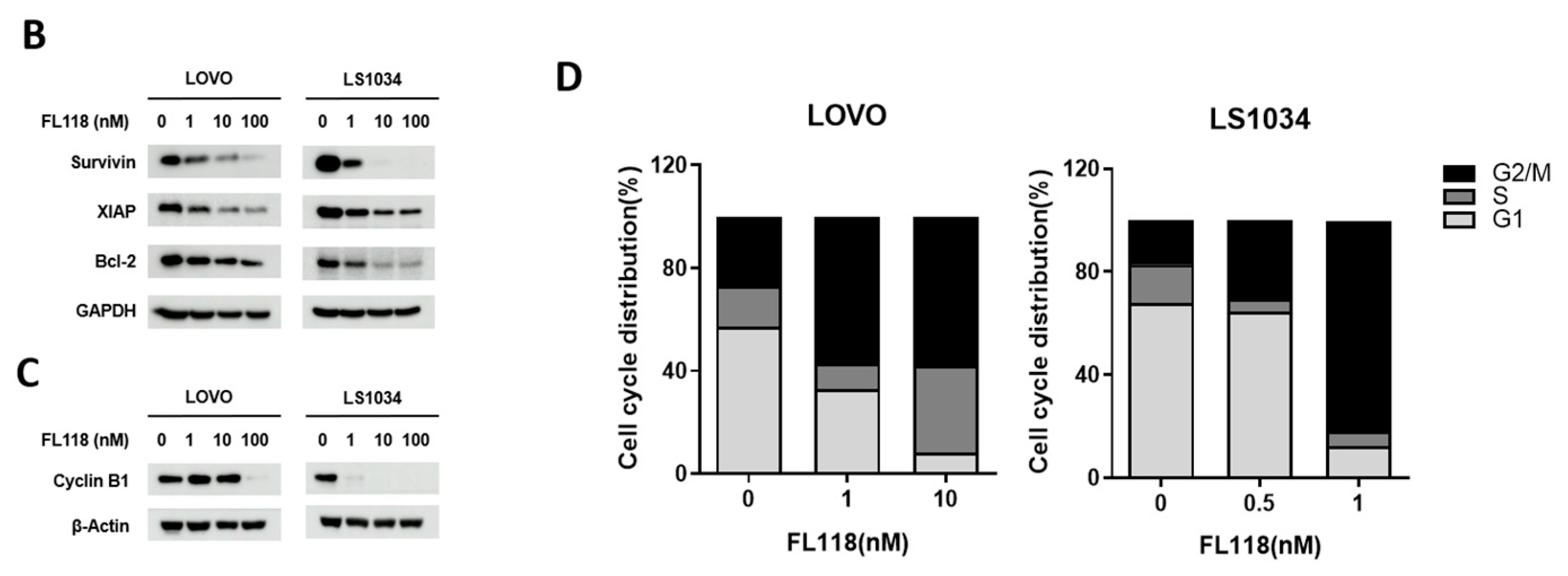
3.1. FL118 Triggered Apoptosis and G2/M Arrest by Suppressing Apoptosis Protein and Cell Cycle Protein
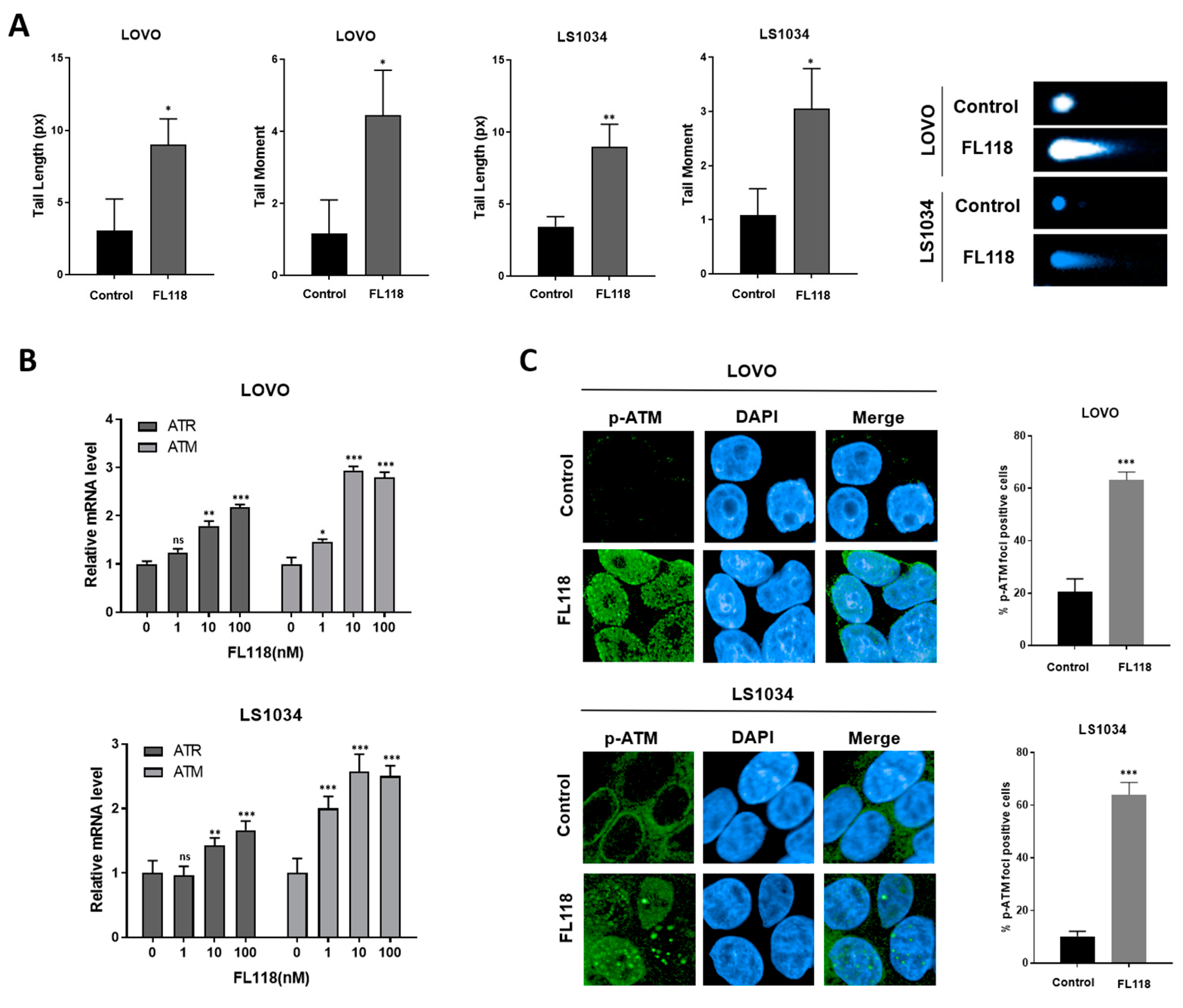
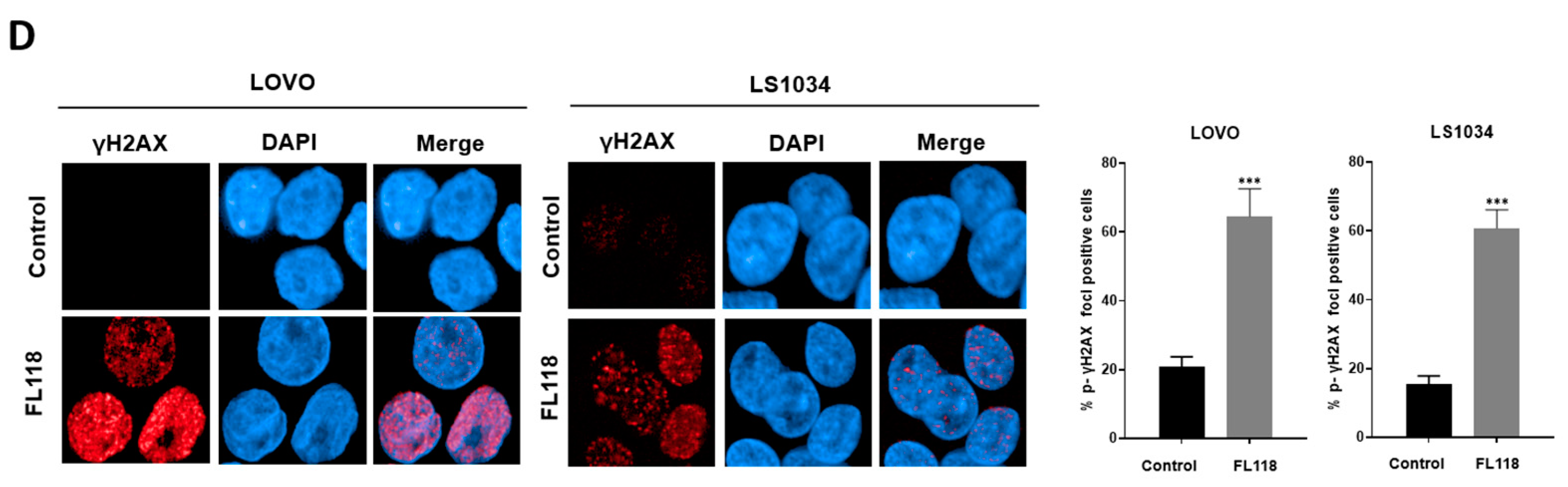
3.2. FL118 Forms DSBs and Elevates Levels of Phosphorylated ATM and Phosphorylated γH2AX
3.3. FL118 Downregulates the Homologous Recombination (HR) Repair Gene RAD51
3.4. FL118-Mediated Survivin Inhibition Downregulated RAD51
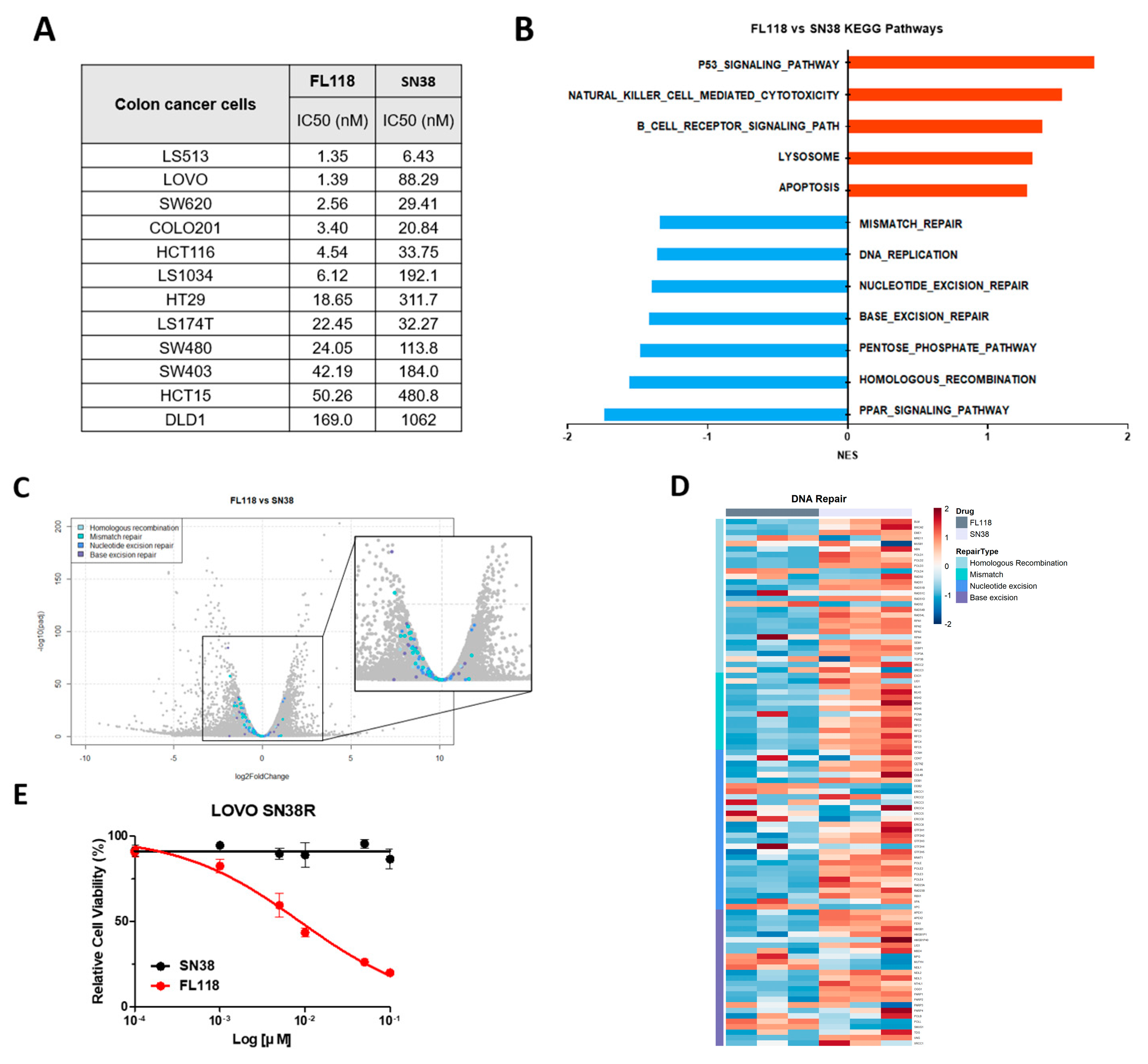
3.5. FL118 Overcomes SN38 Resistance by Effectively Blocking the DNA Repair Mechanism
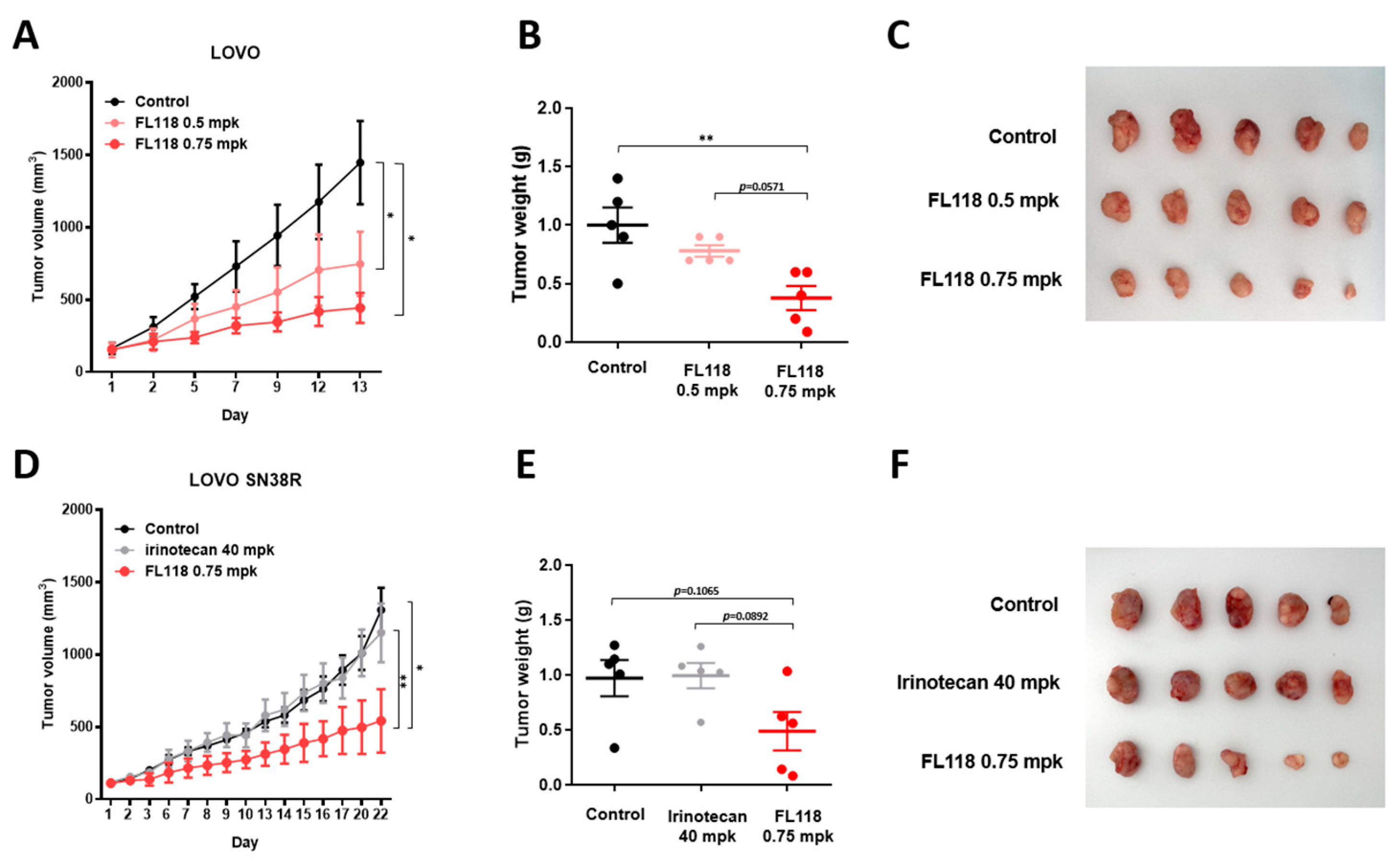
3.6. FL118 Suppresses Tumor Growth in Both LOVO and LOVO SN38R Cell-Derived Xenograft Models
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Marques, R.P.; Duarte, G.S.; Sterrantino, C.; Pais, H.L.; Quintela, A.; Martins, A.P.; Costa, J. Triplet (FOLFOXIRI) versus doublet (FOLFOX or FOLFIRI) backbone chemotherapy as first-line treatment of metastatic colorectal cancer: A systematic review and meta-analysis. Crit. Rev. Oncol. Hematol. 2017, 118, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Marques, R.P.; Godinho, A.R.; Heudtlass, P.; Pais, H.L.; Quintela, A.; Martins, A.P. Cetuximab versus bevacizumab in metastatic colorectal cancer: A comparative effectiveness study. J. Cancer Res. Clin. Oncol. 2020, 146, 1321–1334. [Google Scholar] [CrossRef]
- Panczyk, M. Pharmacogenetics research on chemotherapy resistance in colorectal cancer over the last 20 years. World J. Gastroenterol. 2014, 20, 9775–9827. [Google Scholar] [CrossRef] [PubMed]
- Martino, E.; Della Volpe, S.; Terribile, E.; Benetti, E.; Sakaj, M.; Centamore, A.; Sala, A.; Collina, S. The long story of camptothecin: From traditional medicine to drugs. Bioorg. Med. Chem. Lett. 2017, 27, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Nakatomi, K.; Yoshikawa, M.; Oka, M.; Ikegamib, Y.; Hayasakab, S.; Sanob, K.; Shiozawaa, K.; Kawabataa, S.; Sodaa, H.; Ishikawad, T.; et al. Transport of 7-ethyl-10-hydroxycamptothecin (SN-38) by breast cancer resistance protein ABCG2 in human lung cancer cells. Biochem. Biophys. Res. Comm. 2001, 288, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Ling, X.; Liu, X.; Zhong, K.; Smith, N.; Prey, J.; Li, F. FL118, a novel camptothecin analogue, overcomes irinotecan and topotecan resistance in human tumor xenograft models. Am. J. Transl. Res. 2015, 7, 1765–1781. [Google Scholar] [PubMed]
- Li, F. Anticancer drug FL118 is more than a survivin inhibitor: Where is the Achilles’ heel of cancer? Am. J. Cancer Res. 2014, 4, 304–311. [Google Scholar]
- Han, J.-Y.; Lim, H.-S.; Shin, E.S.; Yoo, Y.-K.; Park, Y.H.; Lee, J.-E.; Jang, I.-J.; Lee, D.H.; Lee, J.S. Comprehensive analysis of UGT1A polymorphisms predictive for pharmacokinetics and treatment outcome in patients with non-small-cell lung cancer treated with irinotecan and cisplatin. J. Clin. Oncol. 2006, 24, 2237–2244. [Google Scholar] [CrossRef]
- Ling, X.; Cao, S.; Cheng, Q.; Keefe, J.T.; Rustum, Y.M.; Li, F. A novel small molecule FL118 that selectively inhibits survivin, Mcl-1, XIAP and cIAP2 in a p53-independent manner, shows superior antitumor activity. PLoS ONE 2012, 7, e45571. [Google Scholar] [CrossRef]
- Rose, M.; Burgess, J.T.; O’byrne, K.; Richard, D.J.; Bolderson, E. PARP inhibitors: Clinical relevance, mechanisms of action and tumor resistance. Front. Cell Dev. Biol. 2020, 8, 564601. [Google Scholar] [CrossRef]
- Véquaud, E.; Desplanques, G.; Jézéquel, P.; Juin, P.; Barillé-Nion, S. Survivin contributes to DNA repair by homologous recombination in breast cancer cells. Breast Cancer Res. Treat. 2016, 155, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Qin, Q.; Cheng, H.; Lu, J.; Zhan, L.; Zheng, J.; Cai, J.; Yang, X.; Xu, L.; Zhu, H.; Zhang, C.; et al. Small-molecule survivin inhibitor YM155 enhances radiosensitization in esophageal squamous cell carcinoma by the abrogation of G2 checkpoint and suppression of homologous recombination repair. J. Hematol. Oncol. 2014, 7, 62. [Google Scholar] [CrossRef] [PubMed]
- Lavin, M.F.; Kozlov, S. ATM activation and DNA damage response. Cell Cycle 2007, 6, 931–942. [Google Scholar] [CrossRef]
- Podhorecka, M.; Skladanowski, A.; Bozko, P. H2AX phosphorylation: Its role in DNA damage response and cancer therapy. J. Nucleic Acids 2010, 2010, 920161. [Google Scholar] [CrossRef]
- Yarchoan, M.; Myzak, M.C.; Johnson, B.A.; De Jesus-Acosta, A.; Le, D.T.; Jaffee, E.M.; Azad, N.S.; Donehower, R.C.; Zheng, L.; Oberstein, P.E.; et al. Olaparib in combination with irinotecan, cisplatin, and mitomycin C in patients with advanced pancreatic cancer. Oncotarget 2017, 8, 44073–44081. [Google Scholar] [CrossRef]
- Augustine, T.; Maitra, R.; Zhang, J.; Nayak, J.; Goel, S. Sensitization of colorectal cancer to irinotecan therapy by PARP inhibitor rucaparib. Investig. New Drugs 2019, 37, 948–960. [Google Scholar] [CrossRef]
- Williams, S.M.G.; Kuznicki, A.M.; Andrade, P.; Dolinski, B.M.; Elbi, C.; O’hagan, R.C.; Toniatti, C. Treatment with the PARP inhibitor, niraparib, sensitizes colorectal cancer cell lines to irinotecan regardless of MSI/MSS status. Cancer Cell Int. 2015, 15, 14. [Google Scholar] [CrossRef] [PubMed]
- Davidson, D.; Wang, Y.; Aloyz, R.; Panasci, L. The PARP inhibitor ABT-888 synergizes irinotecan treatment of colon cancer cell lines. Investig. New Drugs 2013, 31, 461–468. [Google Scholar] [CrossRef]
- Tahara, M.; Inoue, T.; Sato, F.; Miyakura, Y.; Horie, H.; Yasuda, Y.; Fujii, H.; Kotake, K.; Sugano, K. The use of Olaparib (AZD2281) potentiates SN-38 cytotoxicity in colon cancer cells by indirect inhibition of Rad51-mediated repair of DNA double-strand breaks. Mol. Cancer Ther. 2014, 13, 1170–1180. [Google Scholar] [CrossRef]
- Griguolo, G.; Dieci, M.V.; Guarneri, V.; Conte, P. Olaparib for the treatment of breast cancer. Expert Rev. Anticancer. Ther. 2018, 18, 519–530. [Google Scholar] [CrossRef]
- Bixel, K.; Hays, J.L. Olaparib in the management of ovarian cancer. Pharmacogenomics Pers. Med. 2015, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Li, F. Discovery of survivin inhibitors and beyond: FL118 as a proof of concept. Int. Rev. Cell Mol. Biol. 2013, 305, 217–252. [Google Scholar] [CrossRef] [PubMed]
- Kanwar, J.R.; Kamalapuram, S.K.; Kanwar, R.K. Targeting survivin in cancer: The cell-signalling perspective. Drug Discov. Today 2011, 16, 485–494. [Google Scholar] [CrossRef]
- Sforza, V.; Martinelli, E.; Ciardiello, F.; Gambardella, V.; Napolitano, S.; Martini, G.; della Corte, C.; Cardone, C.; Ferrara, M.L.; Reginelli, A.; et al. Mechanisms of resistance to anti-epidermal growth factor receptor inhibitors in metastatic colorectal cancer. World J. Gastroenterol. 2016, 22, 6345–6361. [Google Scholar] [CrossRef]
- Xiao, Q.; Xiao, J.; Liu, J.; Liu, J.; Shu, G.; Yin, G. Metformin suppresses the growth of colorectal cancer by targeting INHBA to inhibit TGF-β/PI3K/AKT signaling transduction. Cell Death Dis. 2022, 13, 202. [Google Scholar] [CrossRef]
- Garcia-Carbonero, R.; Supko, J.G. Current perspectives on the clinical experience, pharmacology, and continued development of the camptothecins. Clin. Cancer Res. 2002, 8, 641–661. [Google Scholar]
- Yaghoubi, S.; Karimi, M.H.; Lotfinia, M.; Gharibi, T.; Mahi-Birjand, M.; Kavi, E.; Hosseini, F.; Sepehr, K.S.; Khatami, M.; Bagheri, N.; et al. Potential drugs used in the antibody–drug conjugate (ADC) architecture for cancer therapy. J. Cell Physiol. 2020, 235, 31–64. [Google Scholar] [CrossRef] [PubMed]
- Starodub, A.N.; Ocean, A.J.; Shah, M.A.; Guarino, M.J.; Picozzi, V.J.; Vahdat, L.T.; Thomas, S.S.; Govindan, S.V.; Maliakal, P.P.; Wegener, W.A.; et al. First-in-human trial of a novel anti-Trop-2 antibody-SN-38 conjugate, sacituzumab govitecan, for the treatment of diverse metastatic solid tumors. Clin. Cancer Res. 2015, 21, 3870–3878. [Google Scholar] [CrossRef]
- Nakada, T.; Masuda, T.; Naito, H.; Yoshida, M.; Ashida, S.; Morita, K.; Miyazaki, H.; Kasuya, Y.; Ogitani, Y.; Yamaguchi, J.; et al. Novel antibody drug conjugates containing exatecan derivative-based cytotoxic payloads. Bioorg. Med. Chem. Lett. 2016, 26, 1542–1545. [Google Scholar] [CrossRef]
- Buck, S.A.; Koolen, S.L.; Mathijssen, R.H.; de Wit, R.; van Soest, R.J. Cross-resistance and drug sequence in prostate cancer. Drug Resist. Updat. 2021, 56, 100761. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.; Jeong, Y.; Shin, Y.M.; Kim, S.E.; Shin, S.J. FL118 Enhances Therapeutic Efficacy in Colorectal Cancer by Inhibiting the Homologous Recombination Repair Pathway through Survivin–RAD51 Downregulation. Cancers 2024, 16, 3385. https://doi.org/10.3390/cancers16193385
Kim J, Jeong Y, Shin YM, Kim SE, Shin SJ. FL118 Enhances Therapeutic Efficacy in Colorectal Cancer by Inhibiting the Homologous Recombination Repair Pathway through Survivin–RAD51 Downregulation. Cancers. 2024; 16(19):3385. https://doi.org/10.3390/cancers16193385
Chicago/Turabian StyleKim, Jungyoun, Yeyeong Jeong, You Me Shin, Sung Eun Kim, and Sang Joon Shin. 2024. "FL118 Enhances Therapeutic Efficacy in Colorectal Cancer by Inhibiting the Homologous Recombination Repair Pathway through Survivin–RAD51 Downregulation" Cancers 16, no. 19: 3385. https://doi.org/10.3390/cancers16193385
APA StyleKim, J., Jeong, Y., Shin, Y. M., Kim, S. E., & Shin, S. J. (2024). FL118 Enhances Therapeutic Efficacy in Colorectal Cancer by Inhibiting the Homologous Recombination Repair Pathway through Survivin–RAD51 Downregulation. Cancers, 16(19), 3385. https://doi.org/10.3390/cancers16193385