
The Ubiquitin-Conjugating Enzyme E2 O (UBE2O) and Its Therapeutic Potential in Human Leukemias and Solid Tumors


Abstract
Simple Summary
Abstract
1. Introduction
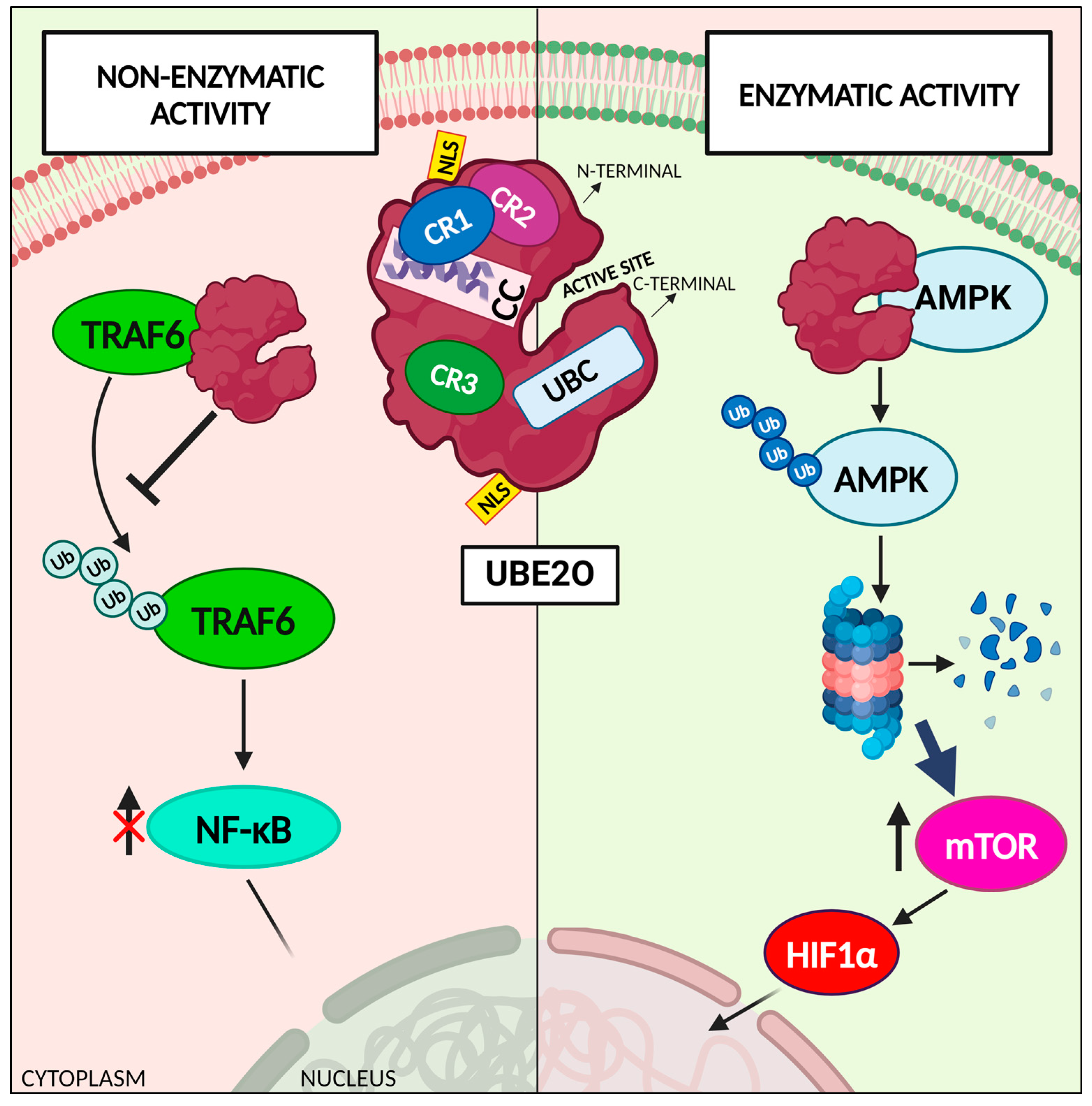
2. The Atypical Ubiquitin-Conjugating Enzyme E2 O: UBE2O
3. Role of UBE2O in the Hematological Field
3.1. UBE2O Regulates Proteome Remodeling during Terminal Erythroid Differentiation
3.2. UBE2O Overexpression Inhibits Acute Myeloid Leukemia Progression
3.3. UBE2O Knockdown Reduces Cell Proliferation in KMT2A Rearranged Leukemias
3.4. UBE2O Induces Apoptosis in Multiple Myeloma Cells
4. UBE2O Acts as a Pro-Oncogenic Factor in Many Human Cancers
5. UBE2O’s Role in Non-Oncologic Diseases
6. Conclusions and Future Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pickart, C.M. Back to the Future with Ubiquitin. Cell 2004, 116, 181–190. [Google Scholar] [CrossRef]
- Wilkinson, K.D. Essay. Cell 2004, 119, 741–745. [Google Scholar] [CrossRef]
- Hershko, A.; Ciechanover, A. THE UBIQUITIN SYSTEM. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef]
- Nalepa, G.; Rolfe, M.; Harper, J.W. Drug Discovery in the Ubiquitin–Proteasome System. Nat. Rev. Drug Discov. 2006, 5, 596–613. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, D.; Riezman, H. Proteasome-Independent Functions of Ubiquitin in Endocytosis and Signaling. Science 2007, 315, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Ullah, K.; Zubia, E.; Narayan, M.; Yang, J.; Xu, G. Diverse Roles of the E2/E3 Hybrid Enzyme UBE 2O in the Regulation of Protein Ubiquitination, Cellular Functions, and Disease Onset. FEBS J. 2019, 286, 2018–2034. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, G.; Scheid, M.; Hammerling, U.; Schlesinger, D.H.; Niall, H.D.; Boyse, E.A. Isolation of a Polypeptide That Has Lymphocyte-Differentiating Properties and Is Probably Represented Universally in Living Cells. Proc. Natl. Acad. Sci. USA 1975, 72, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Dubois, M.-L.; Meller, A.; Samandi, S.; Brunelle, M.; Frion, J.; Brunet, M.A.; Toupin, A.; Beaudoin, M.C.; Jacques, J.-F.; Lévesque, D.; et al. UBB Pseudogene 4 Encodes Functional Ubiquitin Variants. Nat. Commun. 2020, 11, 1306. [Google Scholar] [CrossRef]
- Clague, M.J.; Heride, C.; Urbé, S. The Demographics of the Ubiquitin System. Trends Cell Biol. 2015, 25, 417–426. [Google Scholar] [CrossRef]
- Nijman, S.M.B.; Luna-Vargas, M.P.A.; Velds, A.; Brummelkamp, T.R.; Dirac, A.M.G.; Sixma, T.K.; Bernards, R. A Genomic and Functional Inventory of Deubiquitinating Enzymes. Cell 2005, 123, 773–786. [Google Scholar] [CrossRef]
- Ciechanover, A.; Elias, S.; Heller, H.; Ferber, S.; Hershko, A. Characterization of the Heat-Stable Polypeptide of the ATP-Dependent Proteolytic System from Reticulocytes. J. Biol. Chem. 1980, 255, 7525–7528. [Google Scholar] [CrossRef] [PubMed]
- Kresge, N.; Simoni, R.D.; Hill, R.L. The Discovery of Ubiquitin-Mediated Proteolysis by Aaron Ciechanover, Avram Hershko, and Irwin Rose. J. Biol. Chem. 2006, 281, e32–e36. [Google Scholar] [CrossRef]
- Ciechanover, A. The Ubiquitin-Proteasome Pathway: On Protein Death and Cell Life. EMBO J. 1998, 17, 7151–7160. [Google Scholar] [CrossRef] [PubMed]
- Breitschopf, K. A Novel Site for Ubiquitination: The N-Terminal Residue, and Not Internal Lysines of MyoD, Is Essential for Conjugation and Degradation of the Protein. EMBO J. 1998, 17, 5964–5973. [Google Scholar] [CrossRef]
- Schulman, B.A.; Wade Harper, J. Ubiquitin-like Protein Activation by E1 Enzymes: The Apex for Downstream Signalling Pathways. Nat. Rev. Mol. Cell Biol. 2009, 10, 319–331. [Google Scholar] [CrossRef]
- Sun, Y. E3 Ubiquitin Ligases as Cancer Targets and Biomarkers. Neoplasia 2006, 8, 645–654. [Google Scholar] [CrossRef]
- Stewart, M.D.; Ritterhoff, T.; Klevit, R.E.; Brzovic, P.S. E2 Enzymes: More than Just Middle Men. Cell Res. 2016, 26, 423–440. [Google Scholar] [CrossRef]
- Yokota, T.; Nagai, H.; Harada, H.; Mine, N.; Terada, Y.; Fujiwara, H.; Yabe, A.; Miyazaki, K.; Emi, M. Identification, Tissue Expression, and Chromosomal Position of a Novel Gene Encoding Human Ubiquitin-Conjugating Enzyme E2-230k. Gene 2001, 267, 95–100. [Google Scholar] [CrossRef]
- Wijk, S.J.L.; Timmers, H.T.M. The Family of Ubiquitin-conjugating Enzymes (E2s): Deciding between Life and Death of Proteins. FASEB J. 2010, 24, 981–993. [Google Scholar] [CrossRef]
- Klemperer, N.S.; Berleth, E.S.; Pickart, C.M. A Novel, Arsenite-Sensitive E2 of the Ubiquitin Pathway: Purification and Properties. Biochemistry 1989, 28, 6035–6041. [Google Scholar] [CrossRef]
- Hormaechea-Agulla, D.; Kim, Y.; Song, M.S.; Song, S.J. New Insights into the Role of E2s in the Pathogenesis of Diseases: Lessons Learned from UBE2O. Mol. Cells 2018, 41, 168–178. [Google Scholar] [PubMed]
- Mashtalir, N.; Daou, S.; Barbour, H.; Sen, N.N.; Gagnon, J.; Hammond-Martel, I.; Dar, H.H.; Therrien, M.; Affar, E.B. Autodeubiquitination Protects the Tumor Suppressor BAP1 from Cytoplasmic Sequestration Mediated by the Atypical Ubiquitin Ligase UBE2O. Mol. Cell 2014, 54, 392–406. [Google Scholar] [CrossRef] [PubMed]
- Liang, K.; Volk, A.G.; Haug, J.S.; Marshall, S.A.; Woodfin, A.R.; Bartom, E.T.; Gilmore, J.M.; Florens, L.; Washburn, M.P.; Sullivan, K.D.; et al. Therapeutic Targeting of MLL Degradation Pathways in MLL-Rearranged Leukemia. Cell 2017, 168, 59–72.e13. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Yang, J.; Zhang, Y.; Duan, C.; Liu, Q.; Huang, Z.; Xu, Y.; Zhou, L.; Xu, G. Ubiquitin-Conjugating Enzyme UBE2O Regulates Cellular Clock Function by Promoting the Degradation of the Transcription Factor BMAL1. J. Biol. Chem. 2018, 293, 11296–11309. [Google Scholar] [CrossRef] [PubMed]
- Berleth, E.S.; Pickart, C.M. Mechanism of Ubiquitin Conjugating Enzyme E2-230K: Catalysis Involving a Thiol Relay? Biochemistry 1996, 35, 1664–1671. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, J.; Bauer, A.; Zhang, L.; Selinger, D.W.; Lu, C.X.; Ten Dijke, P. Fine-Tuning BMP7 Signalling in Adipogenesis by UBE2O/E2-230K-Mediated Monoubiquitination of SMAD6. EMBO J. 2013, 32, 996–1007. [Google Scholar] [CrossRef]
- Liu, X.; Ma, F.; Liu, C.; Zhu, K.; Li, W.; Xu, Y.; Li, G.; Niu, Z.; Liu, J.; Chen, D.; et al. UBE2O Promotes the Proliferation, EMT and Stemness Properties of Breast Cancer Cells through the UBE2O/AMPKα2/mTORC1-MYC Positive Feedback Loop. Cell Death Dis. 2020, 11, 10. [Google Scholar] [CrossRef]
- Wang, R.N.; Green, J.; Wang, Z.; Deng, Y.; Qiao, M.; Peabody, M.; Zhang, Q.; Ye, J.; Yan, Z.; Denduluri, S.; et al. Bone Morphogenetic Protein (BMP) Signaling in Development and Human Diseases. Genes Dis. 2014, 1, 87–105. [Google Scholar] [CrossRef]
- Abe, Y.O.; Yoshitane, H.; Kim, D.W.; Kawakami, S.; Koebis, M.; Nakao, K.; Aiba, A.; Kim, J.K.; Fukada, Y. Rhythmic Transcription of Bmal1 Stabilizes the Circadian Timekeeping System in Mammals. Nat. Commun. 2022, 13, 4652. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, J.; Zhang, L.; Van Dam, H.; Ten Dijke, P. UBE2O Negatively Regulates TRAF6-Mediated NF-κB Activation by Inhibiting TRAF6 Polyubiquitination. Cell Res. 2013, 23, 366–377. [Google Scholar] [CrossRef]
- Nakayama, K.I.; Nakayama, K. Ubiquitin Ligases: Cell-Cycle Control and Cancer. Nat. Rev. Cancer 2006, 6, 369–381. [Google Scholar] [CrossRef]
- Korenev, G.; Yakukhnov, S.; Druk, A.; Golovina, A.; Chasov, V.; Mirgayazova, R.; Ivanov, R.; Bulatov, E. USP7 Inhibitors in Cancer Immunotherapy: Current Status and Perspective. Cancers 2022, 14, 5539. [Google Scholar] [CrossRef]
- Drula, R.; Iluta, S.; Gulei, D.; Iuga, C.; Dima, D.; Ghiaur, G.; Buzoianu, A.D.; Ciechanover, A.; Tomuleasa, C. Exploiting the Ubiquitin System in Myeloid Malignancies. From Basic Research to Drug Discovery in MDS and AML. Blood Rev. 2022, 56, 100971. [Google Scholar] [CrossRef] [PubMed]
- Nijman, S.M.B.; Huang, T.T.; Dirac, A.M.G.; Brummelkamp, T.R.; Kerkhoven, R.M.; D’Andrea, A.D.; Bernards, R. The Deubiquitinating Enzyme USP1 Regulates the Fanconi Anemia Pathway. Mol. Cell 2005, 17, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Liu, N.; Xia, X.; Guo, Z.; Li, Y.; Jiang, L.; Zhou, R.; Tang, D.; Huang, H.; Liu, J. USP10 Modulates the SKP2/Bcr-Abl Axis via Stabilizing SKP2 in Chronic Myeloid Leukemia. Cell Discov. 2019, 5, 24. [Google Scholar] [CrossRef] [PubMed]
- Shibata, N.; Ohoka, N.; Tsuji, G.; Demizu, Y.; Miyawaza, K.; Ui-Tei, K.; Akiyama, T.; Naito, M. Deubiquitylase USP25 Prevents Degradation of BCR-ABL Protein and Ensures Proliferation of Ph-Positive Leukemia Cells. Oncogene 2020, 39, 3867–3878. [Google Scholar] [CrossRef]
- Fang, J.; Liu, X.; Bolanos, L.; Barker, B.; Rigolino, C.; Cortelezzi, A.; Oliva, E.N.; Cuzzola, M.; Grimes, H.L.; Fontanillo, C.; et al. A Calcium- and Calpain-Dependent Pathway Determines the Response to Lenalidomide in Myelodysplastic Syndromes. Nat. Med. 2016, 22, 727–734. [Google Scholar] [CrossRef]
- Sarodaya, N.; Karapurkar, J.; Kim, K.-S.; Hong, S.-H.; Ramakrishna, S. The Role of Deubiquitinating Enzymes in Hematopoiesis and Hematological Malignancies. Cancers 2020, 12, 1103. [Google Scholar] [CrossRef]
- Vinchi, F. Erythroid Differentiation: A Matter of Proteome Remodeling. HemaSphere 2018, 2, e26. [Google Scholar] [CrossRef]
- Wefes, I.; Mastrandrea, L.D.; Haldeman, M.; Koury, S.T.; Tamburlin, J.; Pickart, C.M.; Finley, D. Induction of Ubiquitin-Conjugating Enzymes during Terminal Erythroid Differentiation. Proc. Natl. Acad. Sci. USA 1995, 92, 4982–4986. [Google Scholar] [CrossRef]
- Haldeman, M.T.; Finley, D.; Pickart, C.M. Dynamics of Ubiquitin Conjugation during Erythroid Differentiation in Vitro. J. Biol. Chem. 1995, 270, 9507–9516. [Google Scholar] [CrossRef]
- Nguyen, A.T.; Prado, M.A.; Schmidt, P.J.; Sendamarai, A.K.; Wilson-Grady, J.T.; Min, M.; Campagna, D.R.; Tian, G.; Shi, Y.; Dederer, V.; et al. UBE2O Remodels the Proteome during Terminal Erythroid Differentiation. Science 2017, 357, eaan0218. [Google Scholar] [CrossRef] [PubMed]
- Yanagitani, K.; Juszkiewicz, S.; Hegde, R.S. UBE2O Is a Quality Control Factor for Orphans of Multiprotein Complexes. Science 2017, 357, 472–475. [Google Scholar] [CrossRef] [PubMed]
- Estey, E.; Döhner, H. Acute Myeloid Leukaemia. Lancet 2006, 368, 1894–1907. [Google Scholar] [CrossRef]
- Wang, J.; Wang, P.; Zhang, T.; Gao, Z.; Wang, J.; Feng, M.; Yin, R.; Zhang, H. Molecular Mechanisms for Stemness Maintenance of Acute Myeloid Leukemia Stem Cells. Blood Sci. 2019, 1, 77–83. [Google Scholar] [CrossRef]
- Colmone, A.; Amorim, M.; Pontier, A.L.; Wang, S.; Jablonski, E.; Sipkins, D.A. Leukemic Cells Create Bone Marrow Niches That Disrupt the Behavior of Normal Hematopoietic Progenitor Cells. Science 2008, 322, 1861–1865. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Chen, Z.; Wang, L.; Si, J.; Kang, J.; Li, Y.; Zheng, Y.; Gao, Y.; Nuermaimaiti, R.; You, M.J.; et al. Over Expression of Ubiquitin-conjugating Enzyme E2O in Bone Marrow Mesenchymal Stromal Cells Partially Attenuates Acute Myeloid Leukaemia Progression. Br. J. Haematol. 2023, 200, 476–488. [Google Scholar] [CrossRef] [PubMed]
- Tian, C. Low-Expression of UBE2O in Bone Marrow Mesenchymal Stromal Cells Promotes Acute Myeloid Leukemia Progression. Blood 2022, 140, 11423. [Google Scholar] [CrossRef]
- Castiglioni, S.; Di Fede, E.; Bernardelli, C.; Lettieri, A.; Parodi, C.; Grazioli, P.; Colombo, E.A.; Ancona, S.; Milani, D.; Ottaviano, E.; et al. KMT2A: Umbrella Gene for Multiple Diseases. Genes 2022, 13, 514. [Google Scholar] [CrossRef]
- Tkachuk, D.C.; Kohler, S.; Cleary, M.L. Involvement of a Homolog of Drosophila Trithorax by 11q23 Chromosomal Translocations in Acute Leukemias. Cell 1992, 71, 691–700. [Google Scholar] [CrossRef]
- Górecki, M.; Kozioł, I.; Kopystecka, A.; Budzyńska, J.; Zawitkowska, J.; Lejman, M. Updates in KMT2A Gene Rearrangement in Pediatric Acute Lymphoblastic Leukemia. Biomedicines 2023, 11, 821. [Google Scholar] [CrossRef]
- Rossi, J.G.; Bernasconi, A.R.; Alonso, C.N.; Rubio, P.L.; Gallego, M.S.; Carrara, C.A.; Guitter, M.R.; Eberle, S.E.; Cocce, M.; Zubizarreta, P.A.; et al. Lineage Switch in Childhood Acute Leukemia: An Unusual Event with Poor Outcome. Am. J. Hematol. 2012, 87, 890–897. [Google Scholar] [CrossRef] [PubMed]
- For The St. Jude Children’s Research Hospital–Washington University Pediatric Cancer Genome Project; Andersson, A.K.; Ma, J.; Wang, J.; Chen, X.; Gedman, A.L.; Dang, J.; Nakitandwe, J.; Holmfeldt, L.; Parker, M.; et al. The Landscape of Somatic Mutations in Infant MLL-Rearranged Acute Lymphoblastic Leukemias. Nat. Genet. 2015, 47, 330–337. [Google Scholar] [CrossRef]
- Agraz-Doblas, A.; Bueno, C.; Bashford-Rogers, R.; Roy, A.; Schneider, P.; Bardini, M.; Ballerini, P.; Cazzaniga, G.; Moreno, T.; Revilla, C.; et al. Unraveling the Cellular Origin and Clinical Prognostic Markers of Infant B-Cell Acute Lymphoblastic Leukemia Using Genome-Wide Analysis. Haematologica 2019, 104, 1176–1188. [Google Scholar] [CrossRef] [PubMed]
- Prelle, C.; Bursen, A.; Dingermann, T.; Marschalek, R. Secondary Mutations in t(4;11) Leukemia Patients. Leukemia 2013, 27, 1425–1427. [Google Scholar] [CrossRef] [PubMed]
- Lanza, C.; Gaidano, G.; Cimino, G.; Pastore, C.; Nomdedeu, J.; Volpe, G.; Vivenza, C.; Parvis, G.; Mazza, U.; Basso, G.; et al. Distribution of TP53 Mutations among Acute Leukemias with MLL Rearrangements. Genes Chromosom. Cancer 1996, 15, 48–53. [Google Scholar] [CrossRef]
- Poreba, E.; Lesniewicz, K.; Durzynska, J. Aberrant Activity of Histone–Lysine N-Methyltransferase 2 (KMT2) Complexes in Oncogenesis. IJMS 2020, 21, 9340. [Google Scholar] [CrossRef]
- Charliński, G.; Jurczyszyn, A. Non-Secretory Multiple Myeloma: Diagnosis and Management. Adv. Clin. Exp. Med. 2021, 31, 95–100. [Google Scholar] [CrossRef]
- Lv, Y.; Xing, F. Regulatory Roles of an Atypical Ubiquitin Ligase UBE2O in Orphans of Multi-Protein Complexes for Degradation. Turk. J. Biol. 2022, 46, 186–194. [Google Scholar] [CrossRef]
- Hideshima, T.; Bergsagel, P.L.; Kuehl, W.M.; Anderson, K.C. Advances in Biology of Multiple Myeloma: Clinical Applications. Blood 2004, 104, 607–618. [Google Scholar] [CrossRef]
- Zhang, Z.; Tong, J.; Tang, X.; Juan, J.; Cao, B.; Hurren, R.; Chen, G.; Taylor, P.; Xu, X.; Shi, C.; et al. The Ubiquitin Ligase HERC4 Mediates C-Maf Ubiquitination and Delays the Growth of Multiple Myeloma Xenografts in Nude Mice. Blood 2016, 127, 1676–1686. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, Z.; Li, J.; Tong, J.; Cao, B.; Taylor, P.; Tang, X.; Wu, D.; Moran, M.F.; Zeng, Y.; et al. The Ubiquitin-Conjugating Enzyme UBE2O Modulates c-Maf Stability and Induces Myeloma Cell Apoptosis. J. Hematol. Oncol. 2017, 10, 132. [Google Scholar] [CrossRef] [PubMed]
- Hurt, E.M.; Wiestner, A.; Rosenwald, A.; Shaffer, A.L.; Campo, E.; Grogan, T.; Bergsagel, P.L.; Kuehl, W.M.; Staudt, L.M. Overexpression of C-Maf Is a Frequent Oncogenic Event in Multiple Myeloma That Promotes Proliferation and Pathological Interactions with Bone Marrow Stroma. Cancer Cell 2004, 5, 191–199. [Google Scholar] [CrossRef]
- Rice, K.L.; Lin, X.; Wolniak, K.; Ebert, B.L.; Berkofsky-Fessler, W.; Buzzai, M.; Sun, Y.; Xi, C.; Elkin, P.; Levine, R.; et al. Analysis of Genomic Aberrations and Gene Expression Profiling Identifies Novel Lesions and Pathways in Myeloproliferative Neoplasms. Blood Cancer J. 2011, 1, e40. [Google Scholar] [CrossRef]
- Toffoli, S.; Bar, I.; Abdel-Sater, F.; Delrée, P.; Hilbert, P.; Cavallin, F.; Moreau, F.; Van Criekinge, W.; Lacroix-Triki, M.; Campone, M.; et al. Identification by Array Comparative Genomic Hybridization of a New Amplicon on Chromosome 17q Highly Recurrent in BRCA1 Mutated Triple Negative Breast Cancer. Breast Cancer Res. 2014, 16, 466. [Google Scholar] [CrossRef]
- Vila, I.K.; Yao, Y.; Kim, G.; Xia, W.; Kim, H.; Kim, S.-J.; Park, M.-K.; Hwang, J.P.; González-Billalabeitia, E.; Hung, M.-C.; et al. A UBE2O-AMPKα2 Axis That Promotes Tumor Initiation and Progression Offers Opportunities for Therapy. Cancer Cell 2017, 31, 208–224. [Google Scholar] [CrossRef]
- Wang, X.; Li, X.; Cheng, Y.; Sun, X.; Sun, X.; Self, S.; Kooperberg, C.; Dai, J.Y. Copy Number Alterations Detected by Whole-Exome and Whole-Genome Sequencing of Esophageal Adenocarcinoma. Hum. Genom. 2015, 9, 22. [Google Scholar] [CrossRef] [PubMed]
- Briffa, R.; Um, I.; Faratian, D.; Zhou, Y.; Turnbull, A.K.; Langdon, S.P.; Harrison, D.J. Multi-Scale Genomic, Transcriptomic and Proteomic Analysis of Colorectal Cancer Cell Lines to Identify Novel Biomarkers. PLoS ONE 2015, 10, e0144708. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yang, X.; Lu, Y.; Zhao, Y.; Meng, R.; Zhang, S.; Dong, X.; Xu, S.; Wu, G. UBE2O Targets Mxi1 for Ubiquitination and Degradation to Promote Lung Cancer Progression and Radioresistance. Cell Death Differ. 2021, 28, 671–684. [Google Scholar] [CrossRef]
- Schreiber-Agus, N.; Meng, Y.; Hoang, T.; Hou, H.; Chen, K.; Greenberg, R.; Cordon-Cardo, C.; Lee, H.-W.; DePinho, R.A. Role of Mxi1 in Ageing Organ Systems and the Regulation of Normal and Neoplastic Growth. Nature 1998, 393, 483–487. [Google Scholar] [CrossRef]
- McGlynn, K.A.; Petrick, J.L.; El-Serag, H.B. Epidemiology of Hepatocellular Carcinoma. Hepatology 2021, 73, 4–13. [Google Scholar] [CrossRef]
- Ma, M.; Zhang, C.; Cao, R.; Tang, D.; Sang, X.; Zou, S.; Wang, X.; Xu, H.; Liu, G.; Dai, L.; et al. UBE2O Promotes Lipid Metabolic Reprogramming and Liver Cancer Progression by Mediating HADHA Ubiquitination. Oncogene 2022, 41, 5199–5213. [Google Scholar] [CrossRef] [PubMed]
- Chikhalya, A.; Dittmann, M.; Zheng, Y.; Sohn, S.-Y.; Rice, C.M.; Hearing, P. Human IFIT3 Protein Induces Interferon Signaling and Inhibits Adenovirus Immediate Early Gene Expression. mBio 2021, 12, e02829-21. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhou, Y.; Hou, J.; Bai, C.; Li, Z.; Fan, J.; Ng, I.O.L.; Zhou, W.; Sun, H.; Dong, Q.; et al. Hepatic IFIT3 Predicts Interferon-α Therapeutic Response in Patients of Hepatocellular Carcinoma. Hepatology 2017, 66, 152–166. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, Y.; Cheng, C.; Wu, Y.; Liang, S.-H.; Wu, L.; Wang, H.; Tu, C.; Yao, H.-H.; Meng, F.-Z.; et al. UBE2O Reduces the Effectiveness of Interferon-α via Degradation of IFIT3 in Hepatocellular Carcinoma. Cell Death Dis. 2023, 14, 854. [Google Scholar] [CrossRef] [PubMed]
- Vila, I.K.; Park, M.K.; Setijono, S.R.; Yao, Y.; Kim, H.; Badin, P.-M.; Choi, S.; Narkar, V.; Choi, S.-W.; Chung, J.; et al. A Muscle-Specific UBE2O/AMPKα2 Axis Promotes Insulin Resistance and Metabolic Syndrome in Obesity. JCI Insight 2019, 4, e128269. [Google Scholar] [CrossRef]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A Nutrient and Energy Sensor That Maintains Energy Homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef]
- Cheng, J.; Zheng, H.; Liu, C.; Jin, J.; Xing, Z.; Wu, Y. Age-Associated UBE2O Reduction Promotes Neuronal Death in Alzheimer’s Disease. JAD 2023, 93, 1083–1093. [Google Scholar] [CrossRef]
- Popovic, D.; Vucic, D.; Dikic, I. Ubiquitination in Disease Pathogenesis and Treatment. Nat. Med. 2014, 20, 1242–1253. [Google Scholar] [CrossRef]
- Chen, H.; Wu, G.; Gao, S.; Guo, R.; Zhao, Z.; Yuan, H.; Liu, S.; Wu, J.; Lu, X.; Yuan, X.; et al. Discovery of Potent Small-Molecule Inhibitors of Ubiquitin-Conjugating Enzyme UbcH5c from α-Santonin Derivatives. J. Med. Chem. 2017, 60, 6828–6852. [Google Scholar] [CrossRef]
- Morreale, F.E.; Testa, A.; Chaugule, V.K.; Bortoluzzi, A.; Ciulli, A.; Walden, H. Mind the Metal: A Fragment Library-Derived Zinc Impurity Binds the E2 Ubiquitin-Conjugating Enzyme Ube2T and Induces Structural Rearrangements. J. Med. Chem. 2017, 60, 8183–8191. [Google Scholar] [CrossRef] [PubMed]
- Maffeo, B.; Panuzzo, C.; Itri, F.; Savi, A.; Marini, S.; Bracco, E.; Fava, C.; Pergolizzi, B.; Cilloni, D. The Potential Role of UBE2O in the Treatment of Ineffective Erythropoiesis in Patients with Myelodysplastic Syndrome; EHA, European Hematology Association: The Hague, The Netherlands, 2024. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
| UBE2O Regulation | Effects | |
|---|---|---|
| Leukemic diseases | ||
| Myelodysplastic syndromes and β-thalassemia | Upregulation | α-globin ubiquitination; promotion of proteome remodeling during RBC maturation |
| Acute myeloid leukemia | Upregulation | NF-κB pathway inhibition; reduced AML cells’ proliferation |
| Multiple myeloma | Upregulation | c-Maf ubiquitination; increased MM cell death |
| KMT2A rearranged leukemias | Downregulation | Increased WT-KMT2A stability; reduced leukemic cells’ proliferation |
| Solid tumors | ||
| Breast and prostate cancer | Downregulation | Reduced AMPKα2 ubiquitination; reduced activation of mTORC1 pathway; reduced cancer cell proliferation |
| Lung cancer | Downregulation | Decreased Mxi1 ubiquitination; reduced radioresistance; reduced cancer proliferation |
| Hepatocellular carcinoma | Downregulation | Reduced HADHA ubiquitination; increased IFN-α signaling efficacy; reduced cancer cells survival. |
| Non-oncologic diseases | ||
| Metabolic disorders | Downregulation | Reduced AMPKα2 ubiquitination; increased insulin sensitivity |
| Alzheimer diseases | Overexpression | Reduced Aβ deposit; reduced neuronal cells death |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maffeo, B.; Cilloni, D. The Ubiquitin-Conjugating Enzyme E2 O (UBE2O) and Its Therapeutic Potential in Human Leukemias and Solid Tumors. Cancers 2024, 16, 3064. https://doi.org/10.3390/cancers16173064
Maffeo B, Cilloni D. The Ubiquitin-Conjugating Enzyme E2 O (UBE2O) and Its Therapeutic Potential in Human Leukemias and Solid Tumors. Cancers. 2024; 16(17):3064. https://doi.org/10.3390/cancers16173064
Chicago/Turabian StyleMaffeo, Beatrice, and Daniela Cilloni. 2024. "The Ubiquitin-Conjugating Enzyme E2 O (UBE2O) and Its Therapeutic Potential in Human Leukemias and Solid Tumors" Cancers 16, no. 17: 3064. https://doi.org/10.3390/cancers16173064
APA StyleMaffeo, B., & Cilloni, D. (2024). The Ubiquitin-Conjugating Enzyme E2 O (UBE2O) and Its Therapeutic Potential in Human Leukemias and Solid Tumors. Cancers, 16(17), 3064. https://doi.org/10.3390/cancers16173064