


Osteopontin Regulates Treg Cell Stability and Function with Implications for Anti-Tumor Immunity and Autoimmunity
 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Flow Cytometry
2.3. Treg Cell ISOLATION, In-Vitro Stimulation and Suppression Assay
2.4. Cell Transfer Model of Colitis—In Vivo Suppression Assay
2.5. Histology and Assessment of Intestinal Inflammation
2.6. B16-F10 Melanoma Model
2.7. Quantitative PCR
2.8. Statistics
3. Results
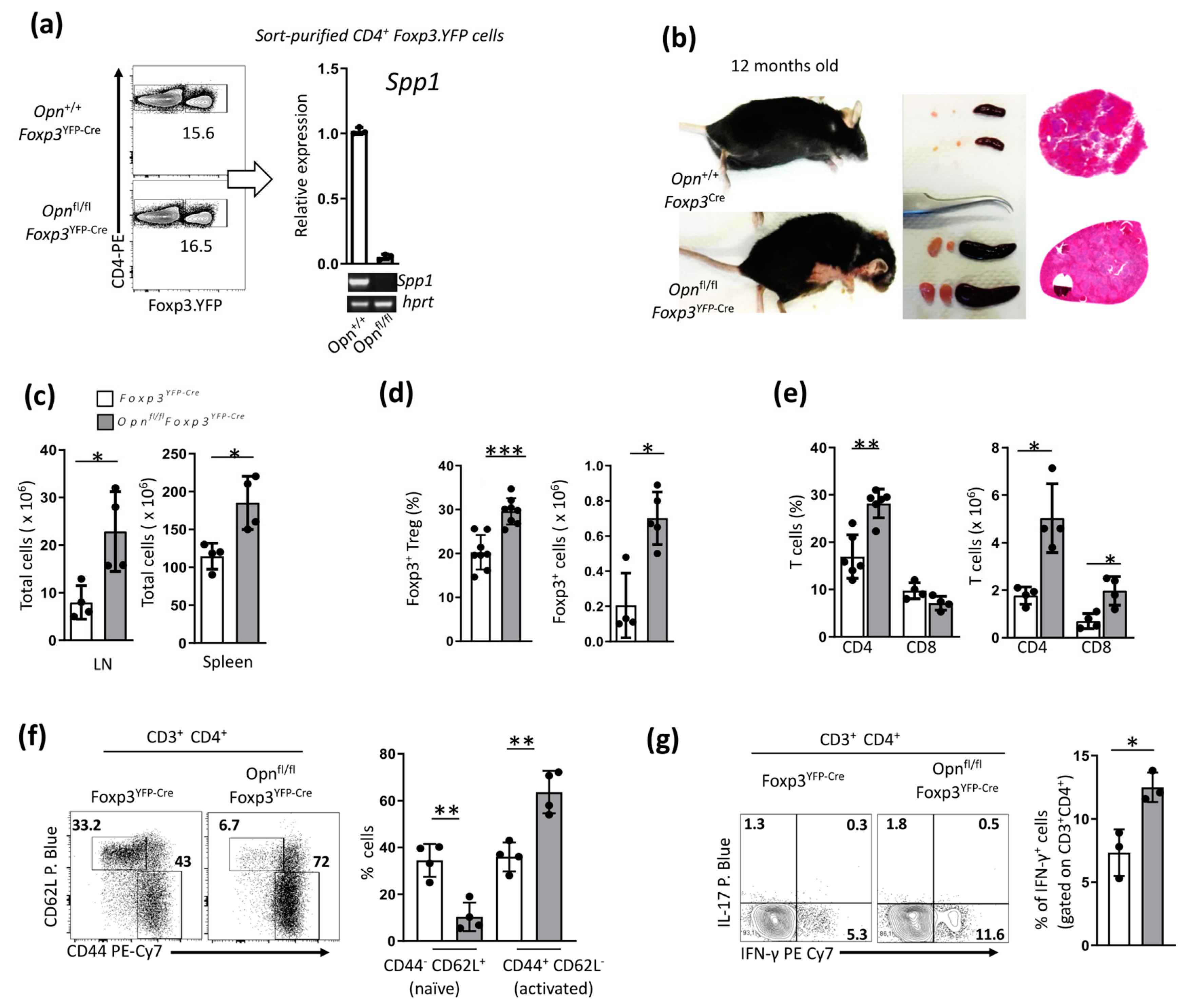
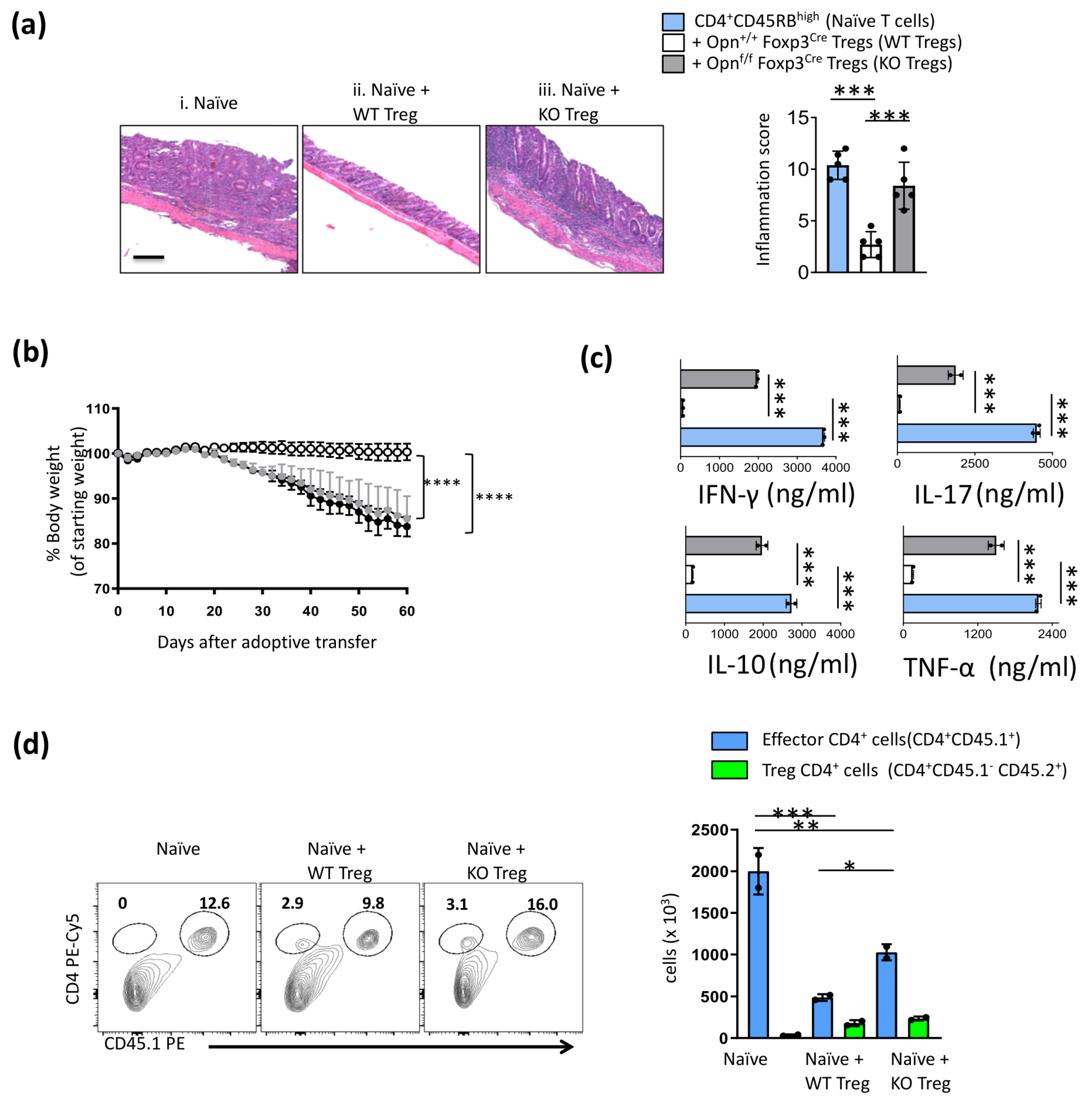
3.1. Opn-Deficient Foxp3+ Tregs Have Impaired In Vivo Function
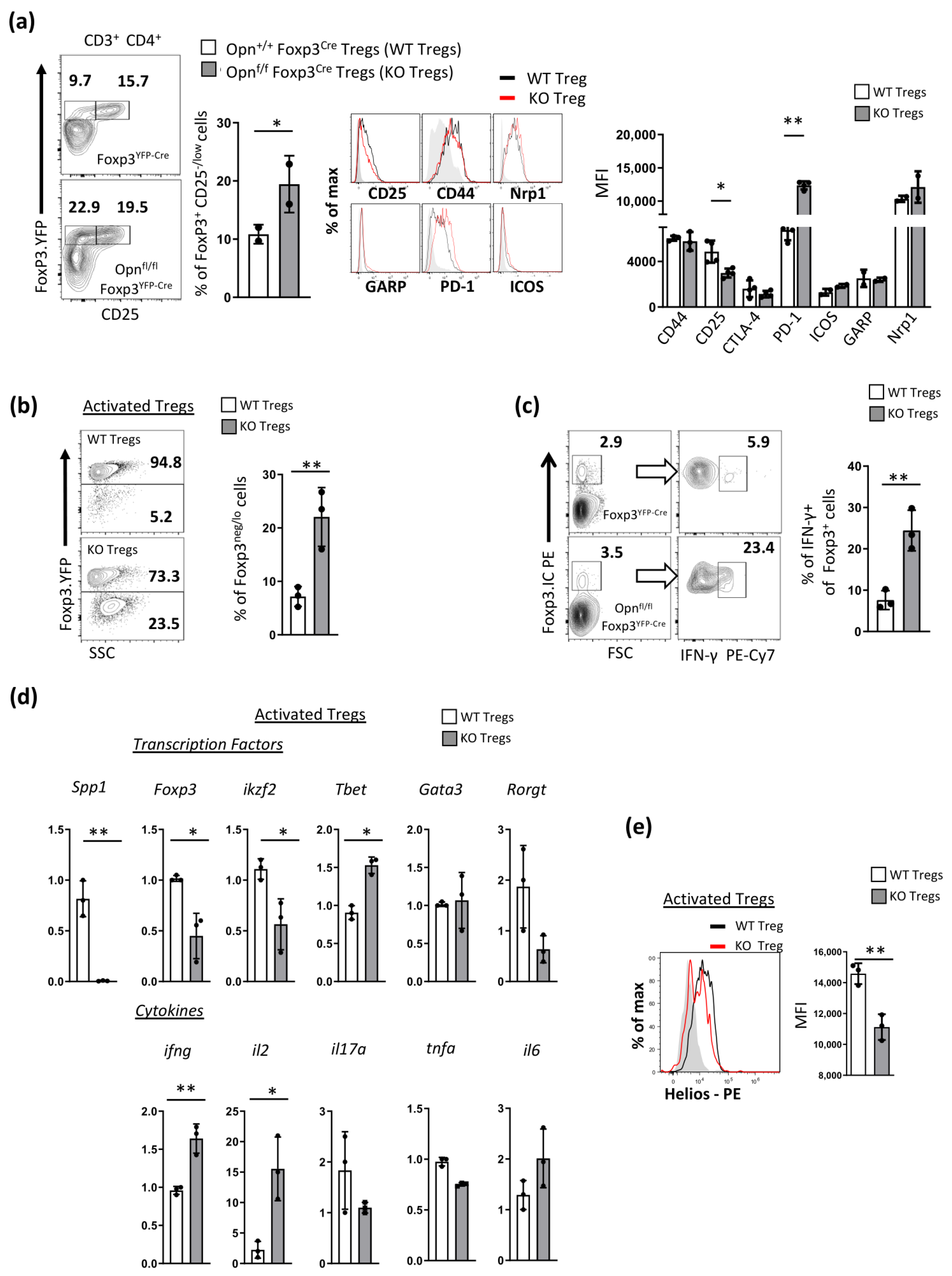
3.2. Opn-Deficient Foxp3+ Tregs Exhibit an Unstable Phenotype
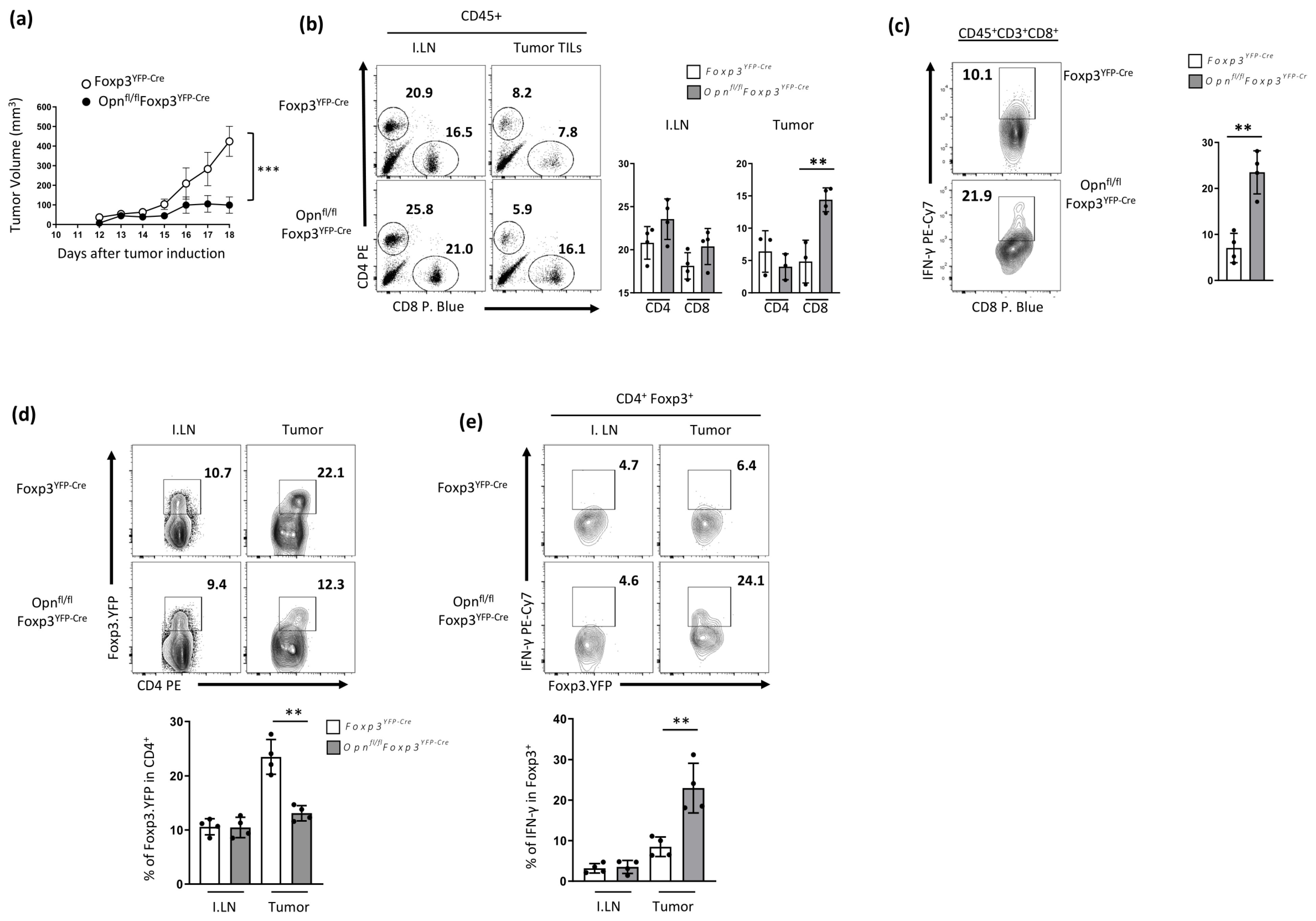
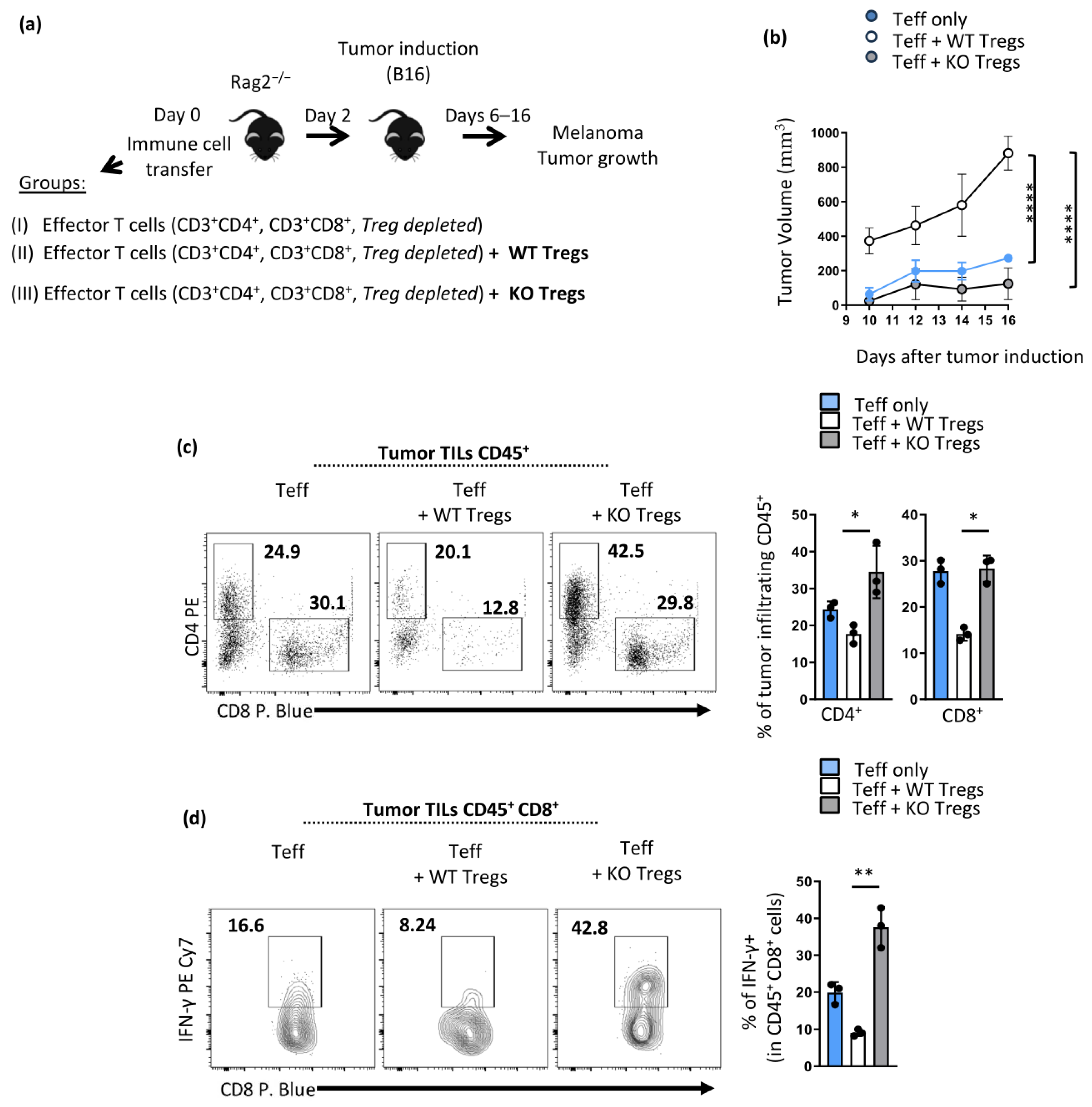
3.3. Mice with Opn-Deficient Foxp3+ Tregs Have Enhanced Anti-Tumor Immunity and Reduced Tumor Burden Associated with an Unstable Treg Phenotype
3.4. Enhanced Anti-Tumor Immunity in Opnfl/fl Foxp3YFP-Cre Is Treg Intrinsic
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kumari, A.; Kashyap, D.; Garg, V.K. Osteopontin in cancer. Adv. Clin. Chem. 2024, 118, 87–110. [Google Scholar] [CrossRef]
- Hur, E.M.; Youssef, S.; Haws, M.E.; Zhang, S.Y.; Sobel, R.A.; Steinman, L. Osteopontin-induced relapse and progression of autoimmune brain disease through enhanced survival of activated T cells. Nat. Immunol. 2007, 8, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Simoes, D.C.M.; Paschalidis, N.; Kourepini, E.; Panoutsakopoulou, V. An integrin axis induces IFN-β production in plasmacytoid dendritic cells. J. Cell Biol. 2022, 221, e202102055. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Uede, T. Osteopontin, intrinsic tissue regulator of intractable inflammatory diseases. Pathol. Int. 2011, 61, 265–280. [Google Scholar] [CrossRef]
- Leavenworth, J.W.; Verbinnen, B.; Wang, Q.; Shen, E.; Cantor, H. Intracellular osteopontin regulates homeostasis and function of natural killer cells. Proc. Natl. Acad. Sci. USA 2015, 112, 494–499. [Google Scholar] [CrossRef] [PubMed]
- Alissafi, T.; Kourepini, E.; Simoes, D.C.M.; Paschalidis, N.; Aggelakopoulou, M.; Sparwasser, T.; Boon, L.; Hammad, H.; Lambrecht, B.N.; Panoutsakopoulou, V. Osteopontin Promotes Protective Antigenic Tolerance against Experimental Allergic Airway Disease. J. Immunol. 2018, 200, 1270–1282. [Google Scholar] [CrossRef] [PubMed]
- Cantor, H.; Shinohara, M.L. Regulation of T-helper-cell lineage development by osteopontin: The inside story. Nat. Rev. Immunol. 2009, 9, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Xanthou, G.; Alissafi, T.; Semitekolou, M.; Simoes, D.C.; Economidou, E.; Gaga, M.; Lambrecht, B.N.; Lloyd, C.M.; Panoutsakopoulou, V. Osteopontin has a crucial role in allergic airway disease through regulation of dendritic cell subsets. Nat. Med. 2007, 13, 570–578. [Google Scholar] [CrossRef]
- Kourepini, E.; Aggelakopoulou, M.; Alissafi, T.; Paschalidis, N.; Simoes, D.C.; Panoutsakopoulou, V. Osteopontin expression by CD103- dendritic cells drives intestinal inflammation. Proc. Natl. Acad. Sci. USA 2014, 111, E856–E865. [Google Scholar] [CrossRef] [PubMed]
- Panda, V.K.; Mishra, B.; Nath, A.N.; Butti, R.; Yadav, A.S.; Malhotra, D.; Khanra, S.; Mahapatra, S.; Mishra, P.; Swain, B.; et al. Osteopontin: A Key Multifaceted Regulator in Tumor Progression and Immunomodulation. Biomedicines 2024, 12, 1527. [Google Scholar] [CrossRef] [PubMed]
- Shurin, M.R. Osteopontin controls immunosuppression in the tumor microenvironment. J. Clin. Investig. 2018, 128, 5209–5212. [Google Scholar] [CrossRef] [PubMed]
- Klement, J.D.; Paschall, A.V.; Redd, P.S.; Ibrahim, M.L.; Lu, C.; Yang, D.; Celis, E.; Abrams, S.I.; Ozato, K.; Liu, K. An osteopontin/CD44 immune checkpoint controls CD8+ T cell activation and tumor immune evasion. J. Clin. Investig. 2018, 128, 5549–5560. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Koh, J.; Ko, J.S.; Kim, H.Y.; Lee, H.; Chung, D.H. Ubiquitin E3 Ligase Pellino-1 Inhibits IL-10-mediated M2c Polarization of Macrophages, Thereby Suppressing Tumor Growth. Immune Netw. 2019, 19, e32. [Google Scholar] [CrossRef]
- Marson, A.; Kretschmer, K.; Frampton, G.M.; Jacobsen, E.S.; Polansky, J.K.; MacIsaac, K.D.; Levine, S.S.; Fraenkel, E.; von Boehmer, H.; Young, R.A. Foxp3 occupancy and regulation of key target genes during T-cell stimulation. Nature 2007, 445, 931–935. [Google Scholar] [CrossRef]
- Sugimoto, N.; Oida, T.; Hirota, K.; Nakamura, K.; Nomura, T.; Uchiyama, T.; Sakaguchi, S. Foxp3-dependent and -independent molecules specific for CD25+CD4+ natural regulatory T cells revealed by DNA microarray analysis. Int. Immunol. 2006, 18, 1197–1209. [Google Scholar] [CrossRef]
- Arvey, A.; van der Veeken, J.; Samstein, R.M.; Feng, Y.; Stamatoyannopoulos, J.A.; Rudensky, A.Y. Inflammation-induced repression of chromatin bound by the transcription factor Foxp3 in regulatory T cells. Nat. Immunol. 2014, 15, 580–587. [Google Scholar] [CrossRef]
- Butti, R.; Kumar, T.V.S.; Nimma, R.; Banerjee, P.; Kundu, I.G.; Kundu, G.C. Osteopontin Signaling in Shaping Tumor Microenvironment Conducive to Malignant Progression. Adv. Exp. Med. Biol. 2021, 1329, 419–441. [Google Scholar] [CrossRef]
- Han, H.; Ge, X.; Komakula, S.S.B.; Desert, R.; Das, S.; Song, Z.; Chen, W.; Athavale, D.; Gaskell, H.; Lantvit, D.; et al. Macrophage-derived Osteopontin (SPP1) Protects From Nonalcoholic Steatohepatitis. Gastroenterology 2023, 165, 201–217. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Song, Z.; Han, H.; Ge, X.; Desert, R.; Athavale, D.; Babu Komakula, S.S.; Magdaleno, F.; Chen, W.; Lantvit, D.; et al. Intestinal Osteopontin Protects from Alcohol-induced Liver Injury by Preserving the Gut Microbiome and the Intestinal Barrier Function. Cell Mol. Gastroenterol. Hepatol. 2022, 14, 813–839. [Google Scholar] [CrossRef]
- Kourepini, E.; Paschalidis, N.; Simoes, D.C.; Aggelakopoulou, M.; Grogan, J.L.; Panoutsakopoulou, V. TIGIT Enhances Antigen-Specific Th2 Recall Responses and Allergic Disease. J. Immunol. 2016, 196, 3570–3580. [Google Scholar] [CrossRef]
- Read, S.; Malmström, V.; Powrie, F. Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25+CD4+ regulatory cells that control intestinal inflammation. J. Exp. Med. 2000, 192, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Overwijk, W.W.; Restifo, N.P. B16 as a mouse model for human melanoma. Curr. Protoc. Immunol. 2000, 39, 20.1.1–20.1.29. [Google Scholar] [CrossRef] [PubMed]
- Rubtsov, Y.P.; Rasmussen, J.P.; Chi, E.Y.; Fontenot, J.; Castelli, L.; Ye, X.; Treuting, P.; Siewe, L.; Roers, A.; Henderson, W.R., Jr.; et al. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity 2008, 28, 546–558. [Google Scholar] [CrossRef] [PubMed]
- Lowther, D.E.; Goods, B.A.; Lucca, L.E.; Lerner, B.A.; Raddassi, K.; van Dijk, D.; Hernandez, A.L.; Duan, X.; Gunel, M.; Coric, V.; et al. PD-1 marks dysfunctional regulatory T cells in malignant gliomas. JCI Insight 2016, 1, e85935. [Google Scholar] [CrossRef] [PubMed]
- Chougnet, C.; Hildeman, D. Helios-controller of Treg stability and function. Transl. Cancer Res. 2016, 5, S338–S341. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Barnitz, R.A.; Kreslavsky, T.; Brown, F.D.; Moffett, H.; Lemieux, M.E.; Kaygusuz, Y.; Meissner, T.; Holderried, T.A.; Chan, S.; et al. Stable inhibitory activity of regulatory T cells requires the transcription factor Helios. Science 2015, 350, 334–339. [Google Scholar] [CrossRef]
- Saito, T.; Nishikawa, H.; Wada, H.; Nagano, Y.; Sugiyama, D.; Atarashi, K.; Maeda, Y.; Hamaguchi, M.; Ohkura, N.; Sato, E.; et al. Two FOXP3+CD4+ T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat. Med. 2016, 22, 679–684. [Google Scholar] [CrossRef]
- Strauss, L.; Bergmann, C.; Szczepanski, M.J.; Lang, S.; Kirkwood, J.M.; Whiteside, T.L. Expression of ICOS on human melanoma-infiltrating CD4+CD25highFoxp3+ T regulatory cells: Implications and impact on tumor-mediated immune suppression. J. Immunol. 2008, 180, 2967–2980. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, B.; Elkord, E. Regulatory T Cells in the Tumor Microenvironment and Cancer Progression: Role and Therapeutic Targeting. Vaccines 2016, 4, 28. [Google Scholar] [CrossRef]
- Qiu, R.; Zhou, L.; Ma, Y.; Zhou, L.; Liang, T.; Shi, L.; Long, J.; Yuan, D. Regulatory T Cell Plasticity and Stability and Autoimmune Diseases. Clin. Rev. Allergy Immunol. 2020, 58, 52–70. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Vignali, D.A.; Rudensky, A.Y.; Niec, R.E.; Waldmann, H. The plasticity and stability of regulatory T cells. Nat. Rev. Immunol. 2013, 13, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Huynh, A.; DuPage, M.; Priyadharshini, B.; Sage, P.T.; Quiros, J.; Borges, C.M.; Townamchai, N.; Gerriets, V.A.; Rathmell, J.C.; Sharpe, A.H.; et al. Control of PI(3) kinase in Treg cells maintains homeostasis and lineage stability. Nat. Immunol. 2015, 16, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Zhang, M.; Zhang, L.; Wang, P.; Song, G.; Liu, B.; Wu, H.; Yin, Z.; Gao, C. Intracellular osteopontin stabilizes TRAF3 to positively regulate innate antiviral response. Sci. Rep. 2016, 6, 23771. [Google Scholar] [CrossRef]
- Zhang, H.; Guo, M.; Chen, J.H.; Wang, Z.; Du, X.F.; Liu, P.X.; Li, W.H. Osteopontin knockdown inhibits alphav,beta3 integrin-induced cell migration and invasion and promotes apoptosis of breast cancer cells by inducing autophagy and inactivating the PI3K/Akt/mTOR pathway. Cell Physiol. Biochem. 2014, 33, 991–1002. [Google Scholar] [CrossRef]
- Chang, J.H.; Hu, H.; Jin, J.; Puebla-Osorio, N.; Xiao, Y.; Gilbert, B.E.; Brink, R.; Ullrich, S.E.; Sun, S.C. TRAF3 regulates the effector function of regulatory T cells and humoral immune responses. J. Exp. Med. 2014, 211, 137–151. [Google Scholar] [CrossRef]
- Moorman, H.R.; Poschel, D.; Klement, J.D.; Lu, C.; Redd, P.S.; Liu, K. Osteopontin: A Key Regulator of Tumor Progression and Immunomodulation. Cancers 2020, 12, 3379. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Huang, C. The role of osteopontin in the development and metastasis of melanoma. Melanoma Res. 2021, 31, 283–289. [Google Scholar] [CrossRef]
- Sangaletti, S.; Tripodo, C.; Sandri, S.; Torselli, I.; Vitali, C.; Ratti, C.; Botti, L.; Burocchi, A.; Porcasi, R.; Tomirotti, A.; et al. Osteopontin shapes immunosuppression in the metastatic niche. Cancer Res. 2014, 74, 4706–4719. [Google Scholar] [CrossRef]
- Shi, L.; Sun, Z.; Su, W.; Xu, F.; Xie, D.; Zhang, Q.; Dai, X.; Iyer, K.; Hitchens, T.K.; Foley, L.M.; et al. Treg cell-derived osteopontin promotes microglia-mediated white matter repair after ischemic stroke. Immunity 2021, 54, 1527–1542.e8. [Google Scholar] [CrossRef]
- Leavenworth, J.W.; Verbinnen, B.; Yin, J.; Huang, H.; Cantor, H. A p85alpha-osteopontin axis couples the receptor ICOS to sustained Bcl-6 expression by follicular helper and regulatory T cells. Nat. Immunol. 2015, 16, 96–106. [Google Scholar] [CrossRef]
- Fei, Y.; Wu, Y.; Chen, L.; Yu, H.; Pan, L. Comprehensive pan-carcinoma analysis of ITGB1 distortion and its potential clinical significance for cancer immunity. Discov. Oncol. 2024, 15, 47. [Google Scholar] [CrossRef] [PubMed]
- Kale, S.; Raja, R.; Thorat, D.; Soundararajan, G.; Patil, T.V.; Kundu, G.C. Osteopontin signaling upregulates cyclooxygenase-2 expression in tumor-associated macrophages leading to enhanced angiogenesis and melanoma growth via alpha9beta1 integrin. Oncogene 2014, 33, 2295–2306. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vakrakou, A.G.; Kourepini, E.; Skordos, I.; Nieto, N.; Panoutsakopoulou, V.; Paschalidis, N. Osteopontin Regulates Treg Cell Stability and Function with Implications for Anti-Tumor Immunity and Autoimmunity. Cancers 2024, 16, 2952. https://doi.org/10.3390/cancers16172952
Vakrakou AG, Kourepini E, Skordos I, Nieto N, Panoutsakopoulou V, Paschalidis N. Osteopontin Regulates Treg Cell Stability and Function with Implications for Anti-Tumor Immunity and Autoimmunity. Cancers. 2024; 16(17):2952. https://doi.org/10.3390/cancers16172952
Chicago/Turabian StyleVakrakou, Aigli G., Evangelia Kourepini, Ioannis Skordos, Natalia Nieto, Vily Panoutsakopoulou, and Nikolaos Paschalidis. 2024. "Osteopontin Regulates Treg Cell Stability and Function with Implications for Anti-Tumor Immunity and Autoimmunity" Cancers 16, no. 17: 2952. https://doi.org/10.3390/cancers16172952
APA StyleVakrakou, A. G., Kourepini, E., Skordos, I., Nieto, N., Panoutsakopoulou, V., & Paschalidis, N. (2024). Osteopontin Regulates Treg Cell Stability and Function with Implications for Anti-Tumor Immunity and Autoimmunity. Cancers, 16(17), 2952. https://doi.org/10.3390/cancers16172952