
Colocalised Genetic Associations Reveal Alternative Splicing Variants as Candidate Causal Links for Breast Cancer Risk in 10 Loci

 , ,
, , {kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
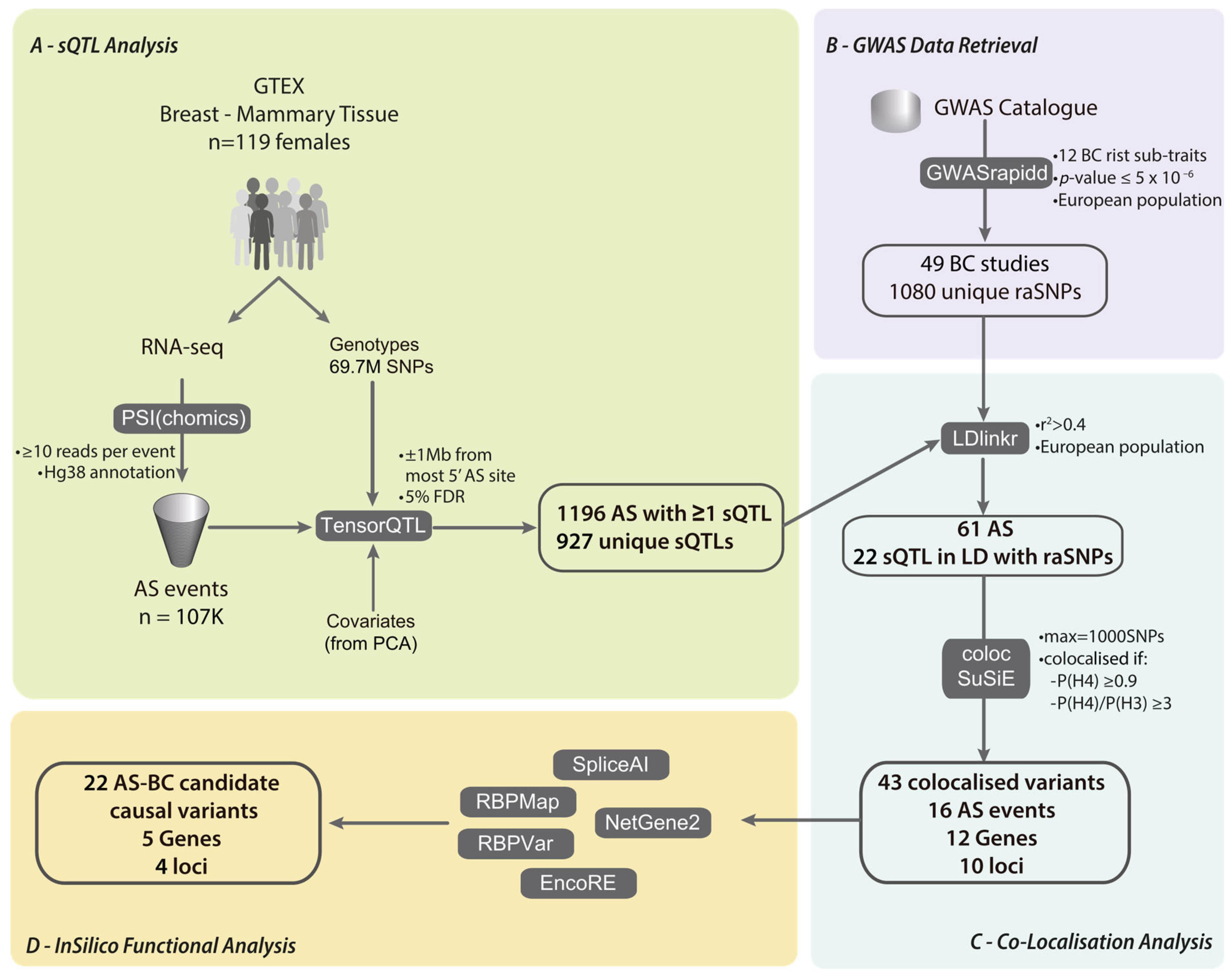
2. Materials and Methods
2.1. Data Sources
2.2. Ancestry Analysis
2.3. RNA-Seq Analysis
2.4. Alternative Splicing Quantification
2.5. sQTL Analysis
2.6. Linkage Disequilibrium Analysis
2.7. Colocalisation Analysis
2.8. In Silico Splicing Analysis
2.9. eQTL Analysis
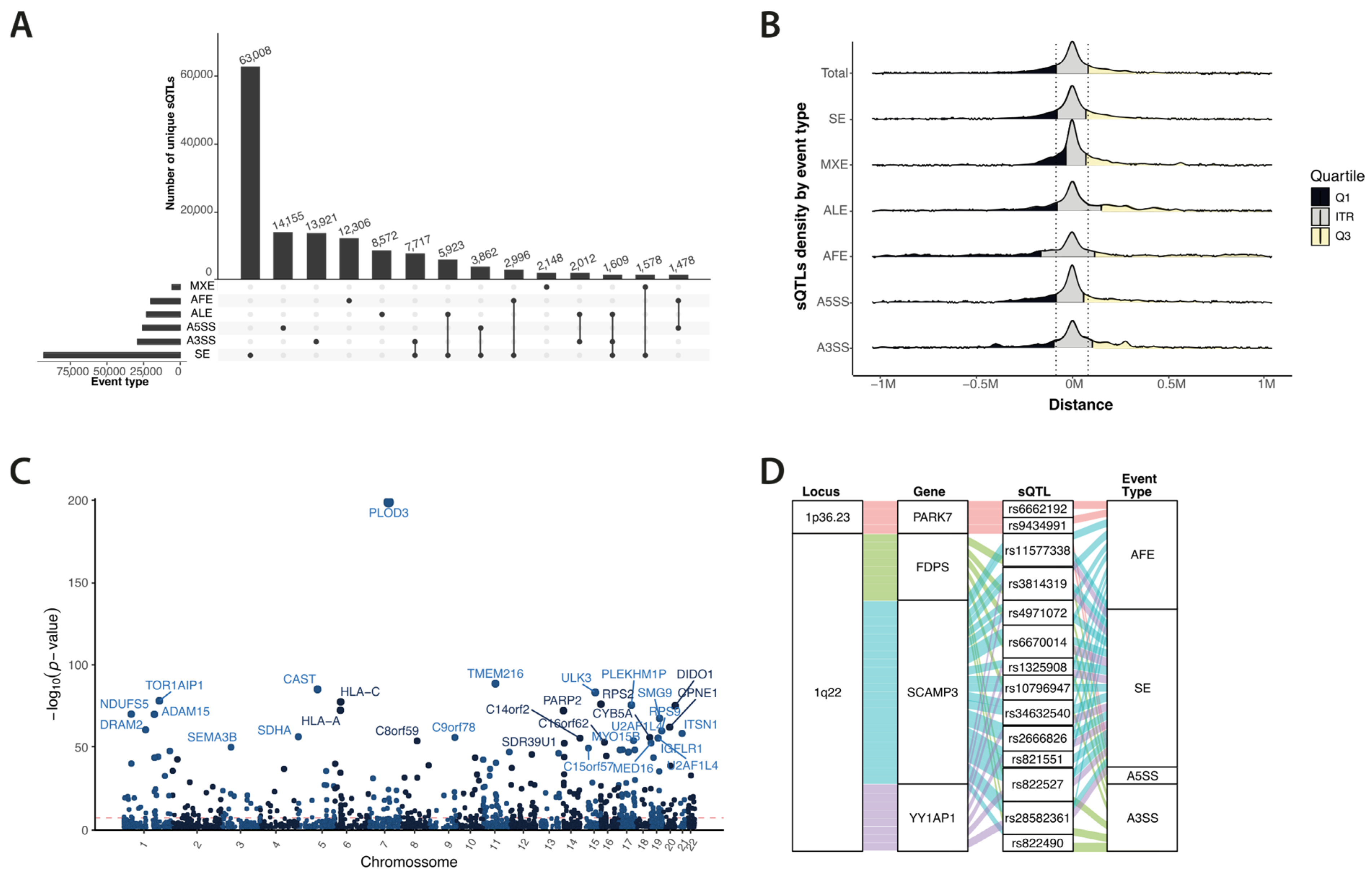
3. Results
3.1. Splicing QTL Hints at Underlying Gene Regulation
3.2. Colocalisation Implicates Splicing Modulation in Risk for BC at Ten Loci
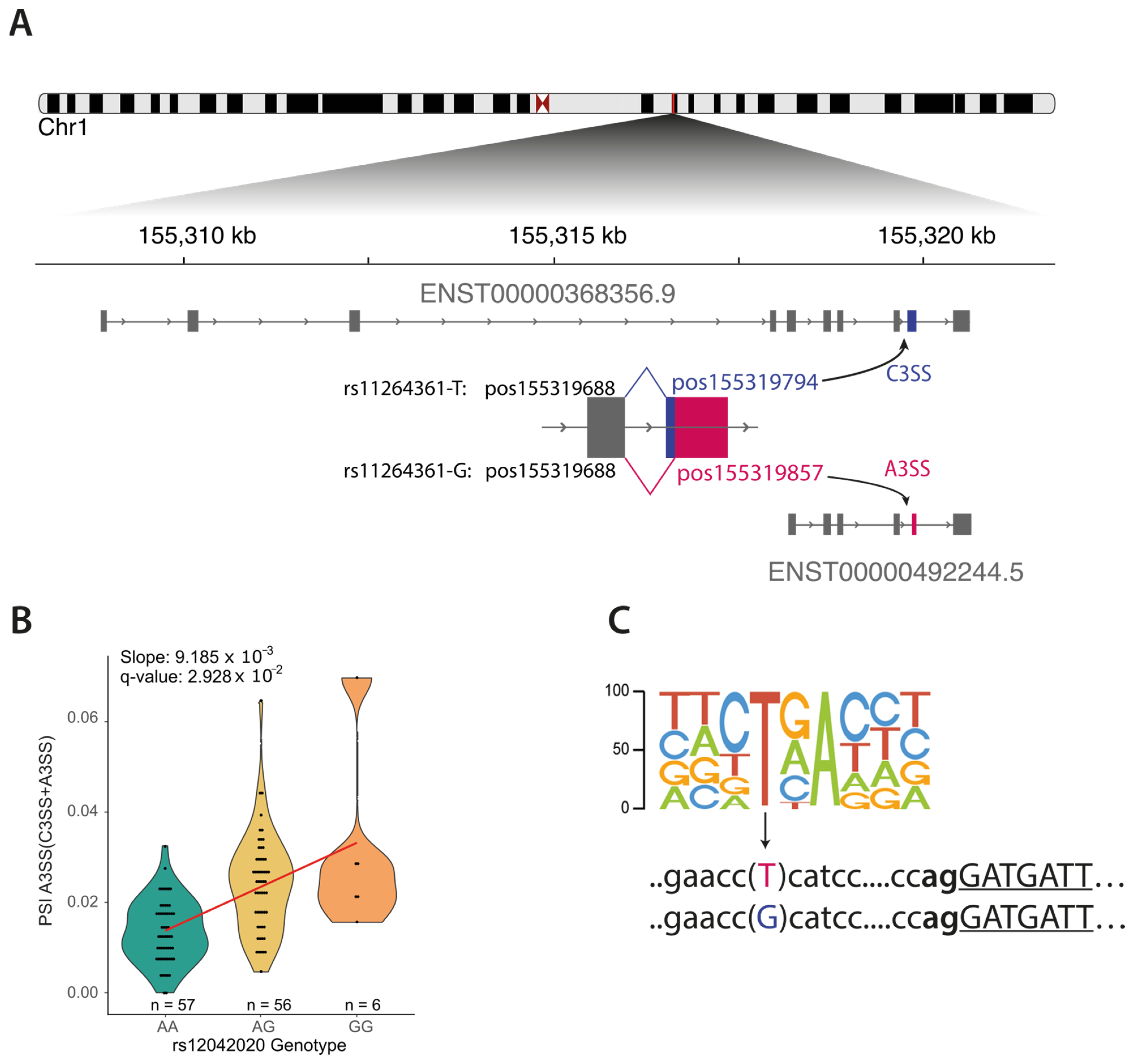
3.3. Linking Risk Variants to Mechanism at Three Loci
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Michailidou, K.; Lindström, S.; Dennis, J.; Beesley, J.; Hui, S.; Kar, S.; Lemaçon, A.; Soucy, P.; Glubb, D.; Rostamianfar, A.; et al. Association Analysis Identifies 65 New Breast Cancer Risk Loci. Nature 2017, 551, 92–94. [Google Scholar] [CrossRef]
- Welter, D.; MacArthur, J.; Morales, J.; Burdett, T.; Hall, P.; Junkins, H.; Klemm, A.; Flicek, P.; Manolio, T.; Hindorff, L.; et al. The NHGRI GWAS Catalog, a Curated Resource of SNP-Trait Associations. Nucleic Acids Res. 2014, 42, D1001–D1006. [Google Scholar] [CrossRef]
- Xavier, J.M.; Magno, R.; Russell, R.; de Almeida, B.P.; Jacinta-Fernandes, A.; Duarte, A.; Dunning, M.; Samarajiwa, S.; O’Reilly, M.; Rocha, C.L.; et al. Mapping of Cis-Regulatory Variants by Differential Allelic Expression Analysis Identifies Candidate Risk Variants and Target Genes of 27 Breast Cancer Risk Loci. medRxiv 2022. [Google Scholar] [CrossRef]
- Jacinta-Fernandes, A.; Xavier, J.M.; Magno, R.; Lage, J.G.; Maia, A.T. Allele-Specific MiRNA-Binding Analysis Identifies Candidate Target Genes for Breast Cancer Risk. NPJ Genom. Med. 2020, 5, 4. [Google Scholar] [CrossRef]
- Kalniņa, Z.; Zayakin, P.; Siliņa, K.; Line, A. Alterations of Pre-MRNA Splicing in Cancer. Genes Chromosomes Cancer 2005, 42, 342–357. [Google Scholar] [CrossRef] [PubMed]
- Baralle, F.E.; Giudice, J. Alternative Splicing as a Regulator of Development and Tissue Identity. Nat. Rev. Mol. Cell Biol. 2017, 18, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Kurtovic-Kozaric, A.; Przychodzen, B.; Singh, J.; Konarska, M.M.; Clemente, M.J.; Otrock, Z.K.; Nakashima, M.; Hsi, E.D.; Yoshida, K.; Shiraishi, Y.; et al. PRPF8 Defects Cause Missplicing in Myeloid Malignancies. Leukemia 2015, 29, 126–136. [Google Scholar] [CrossRef]
- Scotti, M.M.; Swanson, M.S. RNA Mis-Splicing in Disease. Nat. Rev. Genet. 2016, 17, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Jaganathan, K.; Panagiotopoulou, S.K.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548.e24. [Google Scholar] [CrossRef]
- Zhang, J.; Manley, J.L. Misregulation of Pre-MRNA Alternative Splicing in Cancer. Cancer Discov. 2013, 3, 1228–1237. [Google Scholar] [CrossRef]
- Climente-González, H.; Porta-Pardo, E.; Godzik, A.; Eyras, E. The Functional Impact of Alternative Splicing in Cancer. Cell Rep. 2017, 20, 2215–2226. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Chen, C.; Rao, M.; Zhang, M.; Lu, Z.; Cai, Y.; Ying, P.; Li, B.; Wang, H.; Wang, L.; et al. Aberrant RNA Splicing Is a Primary Link between Genetic Variation and Pancreatic Cancer Risk. Cancer Res. 2022, 82, 2084–2096. [Google Scholar] [CrossRef]
- Li, D.; Harlan-Williams, L.M.; Kumaraswamy, E.; Jensen, R.A. BRCA1—No Matter How You Splice It. Cancer Res. 2019, 79, 2091–2098. [Google Scholar] [CrossRef]
- de la Hoya, M.; Soukarieh, O.; López-Perolio, I.; Vega, A.; Walker, L.C.; van Ierland, Y.; Baralle, D.; Santamariña, M.; Lattimore, V.; Wijnen, J.; et al. Combined Genetic and Splicing Analysis of BRCA1 c.[594-2A>C; 641A>G] Highlights the Relevance of Naturally Occurring in-Frame Transcripts for Developing Disease Gene Variant Classification Algorithms. Hum. Mol. Genet. 2016, 25, 2256–2268. [Google Scholar] [CrossRef]
- Dunning, A.M.; Healey, C.S.; Baynes, C.; Maia, A.-T.T.; Scollen, S.; Vega, A.; Rodríguez, R.; Barbosa-Morais, N.L.; Ponder, B.A.J.; Low, Y.-L.L.; et al. Association of ESR1 Gene Tagging SNPs with Breast Cancer Risk. Hum. Mol. Genet. 2009, 18, 1131–1139. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Wang, Z.; Mei, S.; Yang, N.; Yang, Y.; Ke, J.; Zhu, Y.; Gong, Y.; Zou, D.; Peng, X.; et al. CancerSplicingQTL: A Database for Genome-Wide Identification of Splicing QTLs in Human Cancer. Nucleic Acids Res. 2019, 47, D909–D916. [Google Scholar] [CrossRef] [PubMed]
- Caswell, J.L.; Camarda, R.; Zhou, A.Y.; Huntsman, S.; Hu, D.; Brenner, S.E.; Zaitlen, N.; Goga, A.; Ziv, E. Multiple Breast Cancer Risk Variants Are Associated with Differential Transcript Isoform Expression in Tumors. Hum. Mol. Genet. 2015, 24, 7421–7431. [Google Scholar] [CrossRef] [PubMed]
- Magno, R.; Maia, A.-T. Gwasrapidd: An R Package to Query, Download and Wrangle GWAS Catalog Data. Bioinformatics 2020, 36, 649–650. [Google Scholar] [CrossRef]
- Sollis, E.; Mosaku, A.; Abid, A.; Buniello, A.; Cerezo, M.; Gil, L.; Groza, T.; Güneş, O.; Hall, P.; Hayhurst, J.; et al. The NHGRI-EBI GWAS Catalog: Knowledgebase and Deposition Resource. Nucleic Acids Res. 2023, 51, D977–D985. [Google Scholar] [CrossRef]
- 1000 Genome Project Consortium. A Global Reference for Human Genetic Variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve Years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Bonfield, J.K.; Marshall, J.; Danecek, P.; Li, H.; Ohan, V.; Whitwham, A.; Keane, T. HTSlib: C Library for Reading/Writing High-Throughput Sequencing Data. Gigascience 2021, 10, giab007. [Google Scholar] [CrossRef] [PubMed]
- Gay, N.R.; Gloudemans, M.; Antonio, M.L.; Abell, N.S.; Balliu, B.; Park, Y.; Martin, A.R.; Musharoff, S.; Rao, A.S.; Aguet, F.; et al. Impact of Admixture and Ancestry on EQTL Analysis and GWAS Colocalization in GTEx. Genome Biol. 2020, 21, 233. [Google Scholar] [CrossRef] [PubMed]
- SRA-Tools—NCBI. Available online: https://github.com/ncbi/sra-tools/ (accessed on 23 September 2020).
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize Analysis Results for Multiple Tools and Samples in a Single Report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Li, Y.I.; Knowles, D.A.; Humphrey, J.; Barbeira, A.N.; Dickinson, S.P.; Im, H.K.; Pritchard, J.K. Annotation-Free Quantification of RNA Splicing Using LeafCutter. Nat. Genet. 2017, 50, 151–158. [Google Scholar] [CrossRef]
- Saraiva-Agostinho, N.; Barbosa-Morais, N.L. Psichomics: Graphical Application for Alternative Splicing Quantification and Analysis. Nucleic Acids Res. 2019, 47, e7. [Google Scholar] [CrossRef] [PubMed]
- Ongen, H.; Buil, A.; Brown, A.A.; Dermitzakis, E.T.; Delaneau, O. Fast and Efficient QTL Mapper for Thousands of Molecular Phenotypes. Bioinformatics 2015, 32, 1479–1485. [Google Scholar] [CrossRef]
- Taylor-Weiner, A.; Aguet, F.; Haradhvala, N.J.; Gosai, S.; Anand, S.; Kim, J.; Ardlie, K.; Van Allen, E.M.; Getz, G. Scaling Computational Genomics to Millions of Individuals with GPUs. Genome Biol. 2019, 20, 228. [Google Scholar] [CrossRef]
- Machiela, M.J.; Chanock, S.J. LDlink: A Web-Based Application for Exploring Population-Specific Haplotype Structure and Linking Correlated Alleles of Possible Functional Variants. Bioinformatics 2015, 31, 3555–3557. [Google Scholar] [CrossRef]
- Myers, T.A.; Chanock, S.J.; Machiela, M.J. LDlinkR: An R Package for Rapidly Calculating Linkage Disequilibrium Statistics in Diverse Populations. Front. Genet. 2020, 11, 157. [Google Scholar] [CrossRef] [PubMed]
- Wallace, C. A More Accurate Method for Colocalisation Analysis Allowing for Multiple Causal Variants. PLoS Genet. 2021, 17, e1009440. [Google Scholar] [CrossRef] [PubMed]
- Durinck, S.; Moreau, Y.; Kasprzyk, A.; Davis, S.; De Moor, B.; Brazma, A.; Huber, W. BioMart and Bioconductor: A Powerful Link between Biological Databases and Microarray Data Analysis. Bioinformatics 2005, 21, 3439–3440. [Google Scholar] [CrossRef]
- Brunak, S.; Engelbrecht, J.; Knudsen, S. Prediction of Human MRNA Donor and Acceptor Sites from the DNA Sequence. J. Mol. Biol. 1991, 220, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Van Nostrand, E.L.; Freese, P.; Pratt, G.A.; Wang, X.; Wei, X.; Xiao, R.; Blue, S.M.; Chen, J.Y.; Cody, N.A.L.; Dominguez, D.; et al. A Large-Scale Binding and Functional Map of Human RNA-Binding Proteins. Nature 2020, 583, 711–719. [Google Scholar] [CrossRef]
- Zhao, W.; Zhang, S.; Zhu, Y.; Xi, X.; Bao, P.; Ma, Z.; Kapral, T.H.; Chen, S.; Zagrovic, B.; Yang, Y.T.; et al. POSTAR3: An Updated Platform for Exploring Post-Transcriptional Regulation Coordinated by RNA-Binding Proteins. Nucleic Acids Res. 2022, 50, D287–D294. [Google Scholar] [CrossRef] [PubMed]
- Paz, I.; Kosti, I.; Ares, M.; Cline, M.; Mandel-Gutfreund, Y. RBPmap: A Web Server for Mapping Binding Sites of RNA-Binding Proteins. Nucleic Acids Res. 2014, 42, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Desmet, F.-O.O.; Hamroun, D.; Lalande, M.; Collod-Bëroud, G.; Claustres, M.; Béroud, C.; Collod-Béroud, G.; Claustres, M.; Béroud, C. Human Splicing Finder: An Online Bioinformatics Tool to Predict Splicing Signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef]
- Ryan, M.; Wong, W.C.; Brown, R.; Akbani, R.; Su, X.; Broom, B.; Melott, J.; Weinstein, J. TCGASpliceSeq a Compendium of Alternative MRNA Splicing in Cancer. Nucleic Acids Res. 2016, 44, D1018–D1022. [Google Scholar] [CrossRef]
- Shu, X.; Long, J.; Cai, Q.; Kweon, S.-S.; Choi, J.-Y.; Kubo, M.; Park, S.K.; Bolla, M.K.; Dennis, J.; Wang, Q.; et al. Identification of Novel Breast Cancer Susceptibility Loci in Meta-Analyses Conducted among Asian and European Descendants. Nat. Commun. 2020, 11, 1217. [Google Scholar] [CrossRef] [PubMed]
- Ju, D.; Hui, D.; Hammond, D.A.; Wonkam, A.; Tishkoff, S.A. Importance of Including Non-European Populations in Large Human Genetic Studies to Enhance Precision Medicine. Annu. Rev. Biomed. Data Sci. 2022, 5, 321–339. [Google Scholar] [CrossRef] [PubMed]
- Oliva, M.; Muñoz-Aguirre, M.; Kim-Hellmuth, S.; Wucher, V.; Gewirtz, A.D.H.; Cotter, D.J.; Parsana, P.; Kasela, S.; Balliu, B.; Viñuela, A.; et al. The Impact of Sex on Gene Expression across Human Tissues. Science 2020, 369, eaba3066. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Garrido-Martín, D.; Borsari, B.; Calvo, M.; Reverter, F.; Guigó, R. Identification and Analysis of Splicing Quantitative Trait Loci across Multiple Tissues in the Human Genome. Nat. Commun. 2021, 12, 727. [Google Scholar] [CrossRef] [PubMed]
- Glinos, D.A.; Garborcauskas, G.; Hoffman, P.; Ehsan, N.; Jiang, L.; Gokden, A.; Dai, X.; Aguet, F.; Brown, K.L.; Garimella, K.; et al. Transcriptome Variation in Human Tissues Revealed by Long-Read Sequencing. Nature 2022, 608, 353–359. [Google Scholar] [CrossRef]
- Walker, R.L.; Ramaswami, G.; Hartl, C.; Mancuso, N.; Gandal, M.J.; de la Torre-Ubieta, L.; Pasaniuc, B.; Stein, J.L.; Geschwind, D.H. Genetic Control of Expression and Splicing in Developing Human Brain Informs Disease Mechanisms. Cell 2019, 179, 750–771.e22. [Google Scholar] [CrossRef]
- Schwartz, S.; Meshorer, E.; Ast, G. Chromatin Organization Marks Exon-Intron Structure. Nat. Struct. Mol. Biol. 2009, 16, 990–995. [Google Scholar] [CrossRef] [PubMed]
- Spies, N.; Nielsen, C.B.; Padgett, R.A.; Burge, C.B. Biased Chromatin Signatures around Polyadenylation Sites and Exons. Mol. Cell 2009, 36, 245–254. [Google Scholar] [CrossRef]
- Herzel, L.; Ottoz, D.S.M.; Alpert, T.; Neugebauer, K.M. Splicing and Transcription Touch Base: Co-Transcriptional Spliceosome Assembly and Function. Nat. Rev. Mol. Cell Biol. 2017, 18, 637–650. [Google Scholar] [CrossRef]
- Seshacharyulu, P.; Rachagani, S.; Muniyan, S.; Siddiqui, J.A.; Cruz, E.; Sharma, S.; Krishnan, R.; Killips, B.J.; Sheinin, Y.; Lele, S.M.; et al. FDPS Cooperates with PTEN Loss to Promote Prostate Cancer Progression through Modulation of Small GTPases/AKT Axis. Oncogene 2019, 38, 5265–5280. [Google Scholar] [CrossRef]
- Reilly, J.E.; Neighbors, J.D.; Hohl, R.J. Targeting Protein Geranylgeranylation Slows Tumor Development in a Murine Model of Prostate Cancer Metastasis. Cancer Biol. Ther. 2017, 18, 872–882. [Google Scholar] [CrossRef]
- Peall, K.J.; Smith, D.J.; Kurian, M.A.; Wardle, M.; Waite, A.J.; Hedderly, T.; Lin, J.P.; Smith, M.; Whone, A.; Pall, H.; et al. SGCE Mutations Cause Psychiatric Disorders: Clinical and Genetic Characterization. Brain 2013, 136, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Qiu, T.; Jiang, D.; Xu, H.; Zou, L.; Yang, Q.; Chen, C.; Jiao, B. SGCE Promotes Breast Cancer Stem Cells by Stabilizing EGFR. Adv. Sci. 2020, 7, 1903700. [Google Scholar] [CrossRef]
- Bell, J.L.; Hagemann, S.; Holien, J.K.; Liu, T.; Nagy, Z.; Chulte, J.H.; Misiak, D.; Hüttelmaier, S. Identification of Rna-Binding Proteins as Targetable Putative Oncogenes in Neuroblastoma. Int. J. Mol. Sci. 2020, 21, 5098. [Google Scholar] [CrossRef] [PubMed]
- Koc, E.C.; Haciosmanoglu, E.; Claudio, P.P.; Wolf, A.; Califano, L.; Friscia, M.; Cortese, A.; Koc, H. Impaired Mitochondrial Protein Synthesis in Head and Neck Squamous Cell Carcinoma. Mitochondrion 2015, 24, 113–121. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Besouro-Duarte, A.; Carrasqueiro, B.; Sousa, S.; Xavier, J.M.; Maia, A.-T. Colocalised Genetic Associations Reveal Alternative Splicing Variants as Candidate Causal Links for Breast Cancer Risk in 10 Loci. Cancers 2024, 16, 3020. https://doi.org/10.3390/cancers16173020
Besouro-Duarte A, Carrasqueiro B, Sousa S, Xavier JM, Maia A-T. Colocalised Genetic Associations Reveal Alternative Splicing Variants as Candidate Causal Links for Breast Cancer Risk in 10 Loci. Cancers. 2024; 16(17):3020. https://doi.org/10.3390/cancers16173020
Chicago/Turabian StyleBesouro-Duarte, André, Beatriz Carrasqueiro, Sofia Sousa, Joana M. Xavier, and Ana-Teresa Maia. 2024. "Colocalised Genetic Associations Reveal Alternative Splicing Variants as Candidate Causal Links for Breast Cancer Risk in 10 Loci" Cancers 16, no. 17: 3020. https://doi.org/10.3390/cancers16173020
APA StyleBesouro-Duarte, A., Carrasqueiro, B., Sousa, S., Xavier, J. M., & Maia, A.-T. (2024). Colocalised Genetic Associations Reveal Alternative Splicing Variants as Candidate Causal Links for Breast Cancer Risk in 10 Loci. Cancers, 16(17), 3020. https://doi.org/10.3390/cancers16173020