
Expression of Endogenous Retroviral RNA in Prostate Tumors has Prognostic Value and Shows Differences among Americans of African Versus European/Middle Eastern Ancestry

 ,
, 
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Selection
2.2. Locating Ancestry from Sequence Reads (LASER)
2.3. RNA-Seq Analysis of ERVmap
2.4. Transcriptome Analysis of ERVs in Black American and White American PCa Patients
2.5. ERV-Targeted Genes
2.6. PCa Tissue Microarrays and Immunohistochemistry
2.7. Statistical Analysis of ERV Protein Expression in TMAs from BA and WA PCa Patients
2.8. Selection of ERV Biomarkers for Prognostication of Biochemical Relapse
2.9. Risk Evaluation
2.10. Survival Model
3. Results
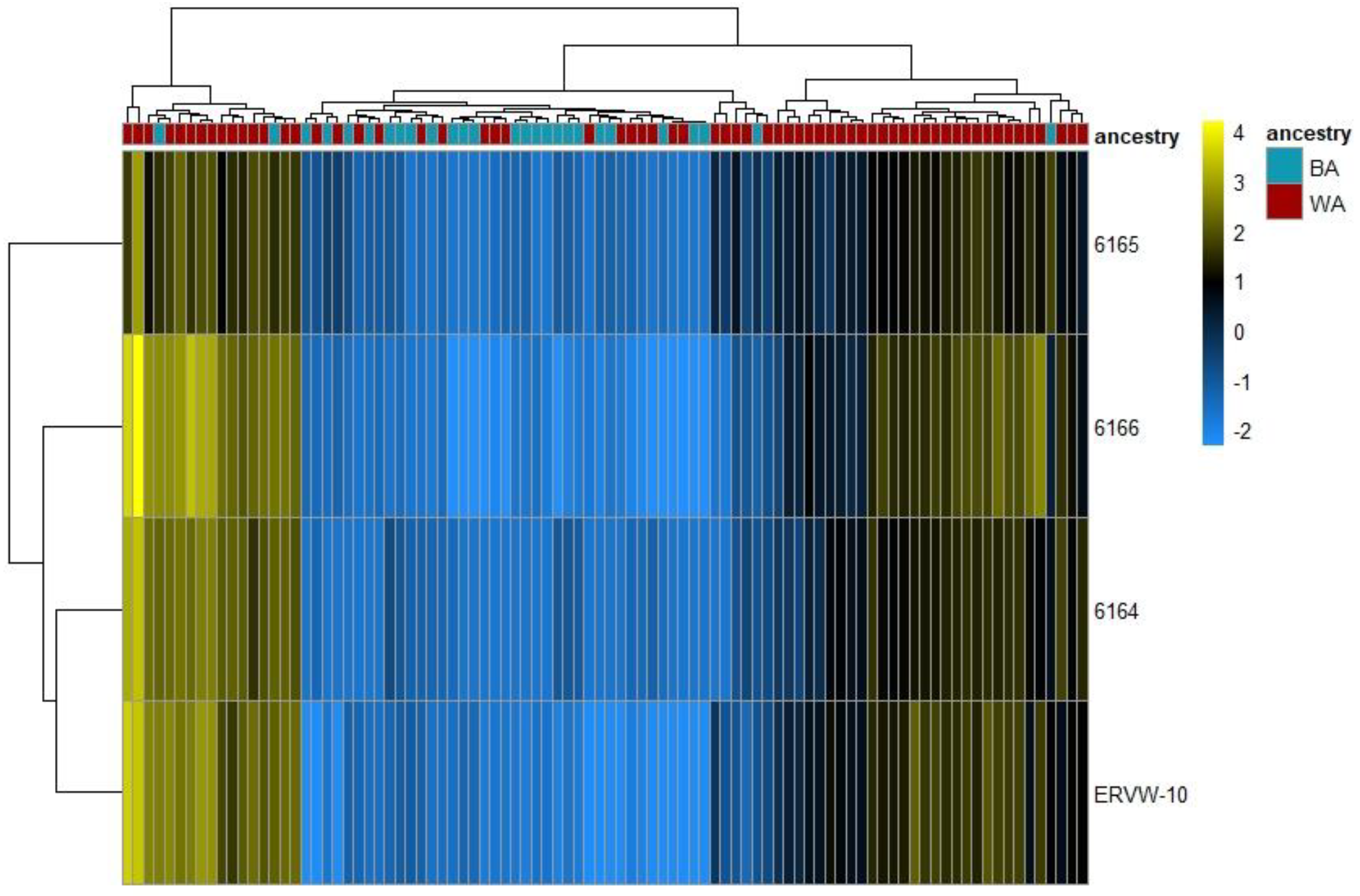
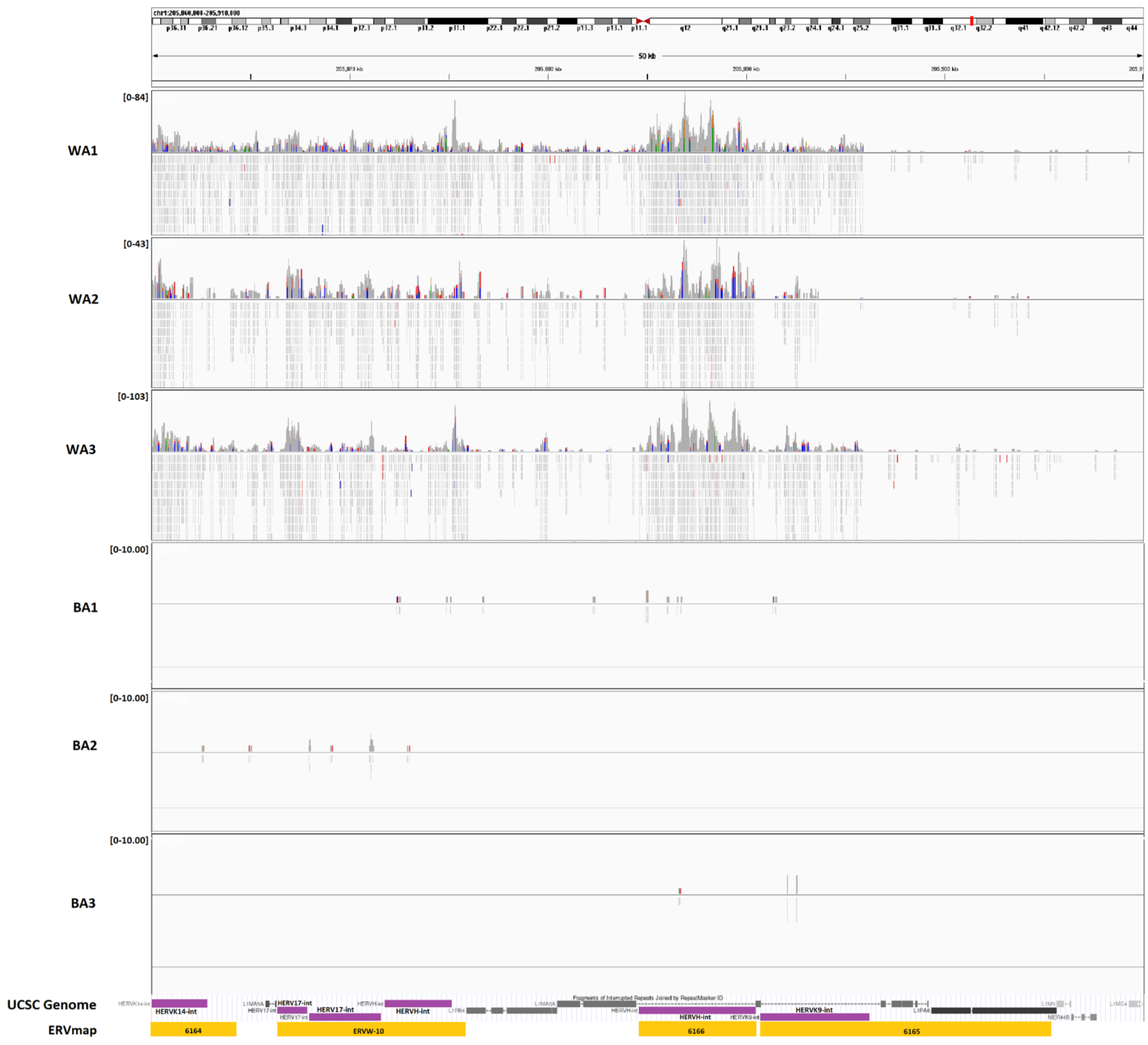
3.1. ERV Expression Profiles of Tumor-Enriched Samples from Black and White American PCa Patients
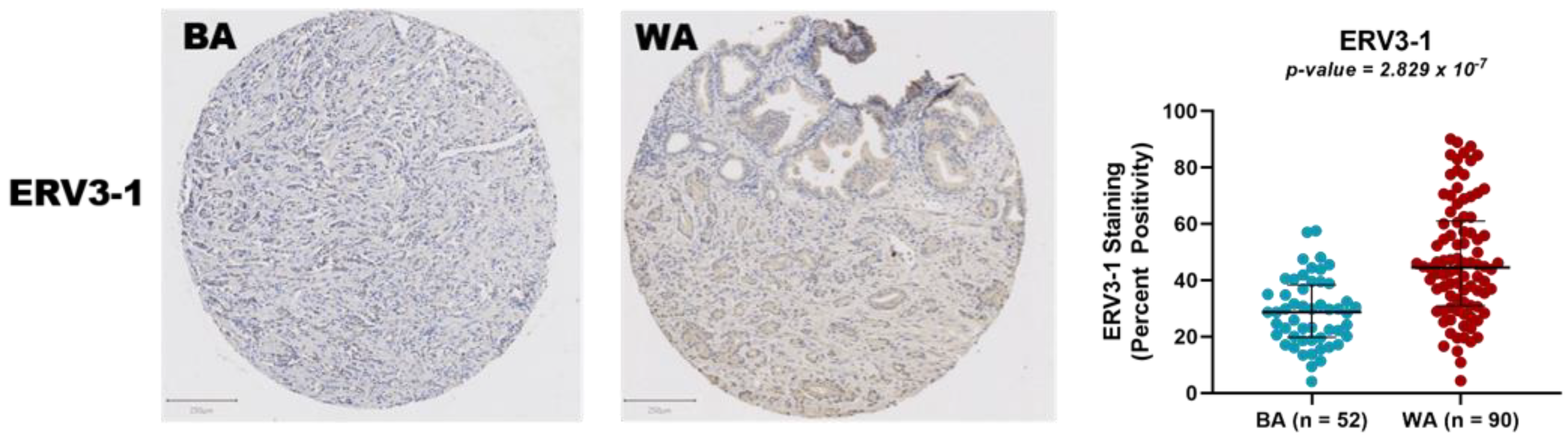
3.2. Protein Expression Analysis of ERV3-1 in PCa of BA and WA Patients Using TMAs
3.3. Expression of Genes Adjacent to Differentially Regulated ERVs
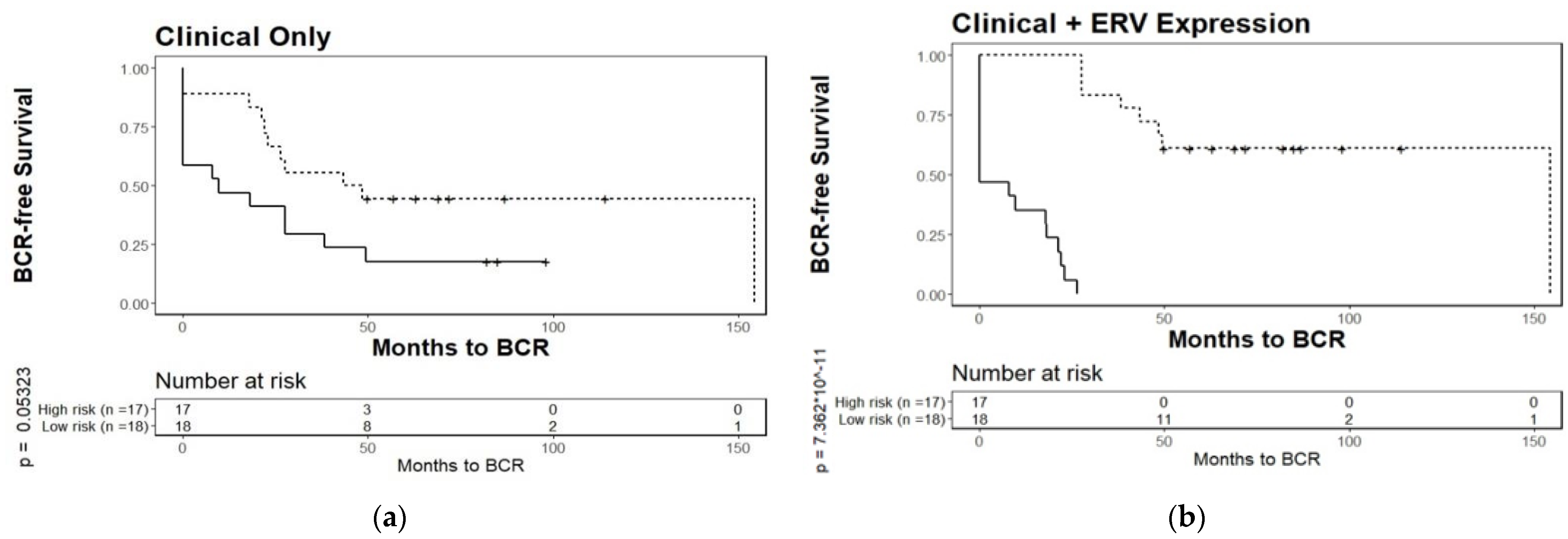
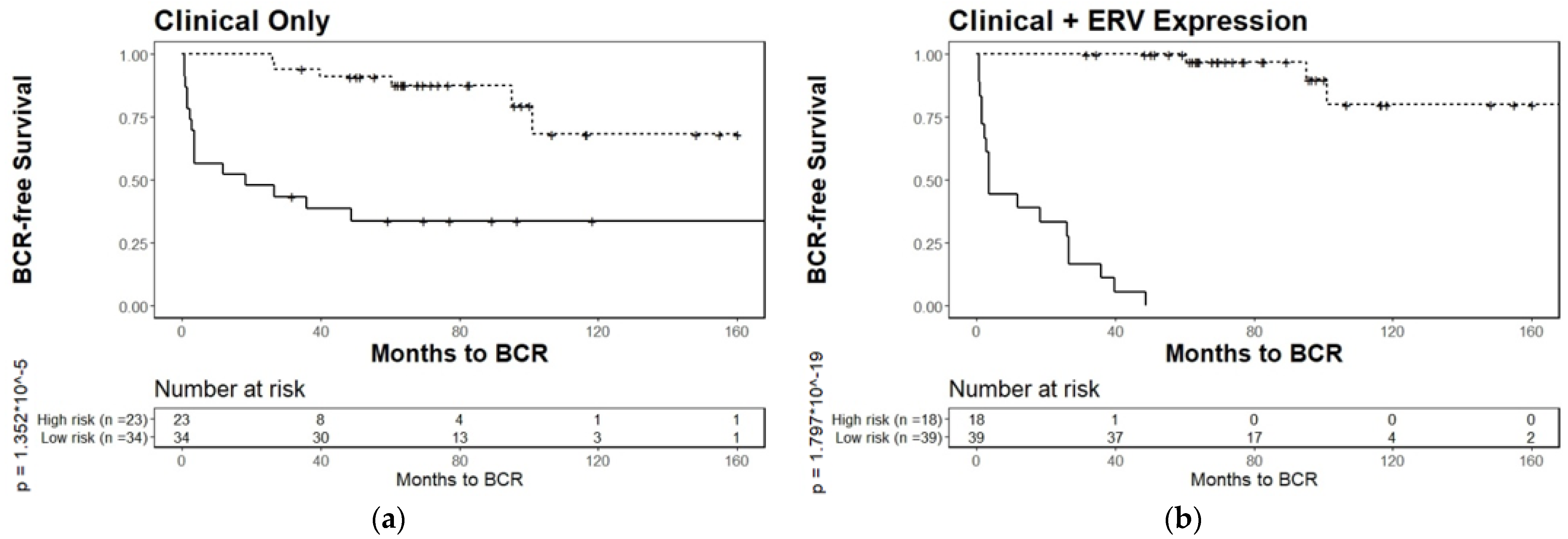
3.4. Biochemical Relapse Risk Assessment of Selected ERVs in PCa Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

References
- Long, Q.; Xu, J.; Osunkoya, A.O.; Sannigrahi, S.; Johnson, B.A.; Zhou, W.; Gillespie, T.; Park, J.Y.; Nam, R.K.; Sugar, L.; et al. Global transcriptome analysis of formalin-fixed prostate cancer specimens identifies biomarkers of disease recurrence. Cancer Res. 2014, 74, 3228–3237. [Google Scholar] [CrossRef]
- Gilligan, T. Prostate cancer: To screen or not to screen? The question is complicated. Cleve. Clin. J. Med. 2020, 88, 17–18. [Google Scholar] [CrossRef] [PubMed]
- Powell, I.J. Epidemiology and pathophysiology of prostate cancer in African-American men. J. Urol. 2007, 177, 444–449. [Google Scholar] [CrossRef]
- Powell, I.J.; Bollig-Fischer, A. Minireview: The molecular and genomic basis for prostate cancer health disparities. Mol. Endocrinol. 2013, 27, 879–891. [Google Scholar] [CrossRef]
- Sundi, D.; Faisal, F.A.; Trock, B.J.; Landis, P.K.; Feng, Z.; Ross, A.E.; Carter, H.B.; Schaeffer, E.M. Reclassification rates are higher among African American men than Caucasians on active surveillance. Urology 2015, 85, 155–160. [Google Scholar] [CrossRef][Green Version]
- Sundi, D.; Ross, A.E.; Humphreys, E.B.; Han, M.; Partin, A.W.; Carter, H.B.; Schaeffer, E.M. African American men with very low-risk prostate cancer exhibit adverse oncologic outcomes after radical prostatectomy: Should active surveillance still be an option for them? J. Clin. Oncol. 2013, 31, 2991–2997. [Google Scholar] [CrossRef] [PubMed]
- Ritch, C.R.; Morrison, B.F.; Hruby, G.; Coard, K.C.; Mayhew, R.; Aiken, W.; Benson, M.C.; McKiernan, J.M. Pathological outcome and biochemical recurrence-free survival after radical prostatectomy in African-American, Afro-Caribbean (Jamaican) and Caucasian-American men: An international comparison. BJU Int. 2013, 111, E186–E190. [Google Scholar] [CrossRef] [PubMed]
- Haiman, C.A.; Chen, G.K.; Blot, W.J.; Strom, S.S.; Berndt, S.I.; Kittles, R.A.; Rybicki, B.A.; Isaacs, W.B.; Ingles, S.A.; Stanford, J.L.; et al. Genome-wide association study of prostate cancer in men of African ancestry identifies a susceptibility locus at 17q21. Nat. Genet. 2011, 43, 570–573. [Google Scholar] [CrossRef] [PubMed]
- Rebbeck, T.R. Prostate Cancer Genetics: Variation by Race, Ethnicity, and Geography. Semin. Radiat. Oncol. 2017, 27, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Schroeck, F.R.; Sun, L.; Freedland, S.J.; Jayachandran, J.; Robertson, C.N.; Moul, J.W. Race and prostate weight as independent predictors for biochemical recurrence after radical prostatectomy. Prostate Cancer Prostatic. Dis. 2008, 11, 371–376. [Google Scholar] [CrossRef]
- Tang, D.; Kryvenko, O.N.; Mitrache, N.; Do, K.C.; Jankowski, M.; Chitale, D.A.; Trudeau, S.; Rundle, A.; Belinsky, S.A.; Rybicki, B.A. Methylation of the RARB gene increases prostate cancer risk in black Americans. J. Urol. 2013, 190, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.D.; Ceniccola, K.; Hwang, S.; Andrawis, R.; Horvath, A.; Freedman, J.A.; Olender, J.; Knapp, S.; Ching, T.; Garmire, L.; et al. Alternative splicing promotes tumour aggressiveness and drug resistance in African American prostate cancer. Nat. Commun. 2017, 8, 15921. [Google Scholar] [CrossRef] [PubMed]
- Farrell, J.; Petrovics, G.; McLeod, D.G.; Srivastava, S. Genetic and molecular differences in prostate carcinogenesis between African American and Caucasian American men. Int. J. Mol. Sci. 2013, 14, 15510–15531. [Google Scholar] [CrossRef] [PubMed]
- Hardiman, G.; Savage, S.J.; Hazard, E.S.; Wilson, R.C.; Courtney, S.M.; Smith, M.T.; Hollis, B.W.; Halbert, C.H.; Gattoni-Celli, S. Systems analysis of the prostate transcriptome in African-American men compared with European-American men. Pharmacogenomics 2016, 17, 1129–1143. [Google Scholar] [CrossRef] [PubMed]
- Reams, R.R.; Agrawal, D.; Davis, M.B.; Yoder, S.; Odedina, F.T.; Kumar, N.; Higginbotham, J.M.; Akinremi, T.; Suther, S.; Soliman, K.F. Microarray comparison of prostate tumor gene expression in African-American and Caucasian American males: A pilot project study. Infect. Agent. Cancer 2009, 4 (Suppl. S1), S3. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed]
- Tokuyama, M.; Kong, Y.; Song, E.; Jayewickreme, T.; Kang, I.; Iwasaki, A. ERVmap analysis reveals genome-wide transcription of human endogenous retroviruses. Proc. Natl. Acad. Sci. USA 2018, 115, 12565–12572. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, H.; Liu, M.; Zhou, W.; Liang, G.; Jones, P.A. Switching roles for DNA and histone methylation depend on evolutionary ages of human endogenous retroviruses. Genome Res. 2018, 28, 1147–1157. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.K.; Pan, D.; Bao, X.; Hu, M.; Li, F.; Li, C.Y. Endogenous Retrovirus Activation as a Key Mechanism of Anti-Tumor Immune Response in Radiotherapy. Radiat. Res. 2020, 193, 305–317. [Google Scholar] [CrossRef]
- Liu, M.; Thomas, S.L.; DeWitt, A.K.; Zhou, W.; Madaj, Z.B.; Ohtani, H.; Baylin, S.B.; Liang, G.; Jones, P.A. Dual Inhibition of DNA and Histone Methyltransferases Increases Viral Mimicry in Ovarian Cancer Cells. Cancer Res. 2018, 78, 5754–5766. [Google Scholar] [CrossRef] [PubMed]
- Kury, P.; Nath, A.; Creange, A.; Dolei, A.; Marche, P.; Gold, J.; Giovannoni, G.; Hartung, H.P.; Perron, H. Human Endogenous Retroviruses in Neurological Diseases. Trends Mol. Med. 2018, 24, 379–394. [Google Scholar] [CrossRef] [PubMed]
- Dear, A.E. Epigenetic Modulators and the New Immunotherapies. N. Engl. J. Med. 2016, 374, 684–686. [Google Scholar] [CrossRef] [PubMed]
- Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A.; et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 2016, 164, 1073. [Google Scholar] [CrossRef] [PubMed]
- Roulois, D.; Loo Yau, H.; Singhania, R.; Wang, Y.; Danesh, A.; Shen, S.Y.; Han, H.; Liang, G.; Jones, P.A.; Pugh, T.J.; et al. DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell 2015, 162, 961–973. [Google Scholar] [CrossRef] [PubMed]
- Steiner, M.C.; Marston, J.L.; Iniguez, L.P.; Bendall, M.L.; Chiappinelli, K.B.; Nixon, D.F.; Crandall, K.A. Locus-Specific Characterization of Human Endogenous Retrovirus Expression in Prostate, Breast, and Colon Cancers. Cancer Res. 2021, 81, 3449–3460. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Radvanyi, L.; Yin, B.; Rycaj, K.; Li, J.; Chivukula, R.; Lin, K.; Lu, Y.; Shen, J.; Chang, D.Z.; et al. Downregulation of Human Endogenous Retrovirus Type K (HERV-K) Viral env RNA in Pancreatic Cancer Cells Decreases Cell Proliferation and Tumor Growth. Clin. Cancer Res. 2017, 23, 5892–5911. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.C.; Beckermann, K.E.; Bortone, D.S.; De Cubas, A.A.; Bixby, L.M.; Lee, S.J.; Panda, A.; Ganesan, S.; Bhanot, G.; Wallen, E.M.; et al. Endogenous retroviral signatures predict immunotherapy response in clear cell renal cell carcinoma. J. Clin. Investig. 2018, 128, 4804–4820. [Google Scholar] [CrossRef] [PubMed]
- Wang-Johanning, F.; Radvanyi, L.; Rycaj, K.; Plummer, J.B.; Yan, P.; Sastry, K.J.; Piyathilake, C.J.; Hunt, K.K.; Johanning, G.L. Human endogenous retrovirus K triggers an antigen-specific immune response in breast cancer patients. Cancer Res. 2008, 68, 5869–5877. [Google Scholar] [CrossRef] [PubMed]
- Tibshirani, R. The lasso method for variable selection in the Cox model. Stat. Med. 1997, 16, 385–395. [Google Scholar] [CrossRef]
- Taliun, D.; Chothani, S.P.; Schonherr, S.; Forer, L.; Boehnke, M.; Abecasis, G.R.; Wang, C. LASER server: Ancestry tracing with genotypes or sequence reads. Bioinformatics 2017, 33, 2056–2058. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhan, X.; Bragg-Gresham, J.; Kang, H.M.; Stambolian, D.; Chew, E.Y.; Branham, K.E.; Heckenlively, J.; Study, F.; Fulton, R.; et al. Ancestry estimation and control of population stratification for sequence-based association studies. Nat. Genet. 2014, 46, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Rahmatpanah, F.; Robles, G.; Lilly, M.; Keane, T.; Kumar, V.; Mercola, D.; Randhawa, P.; McClelland, M. RNA expression differences in prostate tumors and tumor-adjacent stroma between Black and White Americans. Oncotarget 2021, 12, 1457–1469. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Zahn, L.M. ERVs affect brain gene expression. Science 2017, 355, 491–492. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, T.; Zeng, J.; Lowe, C.B.; Sellers, R.G.; Salama, S.R.; Yang, M.; Burgess, S.M.; Brachmann, R.K.; Haussler, D. Species-specific endogenous retroviruses shape the transcriptional network of the human tumor suppressor protein p53. Proc. Natl. Acad. Sci. USA 2007, 104, 18613–18618. [Google Scholar] [CrossRef] [PubMed]
- Brattas, P.L.; Jonsson, M.E.; Fasching, L.; Nelander Wahlestedt, J.; Shahsavani, M.; Falk, R.; Falk, A.; Jern, P.; Parmar, M.; Jakobsson, J. TRIM28 Controls a Gene Regulatory Network Based on Endogenous Retroviruses in Human Neural Progenitor Cells. Cell Rep. 2017, 18, 1–11. [Google Scholar] [CrossRef]
- Bankhead, P.; Loughrey, M.B.; Fernandez, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef]
- Friedman, J.; Hastie, T.; Tibshirani, R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Epstein, J.I.; Zelefsky, M.J.; Sjoberg, D.D.; Nelson, J.B.; Egevad, L.; Magi-Galluzzi, C.; Vickers, A.J.; Parwani, A.V.; Reuter, V.E.; Fine, S.W.; et al. A Contemporary Prostate Cancer Grading System: A Validated Alternative to the Gleason Score. Eur. Urol. 2016, 69, 428–435. [Google Scholar] [CrossRef]
- Uno, H.; Tian, L.; Cai, T.; Kohane, I.S.; Wei, L.J. A unified inference procedure for a class of measures to assess improvement in risk prediction systems with survival data. Stat. Med. 2013, 32, 2430–2442. [Google Scholar] [CrossRef] [PubMed]
- Mineta, K.; Goto, K.; Gojobori, T.; Alkuraya, F.S. Population structure of indigenous inhabitants of Arabia. PLoS Genet. 2021, 17, e1009210. [Google Scholar] [CrossRef] [PubMed]
- Scott, E.M.; Halees, A.; Itan, Y.; Spencer, E.G.; He, Y.; Azab, M.A.; Gabriel, S.B.; Belkadi, A.; Boisson, B.; Abel, L.; et al. Characterization of Greater Middle Eastern genetic variation for enhanced disease gene discovery. Nat. Genet. 2016, 48, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Babaian, A.; Mager, D.L. Endogenous retroviral promoter exaptation in human cancer. Mob. DNA 2016, 7, 24. [Google Scholar] [CrossRef]
- Chuong, E.B.; Elde, N.C.; Feschotte, C. Regulatory evolution of innate immunity through co-option of endogenous retroviruses. Science 2016, 351, 1083–1087. [Google Scholar] [CrossRef] [PubMed]
- Schmid, C.D.; Bucher, P. MER41 repeat sequences contain inducible STAT1 binding sites. PLoS ONE 2010, 5, e11425. [Google Scholar] [CrossRef]
- Zhang, Y.; Yang, H.; Du, Y.; Liu, P.; Zhang, J.; Li, Y.; Shen, H.; Xing, L.; Xue, X.; Chen, J.; et al. Long noncoding RNA TP53TG1 promotes pancreatic ductal adenocarcinoma development by acting as a molecular sponge of microRNA-96. Cancer Sci. 2019, 110, 2760–2772. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Liu, Y.; Liang, P.; Wang, B.; Tan, H.; Zhang, Y.; Gao, X.; Gao, J. TP53TG1 enhances cisplatin sensitivity of non-small cell lung cancer cells through regulating miR-18a/PTEN axis. Cell Biosci. 2018, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.L.; Zhang, Q.; Yuan, X.; Jin, L.; Wang, L.L.; Fang, T.T.; Wang, W.B. Long noncoding RNA RP4 functions as a competing endogenous RNA through miR-7-5p sponge activity in colorectal cancer. World J. Gastroenterol. 2018, 24, 1004–1012. [Google Scholar] [CrossRef]
- Kuhajda, F.P.; Katumuluwa, A.I.; Pasternack, G.R. Expression of haptoglobin-related protein and its potential role as a tumor antigen. Proc. Natl. Acad. Sci. USA 1989, 86, 1188–1192. [Google Scholar] [CrossRef]
- Epelbaum, R.; Shalitin, C.; Segal, R.; Valansi, C.; Arselan, I.; Faraggi, D.; Leviov, M.; Ben-Shahar, M.; Haim, N. Haptoglobin-related protein as a serum marker in malignant lymphoma. Pathol. Oncol. Res. 1998, 4, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.J.; Petersen, S.V.; Jacobsen, C.; Oxvig, C.; Rees, D.; Moller, H.J.; Moestrup, S.K. Haptoglobin-related protein is a high-affinity hemoglobin-binding plasma protein. Blood 2006, 108, 2846–2849. [Google Scholar] [CrossRef] [PubMed]
- Freedland, S.J.; Humphreys, E.B.; Mangold, L.A.; Eisenberger, M.; Dorey, F.J.; Walsh, P.C.; Partin, A.W. Risk of prostate cancer-specific mortality following biochemical recurrence after radical prostatectomy. JAMA 2005, 294, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Brawley, O.W. Trends in prostate cancer in the United States. J. Natl. Cancer Inst. Monogr. 2012, 2012, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Kinseth, M.A.; Jia, Z.; Rahmatpanah, F.; Sawyers, A.; Sutton, M.; Wang-Rodriguez, J.; Mercola, D.; McGuire, K.L. Expression differences between African American and Caucasian prostate cancer tissue reveals that stroma is the site of aggressive changes. Int. J. Cancer 2014, 134, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Nadeau, M.J.; Manghera, M.; Douville, R.N. Inside the Envelope: Endogenous Retrovirus-K Env as a Biomarker and Therapeutic Target. Front. Microbiol. 2015, 6, 1244. [Google Scholar] [CrossRef] [PubMed]
- Woo, W.H.; Shen, L.; Leong, S.M.; Koay, E.S. Prevalence of human endogenous retroviral element associates with Hodgkin’s lymphoma incidence rates. Leuk. Res. Rep. 2014, 3, 1–3. [Google Scholar] [CrossRef] [PubMed][Green Version]
- King Thomas, J.; Mir, H.; Kapur, N.; Singh, S. Racial Differences in Immunological Landscape Modifiers Contributing to Disparity in Prostate Cancer. Cancers 2019, 11, 1857. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Rose, C.M.; Cass, A.A.; Williams, A.G.; Darwish, M.; Lianoglou, S.; Haverty, P.M.; Tong, A.J.; Blanchette, C.; Albert, M.L.; et al. Transposable element expression in tumors is associated with immune infiltration and increased antigenicity. Nat. Commun. 2019, 10, 5228. [Google Scholar] [CrossRef]
- Zapatka, M.; Borozan, I.; Brewer, D.S.; Iskar, M.; Grundhoff, A.; Alawi, M.; Desai, N.; Sultmann, H.; Moch, H.; Pathogens, P.; et al. The landscape of viral associations in human cancers. Nat. Genet. 2020, 52, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Wallace, T.A.; Downey, R.F.; Seufert, C.J.; Schetter, A.; Dorsey, T.H.; Johnson, C.A.; Goldman, R.; Loffredo, C.A.; Yan, P.; Sullivan, F.J.; et al. Elevated HERV-K mRNA expression in PBMC is associated with a prostate cancer diagnosis particularly in older men and smokers. Carcinogenesis 2014, 35, 2074–2083. [Google Scholar] [CrossRef] [PubMed]
- Goering, W.; Schmitt, K.; Dostert, M.; Schaal, H.; Deenen, R.; Mayer, J.; Schulz, W.A. Human endogenous retrovirus HERV-K(HML-2) activity in prostate cancer is dominated by a few loci. Prostate 2015, 75, 1958–1971. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Training Set, n = 57 1 | Test Set, n = 35 1 |
|---|---|---|
| Gleason Value | ||
| 1 (<3) | 3 (5.3%) | 7 (20%) |
| 2 (3 + 4) | 36 (63%) | 15 (43%) |
| 3 (4 + 3) | 10 (18%) | 7 (20%) |
| 4 (>7) | 8 (14%) | 6 (17%) |
| Age | 62 (56, 65) | 61 (57, 66) |
| Pre-PSA | 7 (5, 13) | 8 (6, 13) |
| Ancestry | ||
| BA | 23 (40%) | 4 (11%) |
| WA | 34 (60%) | 31 (89%) |
| Tumor Stage | ||
| 1 | 0 (0%) | 12 (34%) |
| 2 | 45 (79%) | 19 (54%) |
| 3 | 11 (19%) | 4 (11%) |
| 4 | 1 (1.8%) | 0 (0%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, V.; McClelland, M.; Nguyen, J.; De Robles, G.; Ittmann, M.; Castro, P.; Mercola, D.; Jia, Z.; Rahmatpanah, F. Expression of Endogenous Retroviral RNA in Prostate Tumors has Prognostic Value and Shows Differences among Americans of African Versus European/Middle Eastern Ancestry. Cancers 2021, 13, 6347. https://doi.org/10.3390/cancers13246347
Kumar V, McClelland M, Nguyen J, De Robles G, Ittmann M, Castro P, Mercola D, Jia Z, Rahmatpanah F. Expression of Endogenous Retroviral RNA in Prostate Tumors has Prognostic Value and Shows Differences among Americans of African Versus European/Middle Eastern Ancestry. Cancers. 2021; 13(24):6347. https://doi.org/10.3390/cancers13246347
Chicago/Turabian StyleKumar, Vinay, Michael McClelland, James Nguyen, Gabriela De Robles, Michael Ittmann, Patricia Castro, Dan Mercola, Zhenyu Jia, and Farah Rahmatpanah. 2021. "Expression of Endogenous Retroviral RNA in Prostate Tumors has Prognostic Value and Shows Differences among Americans of African Versus European/Middle Eastern Ancestry" Cancers 13, no. 24: 6347. https://doi.org/10.3390/cancers13246347
APA StyleKumar, V., McClelland, M., Nguyen, J., De Robles, G., Ittmann, M., Castro, P., Mercola, D., Jia, Z., & Rahmatpanah, F. (2021). Expression of Endogenous Retroviral RNA in Prostate Tumors has Prognostic Value and Shows Differences among Americans of African Versus European/Middle Eastern Ancestry. Cancers, 13(24), 6347. https://doi.org/10.3390/cancers13246347