1. Introduction
The cure rates for osteosarcoma (OS) using multiagent chemotherapy and surgery have not improved for over 25 years [
1,
2,
3,
4,
5]. Dose intensification for newly diagnosed patients who respond poorly to neoadjuvant chemotherapy failed to improve patient outcomes [
6]. The overall cure rates of 60–65% leave approximately 40% of patients who relapse after initial therapy. These patients, along with those that present with metastases at diagnosis, have an extremely poor prognosis with a long-term survival rate of under 20%. Salvage chemotherapy for these patients has shown limited efficacy and no new therapies were identified in seven recent phase II trials from the Children’s Oncology Group [
7]. The activity of checkpoint inhibitor immunotherapy which targets T-cell activation has been disappointing in sarcoma patients [
8]. However, the demonstrated therapeutic activity of L-MTP-PE (a macrophage activating agent) in relapsed and newly diagnosed OS patients [
9,
10] suggests that other immunotherapies targeting “innate” immune cells should be explored particularly for patients with relapsed/metastatic disease. The success of L-MTP-PE therapy in improving overall survival in newly diagnosed patients and in patients with OS lung metastases also suggests that OS patients can mount an immune response, which further support a cDCV-based therapeutic approach.
Dendritic cells (DCs) are innate immune antigen-presenting cells that link innate and adaptive immunity, present antigen to T-cells, and are pivotal in generating adaptive immunity. Type 1 conventional DCs (cDC1s) are critical in the T-cell-mediated anti-tumor response as well as being essential in the antiviral immune response in the lung. Using DCs to generate cancer vaccines is an emerging immunotherapy focus. DC vaccines derived from monocytes (MoDCVs) have been shown to be well tolerated with no significant toxicity in children, adolescents, and young adults with relapsed OS [
11]. While safety was established and no maximum tolerated dose achieved, therapeutic activity and the induction of an immune T-cell response were seen in only 3 of 12 patients. Immunotherapies are typically most effective against minimal residual disease, as was the case for L-MTP-PE [
9,
10], which may explain the low anti-tumor efficacy in this trial. However, patients with refractory bone and soft tissue sarcomas with metastases in the shoulder, spine, pelvis, and lung showed sclerotic changes in these lesions after MoDCV (a sign of response in sarcoma) with stable disease for 5 months [
12]. Additional support for the potential of DC vaccine therapy for relapsed/metastatic sarcoma patients is provided by a Phase I trial where 18 sarcoma patients received activated DCs intratumorally. Eleven of the eighteen patients were alive and disease-free at 2–8 years [
13].
We developed a unique DC vaccine using in vitro-cultured CD103⁺DCs (cDCVs). When injected
intratumorally, this novel cDCV was superior to the MoDCV in inducing an anti-tumor immune response and in activating a
systemic T-cell immune response [
14,
15]. Using an
experimental OS metastasis model, we demonstrated that a cDCV generated using OS tumor cell lysates injected 30 days
prior to the intravenous injection of OS cells significantly reduced the formation of lung metastases with increased infiltration of IFNɣ
+CD8⁺ T-cells into tumors, spleen, and tumor-draining lymph nodes (TdLNs) [
14]. These data show that the cDCV was effective in
preventing metastatic development [
14]. However, these studies did
not evaluate cDCV efficacy against OS tumors that had already metastasized to the lung. Since our novel cDCV was shown to be superior to a MoDCV in anti-tumor activity, particularly in inducing T-cell activation and the infiltration of tumor antigen-specific CD8⁺ T-cells [
14], our prior findings support the concept of investigating the potential of a DC vaccine derived from CD103⁺cDC1 cells for activity against
established OS lung metastases, which is the area of need in OS as 90% of newly diagnosed patients have micro-metastases in the lung at the time of diagnosis.
An orthotopic model where OS cells are injected into the bone and spread to the lung from the primary tumor is the optimal model to investigate therapeutic efficacy against established lung metastases as this model is more reflective of the clinical disease. In this model, OS cells spread from the bone to the lung rather than being injected intravenously. Using this model, the studies described herein show that cDCV therapy injected systemically following removal of the primary tumor at a time when lung metastases are present induced regression of the established lung metastases and the infiltration of T-cells into the lung tumor and TdLN. We also demonstrated that this anti-tumor activity was enhanced by anti-CTLA-4. Our studies indicate that cDCV therapy may provide a novel therapeutic approach for relapsed OS patients with metastases in the lung, thereby improving survival for this difficult patient population with few alternative therapies.
2. Materials and Methods
2.1. Mice, Cell Line, and Reagents
Balb/c mice were purchased from Charles River (Wilmington, MA, USA) and maintained at the University of Texas MD Anderson Cancer Center in a specific pathogen-free animal facility approved by the American Association for Accreditation of Laboratory Animal Care. Age-matched mice were chosen randomly for treatment groups and were used in accordance with MD Anderson Cancer Center’s Institutional Animal Care and Use Committee-approved protocol (No. 00000896).
The mouse metastatic K7M3 cell line was derived from K7M2 cells in our laboratory [
16] and cultured for no more than 30 passages. The unique signature identification of the K7M3 cells was confirmed by a short-tandem-repeat DNA microsatellite fingerprinting analysis carried out by IDEXX BioAnalytics (Columbia, MO, USA). The cell culture was tested for mycoplasma species every other month using the MycoAlert Mycoplasma Detection Kit (Lonza Bioscience, Houston, TX, USA) and was confirmed to be negative.
CTLA-4 antibody (clone 9H10) was purchased from Bio X-Cell company (West Lebanon, NH, USA) and injected intraperitoneally (i.p.) (200 µg/mouse for the first treatment, 100 µg/mouse for subsequent treatments) twice a week to the mice one week after DCV treatment for a total of four weeks of treatment.
2.2. Isolation of CD103+ cDC1 Cells
Ex vivo generation and expansion of CD103
+ cDC1s cells from Balb/c mice bone marrow was based on our previous studies [
14] and optimized for maximal production efficiency and high yields of CD103
+ cDC1s cells. Bone marrow cells were collected from the femurs of 6-to-8-week-old Balb/c mice and cultured in RPMI 1640 complete medium supplemented with 50 µM β-mercaptoethanol, 50 ng/mL human FMS-like tyrosine kinase 3 ligand (Flt-3L, PeproTech, Rocky Hill, NJ, USA) plus 0.5 ng/mL murine granulocyte-macrophage colony-stimulating factor (GM-CSF, PeproTech, Rocky Hill, NJ, USA) at 1.5 × 10
6 cells/mL. Fresh full medium was supplemented on day 5 and day 7; suspension cells were collected at day 9 and cultured in fresh medium supplemented with 50 µM β-mercaptoethanol, 50 ng/mL Flt-3L plus 2 ng/mL murine GM-CSF at 0.5 × 10
6 cells/mL, and fresh full medium was supplemented at day 13 once to maintain the culture. Non-adherent cells were collected at 17 days (
Supplemental Figure S1A) and stained with CD11b, CD11c, CD103, CD172α, and CD24 antibodies together with GhostDye Violet 510 (Tonbo Biosciences, San Diego, CA, USA). CD103
+ cDC1s (CD11c
+, CD24
+, CD172α
-, and CD103
+ cells) populations were sorted using a FACSAria Fusion Cell Sorter (BD Biosciences, Palo Alto, CA, USA) (
Supplemental Figure S1B).
2.3. Generation of the CD103+ cDC1 Vaccine (cDCV) Using K7M3 Tumor Lysates
Log phase cultured K7M3 cells were washed and resuspended in PBS at 50 × 106 cells/mL. The cell suspension was frozen and thawed ×3 and then ultrasonically disrupted for 50 s three times on ice. Complete disruption of the cells was confirmed by trypan blue staining. CD103+ cDC1s were incubated with K7M3 tumor cell lysate and polyinosinic–polycytidylic acid (poly-I:C) to stimulate cDCV maturation and antigen loading using a ratio of two tumor cells to one CD103+ cDC1 cell and incubated for 4 h at 37 °C. These poly-I:C-stimulated and antigen-loaded CD103+ cDC1 vaccine cells (cDCV) were washed twice with phosphate-buffered saline (PBS) to eliminate the tumor lysate debris and resuspended in sterile PBS. Therapy consisted of 2 × 106 cDCV cells per treatment.
2.4. cDCV Therapy for Primary and Metastatic Osteosarcoma
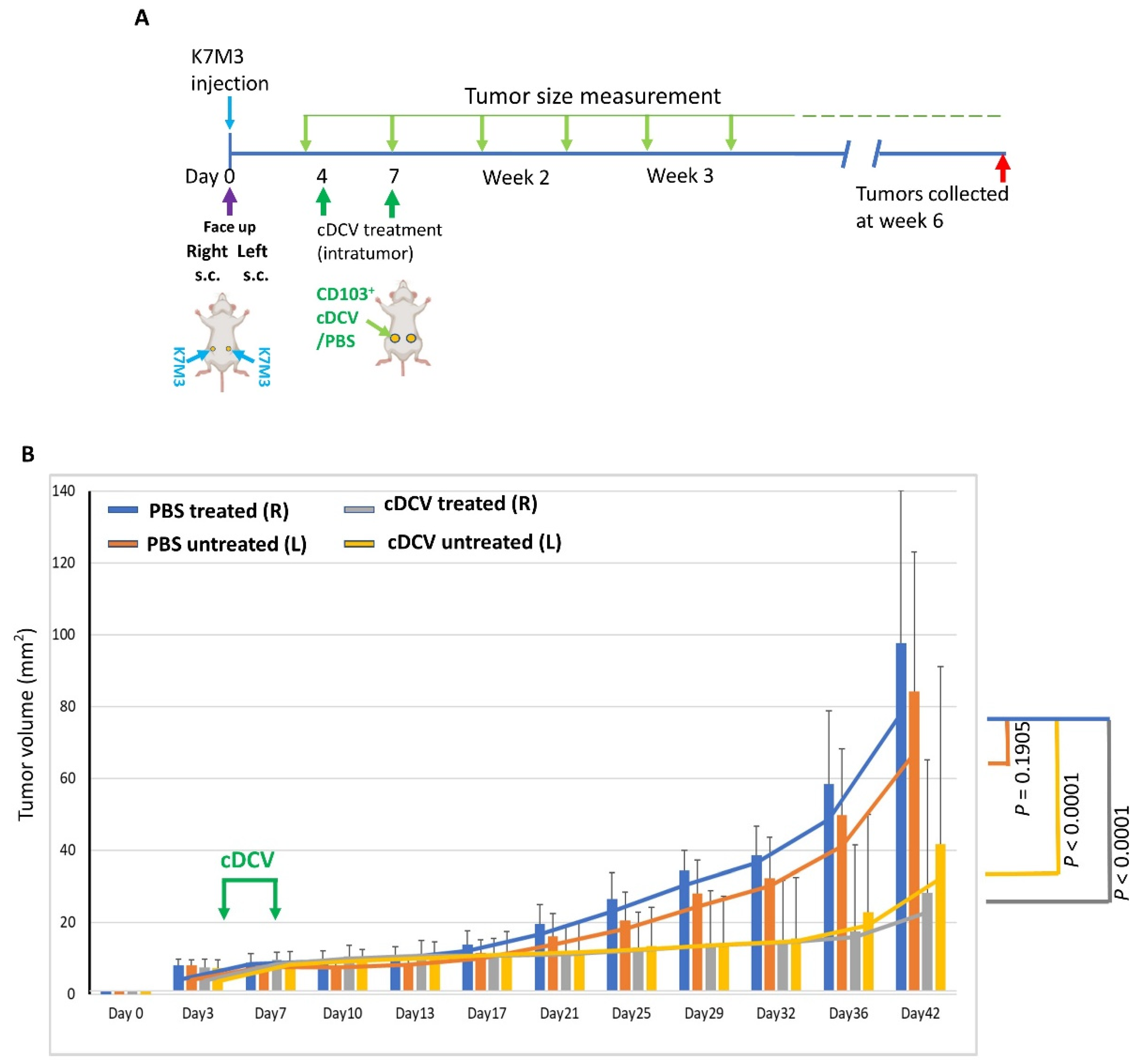
A quantity of 2 × 106 K7M3 cells was subcutaneously (s.c.) injected on both sides of the mice’s abdomens. Tumors on the right were treated intratumorally (i.t.) with cDCV or PBS on days 4 and 7 following implantation, a time when the tumors were palpable. The tumor size (length × width) was measured every 3–4 days using a caliper. Mice were euthanized 5 weeks after therapy.
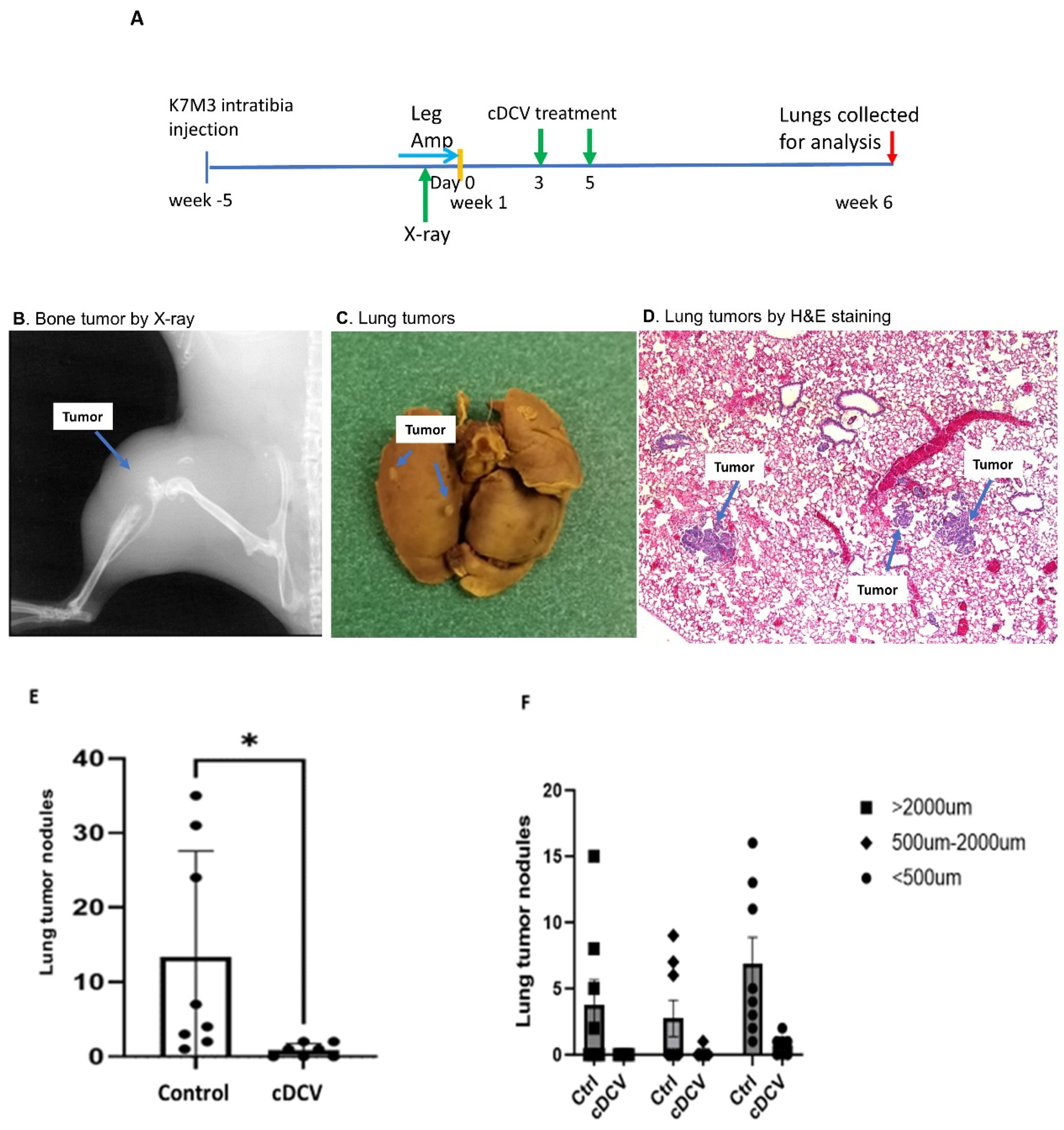
We used our intra-bone injection OS model [
16] to evaluate therapy activity of cDCV against spontaneous lung metastasis. Mice were injected with 0.25 × 10
6 K7M3 cells into the right tibia. The tumors were verified by X-ray imaging at 5 weeks and a leg amputation was performed 24 h later. This prevents further seeding of tumor cells from the leg to the lungs. Three days after amputation the mice were injected intravenously (i.v.) with 2 × 10
6 cDCV cells twice in the week. Mice were euthanized 5 weeks later. Lungs, spleens, and tumor draining lymph nodes (TdLNs) were collected for analysis.
2.5. Immune Profiling of TdLNs and Spleens by Flow Cytometry Analysis
Single-cell suspensions were generated from spleens and inguinal TdLNs, which were closing to the subcutaneous tumor formation as previously described [
14]. The inguinal lymph nodes were also analyzed for immune cell infiltration in the orthotopic investigation where the primary tumor was in the tibia. Cell surface Fc receptors were blocked with rat anti-mouse CD16/32 (Tonbo Biosciences) for 15 min at 4 °C. For the T-cell subset analysis, T-cells were stimulated with 0.5 μg/mL ionomycin (Sigma, St Louis, MI, USA) and 50 ng/mL phorbol 12-myristate 13 acetate (PMA) (Sigma, St Louis, MI, USA) for 4 h in the presence of GolgiStop (BD Biosciences, Palo Alto, CA, USA). For surface maker staining, isolated cells were incubated with fluorescent dye-conjugated antibodies for 30 min at 4 °C; for intracellular protein staining, T-cells were fixed and permeabilized using Intracellular Fixation and Permeabilization Buffer (eBiosciences, San Diego, CA, USA) first, and stained with antibodies against intracellular proteins for 30 min at 4 °C. The following antibodies were used: PE-Cy7-conjugated CD11c (N418) or MHC-II (M5/114.15.2); PE-conjugated CD103 (2E7) or IFN-γ (XMG 1.2) antibodies; APC-Cy7-conjugated CD4 (GK1.5) or CD45 (30-F11) antibodies; PerCP-Cy5.5-conjugated Foxp3 (FJK-16S), CD19 (1D3), or CD11b (M1/70) antibodies; FITC-conjugated CD172α (P84), MHC-I (AF6-88.5), or Ly6G (1A8) antibodies; BV605-conjugated CD3e(17A2) antibody; BV711-conjugated CD8α (53–6.7) or F4/80 (BM8) antibodies; BV421-conjugated IL-4 (11B11) or XCR1 (ZET) antibodies. All antibodies were purchased from BioLegend (San Diego, CA, USA), eBioscience (San Diego, CA, USA), BD Biosciences, or Tonbo Biosciences. Dead cells were eliminated in all experiments using Ghost DyeTM Violet 510 (Tonbo Biosciences). An LSRFortessa flow cytometer (BD) was used to perform the flow cytometry analysis. Gating was performed as previously described [
14]. Data analysis was performed on viable single cells (Ghost Dye) using FlowJo software (version 10, TreeStar, Ashland, OR, USA).
2.6. Immunofluorescence Staining
Slides of frozen tumor sections (5 µm) were fixed in cold Acetone for 5 min then cold Acetone + Chloroform (1:1) for 5 min; followed by cold Acetone for 5 min, and finally permeabilized by 0.25% TritonX-100 in PBS for 5 min. After three PBS washes, the sections were blocked by 5% normal goat serum plus 1% normal horse serum in PBS for 30 min and by 4% fish gel in PBS for another 30 min at room temperature. In a humid chamber box, sections were incubated with anti-CD3-AF488 (1:200), CD8α-AF594 (1:200), CD4-AF647 (1:100), or anti-CD103-AF488(1:100), and CD11c-AF594 (1:200) at 4 °C overnight, then washed ×3 with PBS; the slides were then counterstained with Fluoro-Gel II with DAPI and mounted with a glass cover slip. Three tumor frozen samples from each treatment group were sectioned for CD3/CD4/CD8 staining and CD11c/CD103 staining. Tumor areas from each section were imaged at 400× by Leica DMi8 microscope (Leica Mi-crosystems, Wetzlar, Germany). Positive staining was quantified in eight random microscopy fields from three tumor samples using Simple PCI software (version 6.6, Hamamatsu, Sewickley, PA, USA), and the average expression was calculated as average ± SEM.
Primary antibodies anti-CD86 (1:200) and anti-CD163 (1:200) (CD86, NeoBiotechnologies, Union City, CA, USA; CD163, Abcam, Waltham, MA, USA) were used for the macrophage markers. After stained at 4 °C overnight and washed ×3 with PBS, the sections were incubated with the secondary antibody (1:500, Alexa Fluor 488 goat anti-rabbit, Invitrogen, Waltham, MA, USA) at room temperature for 1 h in the dark and washed ×3 with PBS. Slides were then counterstained with Fluoro-Gel II with DAPI and mounted as above. Tumor areas from each section were imaged at 400× by Leica DMi8 microscope (Leica Microsystems, Wetzlar, Germany). Positive staining was quantified in eight random microscopy fields from three tumor samples using Simple PCI software (Hamamatsu, Sewickley, PA, USA), and the average expression was calculated as average ± SEM.
2.7. Statistical Analysis
All values reported represent means ± SEMs. A 2-tailed Student t test was used to determine significance. For the mouse experiments, the statistical significance was evaluated using the Mann–Whitney U-test and the one-way ANOVA by GraphPad Prism 9 (GraphStats, Bangalore, India). For the tumor growth experiments, a linear mixed effects model was used to compare the tumor growth rates among different groups. The tumor size was log-transformed to ensure normally distributed residuals and homogeneity of variance over time in the model. The analysis was performed using R package (R Development Core Team). p values less than 0.05 were considered statistically significant.
4. Discussion
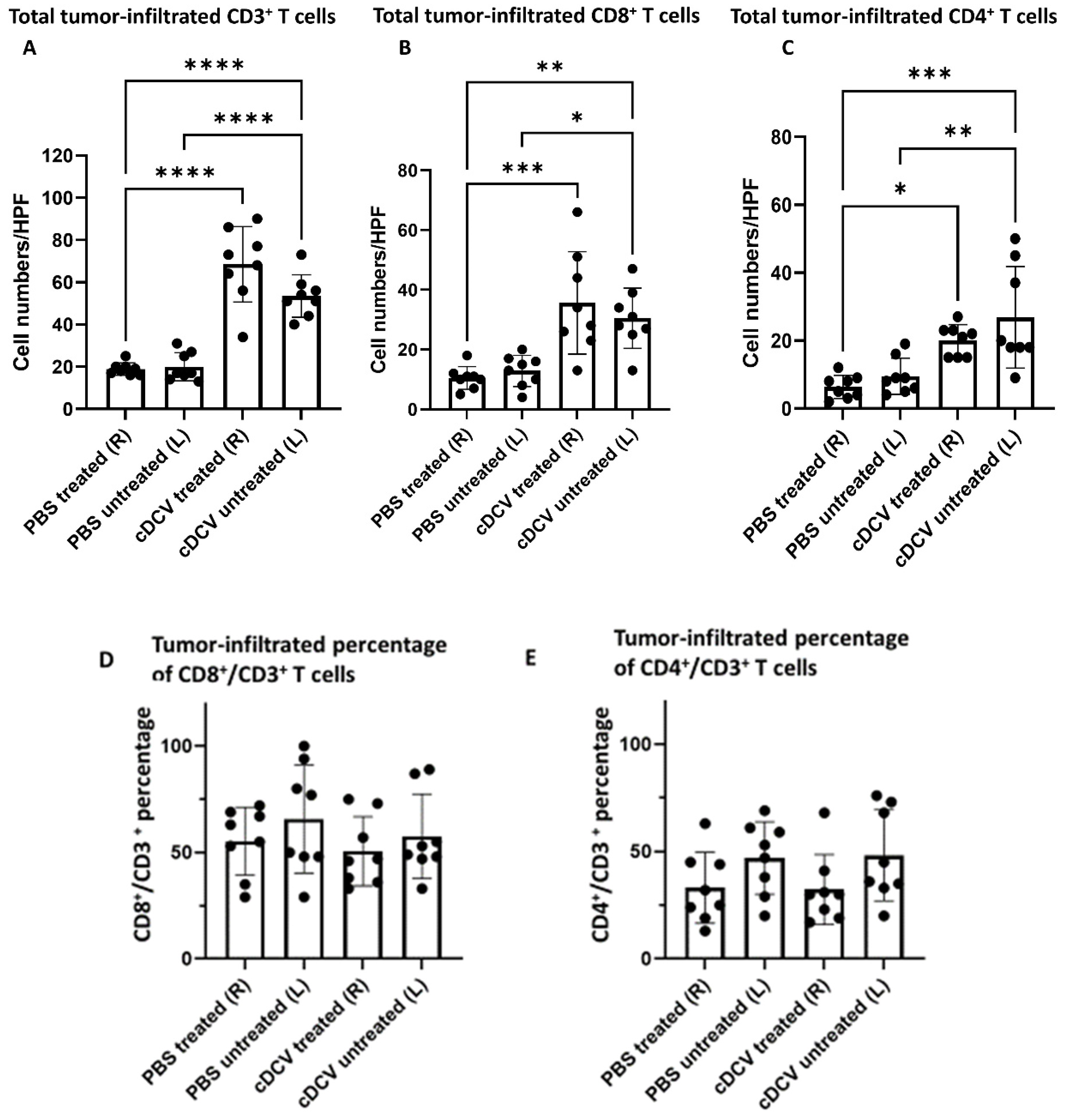
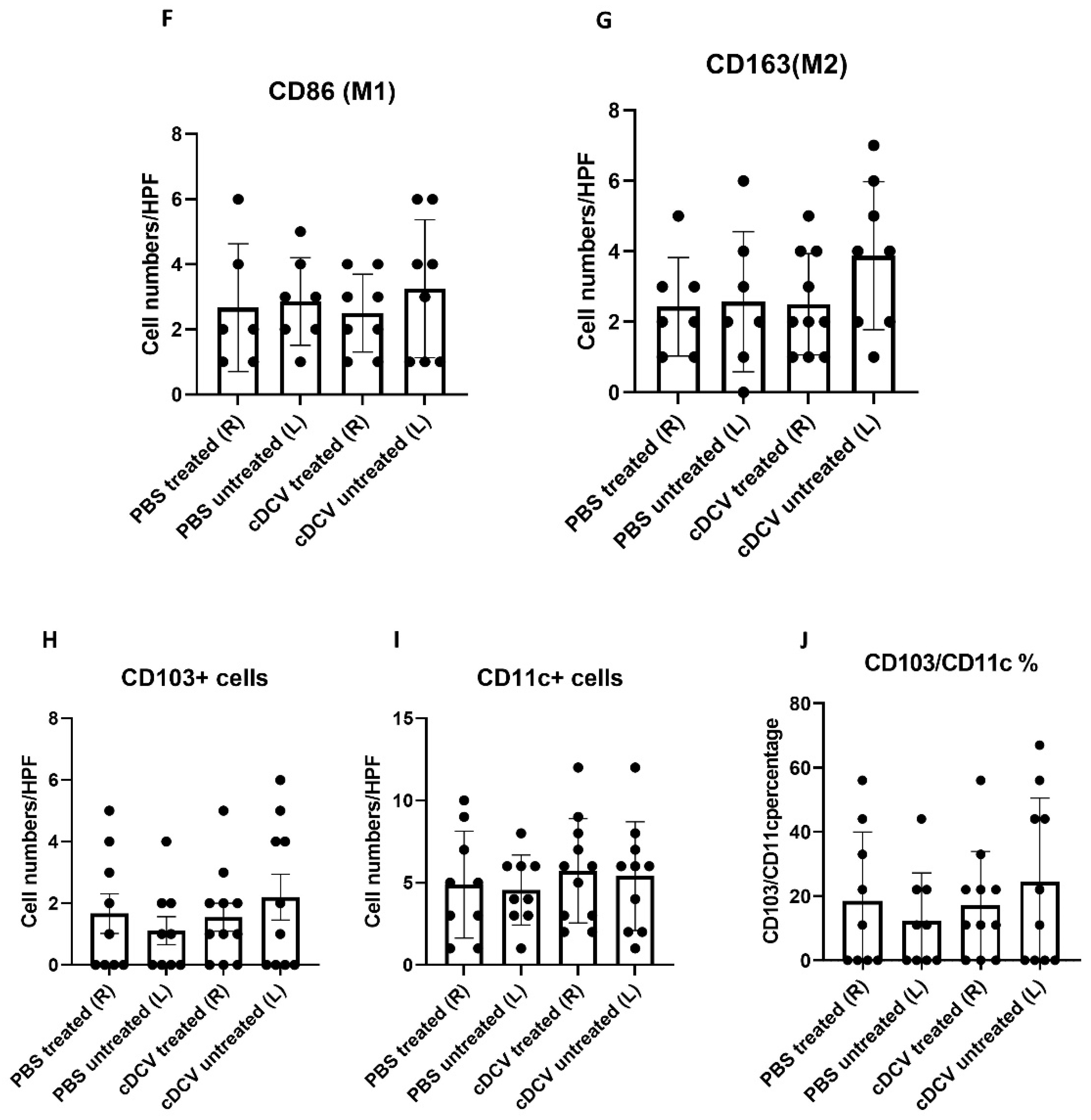
In summary, we demonstrated that a DC vaccine generated using the CD103⁺cDC1 subset and loaded with OS cell lysates as a source of tumor antigens induced tumor regression when injected into the primary tumor and stimulated a systemic immune response resulting in the regression of the untreated tumor on the contralateral side. The activity of the cDCV was accompanied by an increased infiltration of CD8⁺ and CD4⁺ cells into both the treated and untreated tumors. The finding of increased infiltration of CD8⁺ and CD4⁺ cells in the untreated contralateral tumor supports the generation of a systemic response. By contrast, there was no change in the number of intratumor M1 or M2 macrophages. While the total number of CD4⁺ and CD8⁺ T-cells increased, their percentage to the total T-cell number (CD3⁺ cells) did not change. The findings of no change in M1 and M2 macrophages is important, as we previously showed that the activity of anti-PD1 therapy against OS lung metastases was mediated by the increased infiltration of M1 macrophages into the tumor and a decrease in M2 macrophages [
18]. Furthermore, M2 macrophages suppress the immune response and are a negative prognostic factor for OS [
19,
20,
21]. Taken together, these data suggest that the anti-tumor activity of the cDCV therapy was mediated by the activation and infiltration of cytotoxic T-cells.
While we did not demonstrate complete tumor eradication, this is not surprising, as only 2 cDCV injections were given and mice were
untreated for ~5 weeks. Curative therapy will most likely require multiple cDCV injections for longer periods of time, similar to what was used clinically with other immunotherapies targeting
innate immune cells, i.e., L-MTP-PE where therapy continued 1–2 times weekly for 6–9 months [
9,
10,
22]. The failure of a pervious MoDCV in relapsed OS patients may be secondary to the short 3-week treatment period [
11], in addition to the inferior activity of MoDCVs compared to DCVs generated using CD103
+ cells [
14]. This will be important when translating this therapeutic approach. In addition, we anticipate that improved cDCV therapeutic efficacy will be achieved by initiating therapy in the setting of residual
microscopic disease, as is the case in newly diagnosed OS patients following primary tumor resection or patients with lung metastases that have been resected. As we showed with L-MTP-PE, we also anticipate that cDCV therapy can be administered together with the chemotherapy agents used to treat OS [
10,
22,
23,
24].
Having demonstrated the successful anti-tumor activity of the cDCV following
intratumor injection with the generation of a systemic response, we evaluated
systemic vaccine effectiveness against
established lung metastases that arose from the primary tumor. This was carried out using our orthotopic mouse model, which is more relevant to the patient population where 90% of newly diagnosed patients have microscopic spread to the lung at the time of diagnosis prior to chemotherapy administration, with only a 65% 5-year survival rate following combination chemotherapy. In this model, OS cells are injected into the tibia and a tumor is allowed to grow for 5 weeks, at which time there are positive X-ray findings documenting primary tumor formation and evidence of lung metastases. Thus, this is a model of
established, as opposed to
experimental metastases [
16], and more importantly represents
spontaneous metastatic spread from the primary tumor in the leg into the lungs. Amputation of the affected limb prevents any further metastatic spread, allowing for the assessment of systemic therapeutic activity against established metastases. Using this clinically relevant model, we showed that intravenous cDCV therapy decreased the number and size of the lung metastases, with three of seven mice having no evidence of disease. The findings that the sizes of the metastases in the cDCV-treated mice were significantly smaller and that three of seven mice had no evidence of either macro- or microscopic tumors further supports the successful anti-tumor activity of the cDCV therapy. We quantified T-cell numbers in these small tumors. As seen after intratumor cDCV therapy, there was a significant increase in CD8⁺ T-cells in the mice treated with systemic cDCV, which again supports the induction of an immune response and is significant, as cDCV efficacy has been shown to be associated with CD8⁺ T-cell activation and tumor infiltration [
14].
The results of our investigations demonstrate that both the intratumor and intravenous route of cDCV delivery are effective in generating an effective anti-tumor response. The intratumor delivery resulted in both a local and systemic response in primary tumors (untreated and treated) while the intravenous cDCV delivery was effective against
established lung metastases. Since we are targeting established lung metastases, systemic cDCV delivery is required. Intratumor vaccination is not appropriate. This study is the
first to demonstrate the efficacy of systemic cDCV therapy against
established OS lung metastases that arise from the primary tumor in the bone. The prior studies evaluated the effect of
intratumor cDCV administration on the growth of a subcutaneous tumor and whether pretreating mice 1 month
before tumor cell injection
prevented the development of lung metastases. These studies did not evaluate the effect of cDCV intratumor therapy on an
untreated OS tumor on the contralateral side of the mouse, or more importantly its activity against established lung metastases that were present
before systemic therapy is initiated. The prior studies support the
concept of using cDCV therapy for relapsed OS but are not sufficient to justify
translation of cDCV therapy for the treatment of OS lung metastases. Demonstrating that a therapy can
prevent the formation of metastases in the lung is not therapeutically equivalent to inducing the
regression of metastatic tumors that are already growing in the lung. The lung tumor microenvironment (TME) has been shown to play a critical role in the successful growth of the OS cells once the cells have metastasized to the lung and to therapy response [
25]. Eliminating the circulating OS cells
before they reach the lung or interfering with the early metastatic process
before a tumor is formed in the lung does not mean that the therapy will be effective in patients who have OS lung metastases. Therefore, it is imperative that the potential of any new therapy aimed at treating patients with OS lung metastases be evaluated in the context of the lung TME.
As stated above, the majority of newly diagnosed OS patients have
microscopic disease at the time of diagnosis. Standard three-drug chemotherapy cures ~65% of OS patients. Unfortunately, neither dose intensification nor the addition of additional chemotherapy agents has been successful in increasing this 65% survival rate [
6]. The prognosis for patients who relapse with OS lung metastases is dismal (~20% 2-year survival) and has not changed in >25 years. The majority of patients with lung metastases can have these tumors excised putting them in the setting of minimal residual/microscopic disease. However, 85% of patients will relapse again within 1 year. It has been shown that DC vaccine therapy is best for patients with low tumor burden, thereby providing additional justification for its use in this patient population [
26]. Patients with bone metastases have an even poorer prognosis. We demonstrated the induction of systemic immunity in addition to activity against lung metastases. Therefore, cDC-based vaccines using antigen-loaded CD103⁺cDC1 cells may be an alternative therapy for patients with disseminated, relapsed, or metastatic OS. The ability to inject one tumor site and induce systemic anti-tumor immunity may make cDCV-based therapy appropriate for OS patients with skip bone lesions and metastases outside the lung. These patients have the worst prognosis with no effective therapies at the present time. Although these cases are rare, the lack of effective therapeutic options necessitates the identification of newer approaches.
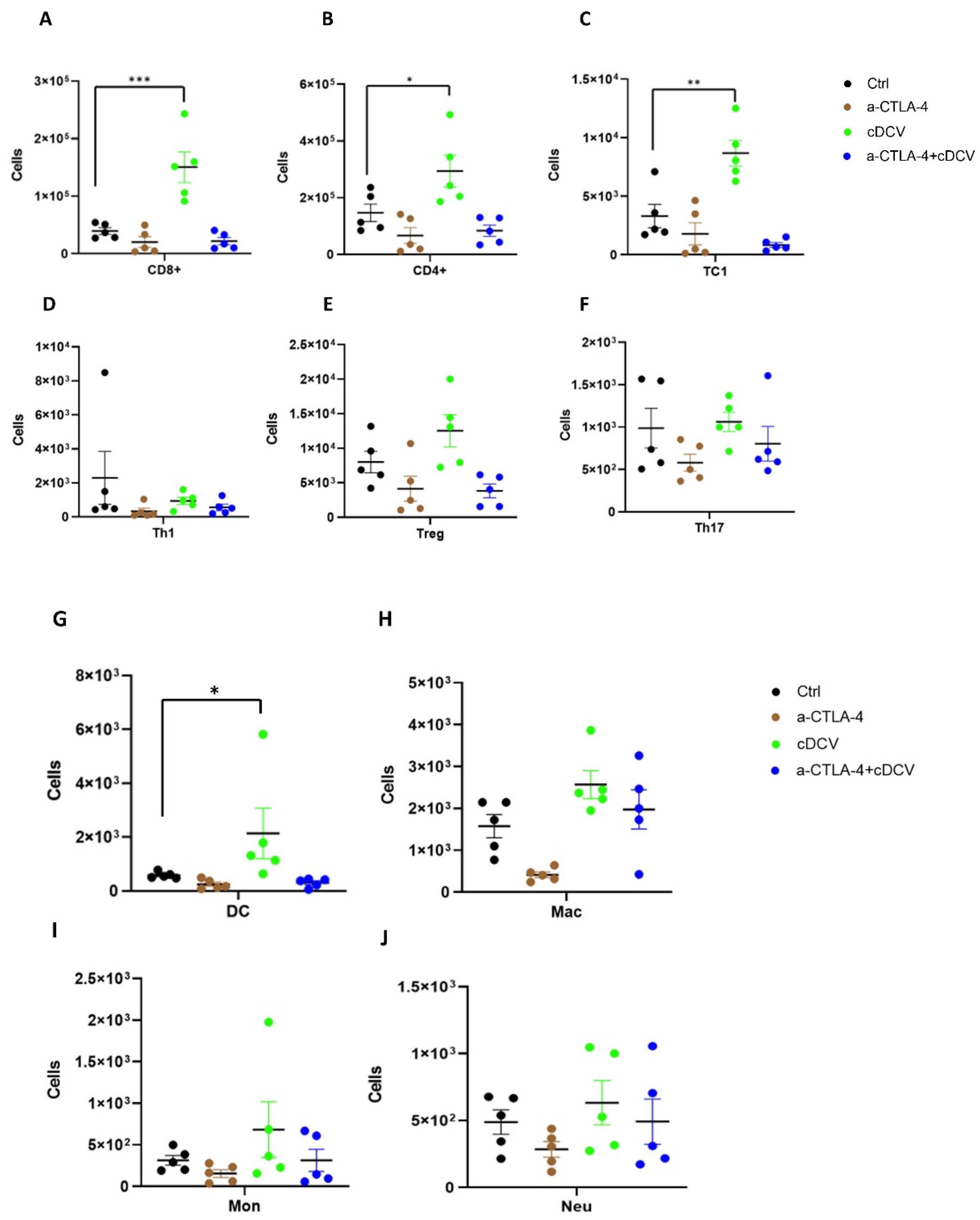
We demonstrated an increase in the number of CD8⁺ T-cells in tumors from cDCV-treated mice, which was associated with the positive therapeutic response. Successful cDCV anti-tumor efficacy has been shown to be linked to CD8⁺ T-cell activation and infiltration into the tumor and the TdLNs [
11,
12,
14]. Anti-CTLA-4 is a checkpoint inhibitor that enhances T-cell priming in the lymph nodes and increases T-cell activation and proliferation [
27]. We speculated that adding anti-CTLA-4 may improve cDCV efficacy by increasing the number of activated CD8⁺ T-cells in the tumors. Our data indeed demonstrated that combining cDCV with anti-CTLA-4 increased the therapeutic activity against established lung metastases, however there was no significant increase in the number of CD8⁺ cells in the tumors compared to cDCV-treatment alone. This may be due to the fact that tumors from both treatment regimens were small, making it difficult to see significant changes.
Since T-cell penetration into the tumor is critical for immunotherapy success, in addition to quantifying the number of CD8+ cells, we also examined the location of the T-cells. The control tumors showed T-cells primarily located around the periphery of the tumor. By contrast, T-cells were found within the tumor in the cDCV-treated mice. Intratumor T-cells were also seen within the tumors from mice treated with combination therapy but not those treated with anti-CTLA-4 alone. There was no detectable difference in the number of intratumor T-cells in tumors from mice treated with combination therapy versus cDCV alone. It is noteworthy that only tumors from mice that had received cDCV as part of their therapy (cDCV alone or in combination with anti-CTLA-4) showed this pattern of CD8+ T-cell infiltration, suggesting that this effect was mediated by the cDCV. This also suggests that the therapeutic activity of other immunotherapies that depend on T-cell activation and tumor infiltration may be enhanced by combining with a cDC1 vaccine. At the present time, we do not have an explanation for the enhanced activity of the combination therapy compared to cDCV therapy alone in terms of T-cell numbers or penetration into the tumor. Our analysis was performed 6 weeks after therapy completion. Earlier analysis may reveal differences between the combination and single therapies that can explain the therapeutic differences.
In addition to the infiltration of activated T-cells into the tumor, T-cell activation in the TdLNs is critical for effective induction of cDCV-anti-tumor immunity and efficacy [
28]. We also found a significant increase in CD8⁺, CD4⁺, TC1⁺ (activated T-cells), and DCs in the TdLNs from cDCV-treated mice. This was not seen in mice treated with anti-CTLA-4 alone or combination therapy. cDC1 cells are critical for cross-presenting tumor antigens to activate CD8
+ T-cells [
29]. For example, OVA-derived specific peptide SIINFEKL can be used to show activation of T-cells by cDCV cells primed by melanoma cells expressing OVA (B16-OVA cells) [
14]. cDC1 cells process and present antigen peptide to activate the adaptive immune response typically by inducing T-cell gene expression such as IFN-γ, granzyme B, perforin, etc. [
30]. cDC1 cells are required to generate broad CD8 responses against a range of diverse neoAgs [
31]. While there is currently no
specific osteosarcoma antigen(s) reported that can be used to rapidly generate cDCVs for patients with metastatic disease or investigate the genes that are activated in the T-cells following exposure to cDCs, we speculate that there will be induction of the IFN-g, granzyme B, and perforin genes by our cDCV. Recently, we demonstrated that CD70 is overexpressed in multiple OS cell lines and patient-derived xenografts [
32,
33]. We are currently investigating the use of CD70 as an OS specific antigen for activation of OS-specific cDCVs. This may provide the needed tool to investigate the mechanism of T-cell activation; we therefore expect that T-cells activated by CD70 peptide presented by cDC1 cells will show induction in IFN-γ, granzyme B, and perforin gene expression as well.
There was no significant increase in Th1, T-regs, TH17, macrophages, monocytes, or neutrophils in the TdLNs from any of the treatment groups. As we failed to show an improved immune cell profile in the tumors and TdLNs of mice treated with combination therapy, further studies are required to identify the mechanism for the enhanced therapeutic response. However, our results suggest the potential for combination therapy against OS lung metastases.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}