
Unveil Intrahepatic Cholangiocarcinoma Heterogeneity through the Lens of Omics and Multi-Omics Approaches

 , , , ,
, , , ,  and
and
Abstract
Simple Summary
Abstract
1. Introduction
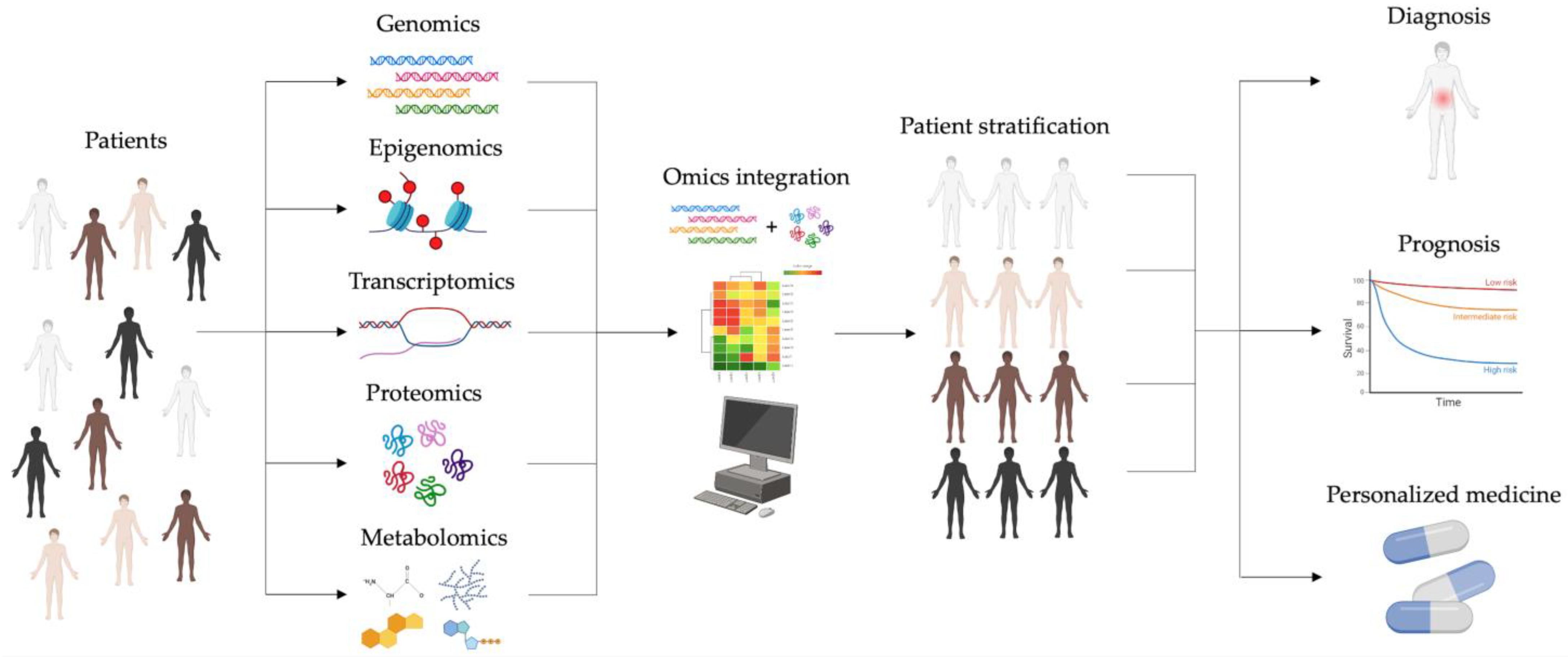
2. Omics Cancer Era: From Single- to Multi-Omics Approaches
2.1. Single-Based Omics
2.2. Multi-Omics Approaches
3. Sequencing-Based Omics in iCCA
3.1. Genomics
3.2. Epigenomics
3.3. Transcriptomics
3.4. Epitranscriptomics
4. A Landscape Still Undiscovered: Proteomics and Metabolomics of iCCA
4.1. Proteomics
4.2. Metabolomics
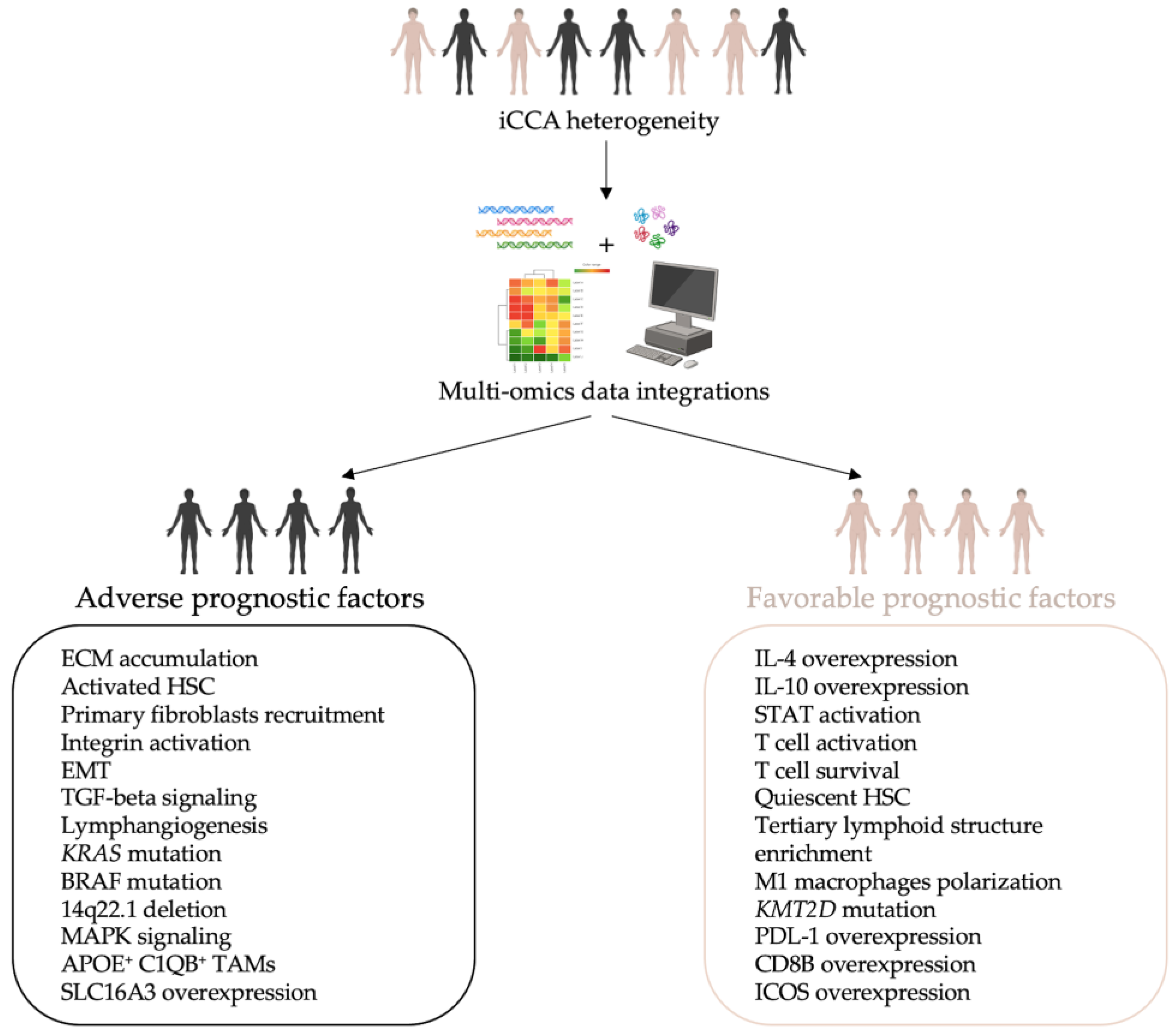
5. Multi-Omics Approaches in iCCA: A New Model of Knowledge
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Khan, S.A.; Thomas, H.C.; Davidson, B.R.; Taylor-Robinson, S.D. Cholangiocarcinoma. Lancet 2005, 366, 1303–1314. [Google Scholar] [CrossRef] [PubMed]
- Blechacz, B.; Gores, G.J. Cholangiocarcinoma: Advances in pathogenesis, diagnosis, and treatment. Hepatology 2008, 48, 308–321. [Google Scholar] [CrossRef]
- Fan, B.; Malato, Y.; Calvisi, D.F.; Naqvi, S.; Razumilava, N.; Ribback, S.; Gores, G.J.; Dombrowski, F.; Evert, M.; Chen, X.; et al. Cholangiocarcinomas can originate from hepatocytes in mice. J. Clin. Investig. 2012, 122, 2911–2915. [Google Scholar] [CrossRef]
- Zhu, Y.; Kwong, L.N. Insights into the Origin of Intrahepatic Cholangiocarcinoma from Mouse Models. Hepatology 2020, 72, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Razumilava, N.; Gores, G.J. Cholangiocarcinoma. Lancet 2014, 383, 2168–2179. [Google Scholar] [CrossRef]
- Valle, J.; Wasan, H.; Palmer, D.H.; Cunningham, D.; Anthoney, A.; Maraveyas, A.; Madhusudan, S.; Iveson, T.; Hughes, S.; Pereira, S.P.; et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N. Engl. J. Med. 2010, 362, 1273–1281. [Google Scholar] [CrossRef]
- Rahnemai-Azar, A.A.; Weisbrod, A.; Dillhoff, M.; Schmidt, C.; Pawlik, T.M. Intrahepatic cholangiocarcinoma: Molecular markers for diagnosis and prognosis. Surg. Oncol. 2017, 26, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Kelley, R.K.; Bridgewater, J.; Gores, G.J.; Zhu, A.X. Systemic therapies for intrahepatic cholangiocarcinoma. J. Hepatol. 2020, 72, 353–363. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.A.; Taylor-Robinson, S.D.; Toledano, M.B.; Beck, A.; Elliott, P.; Thomas, H.C. Changing international trends in mortality rates for liver, biliary and pancreatic tumours. J. Hepatol. 2002, 37, 806–813. [Google Scholar] [CrossRef]
- Yao, K.J.; Jabbour, S.; Parekh, N.; Lin, Y.; Moss, R.A. Increasing mortality in the United States from cholangiocarcinoma: An analysis of the National Center for Health Statistics Database. BMC Gastroenterol. 2016, 16, 117. [Google Scholar] [CrossRef]
- Bridgewater, J.; Galle, P.R.; Khan, S.A.; Llovet, J.M.; Park, J.W.; Patel, T.; Pawlik, T.M.; Gores, G.J. Guidelines for the diagnosis and management of intrahepatic cholangiocarcinoma. J. Hepatol. 2014, 60, 1268–1289. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Chen, H.D.; Yu, Y.W.; Li, N.; Chen, W.Q. Changing profiles of cancer burden worldwide and in China: A secondary analysis of the global cancer statistics 2020. Chin. Med. J. 2021, 134, 783–791. [Google Scholar] [CrossRef]
- Banales, J.M.; Marin, J.J.G.; Lamarca, A.; Rodrigues, P.M.; Khan, S.A.; Roberts, L.R.; Cardinale, V.; Carpino, G.; Andersen, J.B.; Braconi, C.; et al. Cholangiocarcinoma 2020: The next horizon in mechanisms and management. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 557–588. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, V. Classifications and misclassification in cholangiocarcinoma. Liver Int. 2019, 39, 260–262. [Google Scholar] [CrossRef]
- Gupta, A.; Dixon, E. Epidemiology and risk factors: Intrahepatic cholangiocarcinoma. Hepatobiliary Surg. Nutr. 2017, 6, 101–104. [Google Scholar] [CrossRef]
- Maithel, S.K.; Gamblin, T.C.; Kamel, I.; Corona-Villalobos, C.P.; Thomas, M.; Pawlik, T.M. Multidisciplinary approaches to intrahepatic cholangiocarcinoma. Cancer 2013, 119, 3929–3942. [Google Scholar] [CrossRef]
- Witjes, C.D.; Karim-Kos, H.E.; Visser, O.; de Vries, E.; IJzermans, I.J.; de Man, R.A.; Coebergh, J.W.; Verhoef, C. Intrahepatic cholangiocarcinoma in a low endemic area: Rising incidence and improved survival. HPB 2012, 14, 777–781. [Google Scholar] [CrossRef]
- Beal, E.W.; Tumin, D.; Moris, D.; Zhang, X.F.; Chakedis, J.; Dilhoff, M.; Schmidt, C.M.; Pawlik, T.M. Cohort contributions to trends in the incidence and mortality of intrahepatic cholangiocarcinoma. Hepatobiliary Surg. Nutr. 2018, 7, 270–276. [Google Scholar] [CrossRef]
- Antwi, S.O.; Mousa, O.Y.; Patel, T. Racial, Ethnic, and Age Disparities in Incidence and Survival of Intrahepatic Cholangiocarcinoma in the United States; 1995–2014. Ann. Hepatol. 2018, 17, 604–614. [Google Scholar] [CrossRef]
- Brito, A.F.; Abrantes, A.M.; Encarnacao, J.C.; Tralhao, J.G.; Botelho, M.F. Cholangiocarcinoma: From molecular biology to treatment. Med. Oncol. 2015, 32, 245. [Google Scholar] [CrossRef] [PubMed]
- Dodson, R.M.; Weiss, M.J.; Cosgrove, D.; Herman, J.M.; Kamel, I.; Anders, R.; Geschwind, J.F.; Pawlik, T.M. Intrahepatic cholangiocarcinoma: Management options and emerging therapies. J. Am. Coll. Surg. 2013, 217, 736–750.e734. [Google Scholar] [CrossRef]
- Anderson, C.D.; Pinson, C.W.; Berlin, J.; Chari, R.S. Diagnosis and treatment of cholangiocarcinoma. Oncologist 2004, 9, 43–57. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver. EASL-ILCA Clinical Practice Guidelines on the management of intrahepatic cholangiocarcinoma. J. Hepatol. 2023, 79, 181–208. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Swanton, C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell 2015, 27, 15–26. [Google Scholar] [CrossRef]
- Baslan, T.; Hicks, J. Unravelling biology and shifting paradigms in cancer with single-cell sequencing. Nat. Rev. Cancer 2017, 17, 557–569. [Google Scholar] [CrossRef] [PubMed]
- Davidson, N.E.; Armstrong, S.A.; Coussens, L.M.; Cruz-Correa, M.R.; DeBerardinis, R.J.; Doroshow, J.H.; Foti, M.; Hwu, P.; Kensler, T.W.; Morrow, M.; et al. AACR Cancer Progress Report 2016. Clin. Cancer Res. 2016, 22 (Suppl. S19), S1–S137. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2016, 2, 16018. [Google Scholar] [CrossRef] [PubMed]
- Tyson, G.L.; Ilyas, J.A.; Duan, Z.; Green, L.K.; Younes, M.; El-Serag, H.B.; Davila, J.A. Secular trends in the incidence of cholangiocarcinoma in the USA and the impact of misclassification. Dig. Dis. Sci. 2014, 59, 3103–3110. [Google Scholar] [CrossRef]
- Nagtegaal, I.D.; Odze, R.D.; Klimstra, D.; Paradis, V.; Rugge, M.; Schirmacher, P.; Washington, K.M.; Carneiro, F.; Cree, I.A.; WHO Classification of Tumours Editorial Board. The 2019 WHO classification of tumours of the digestive system. Histopathology 2020, 76, 182–188. [Google Scholar] [CrossRef]
- Gonzalez, R.S.; Raza, A.; Propst, R.; Adeyi, O.; Bateman, J.; Sopha, S.C.; Shaw, J.; Auerbach, A. Recent Advances in Digestive Tract Tumors: Updates from the 5th Edition of the World Health Organization “Blue Book”. Arch. Pathol. Lab. Med. 2021, 145, 607–626. [Google Scholar] [CrossRef]
- Kendall, T.; Verheij, J.; Gaudio, E.; Evert, M.; Guido, M.; Goeppert, B.; Carpino, G. Anatomical, histomorphological and molecular classification of cholangiocarcinoma. Liver Int. 2019, 39 (Suppl. S1), 7–18. [Google Scholar] [CrossRef]
- Rodrigues, P.M.; Olaizola, P.; Paiva, N.A.; Olaizola, I.; Agirre-Lizaso, A.; Landa, A.; Bujanda, L.; Perugorria, M.J.; Banales, J.M. Pathogenesis of Cholangiocarcinoma. Annu. Rev. Pathol. 2021, 16, 433–463. [Google Scholar] [CrossRef]
- Ilyas, S.I.; Khan, S.A.; Hallemeier, C.L.; Kelley, R.K.; Gores, G.J. Cholangiocarcinoma—Evolving concepts and therapeutic strategies. Nat. Rev. Clin. Oncol. 2018, 15, 95–111. [Google Scholar] [CrossRef]
- Brivio, S.; Cadamuro, M.; Strazzabosco, M.; Fabris, L. Tumor reactive stroma in cholangiocarcinoma: The fuel behind cancer aggressiveness. World J. Hepatol. 2017, 9, 455–468. [Google Scholar] [CrossRef]
- Colasanti, T.; Vakifahmetoglu-Norberg, H.; Mancone, C. Expression and function of collagens in intrahepatic cholangiocarcinoma. Hepatoma Res. 2023, 9, 19. [Google Scholar] [CrossRef]
- Gentilini, A.; Pastore, M.; Marra, F.; Raggi, C. The Role of Stroma in Cholangiocarcinoma: The Intriguing Interplay between Fibroblastic Component, Immune Cell Subsets and Tumor Epithelium. Int. J. Mol. Sci. 2018, 19, 2885. [Google Scholar] [CrossRef]
- Fabris, L.; Perugorria, M.J.; Mertens, J.; Bjorkstrom, N.K.; Cramer, T.; Lleo, A.; Solinas, A.; Sanger, H.; Lukacs-Kornek, V.; Moncsek, A.; et al. The tumour microenvironment and immune milieu of cholangiocarcinoma. Liver Int. 2019, 39 (Suppl. S1), 63–78. [Google Scholar] [CrossRef]
- Carpino, G.; Cardinale, V.; Di Giamberardino, A.; Overi, D.; Donsante, S.; Colasanti, T.; Amato, G.; Mennini, G.; Franchitto, M.; Conti, F.; et al. Thrombospondin 1 and 2 along with PEDF inhibit angiogenesis and promote lymphangiogenesis in intrahepatic cholangiocarcinoma. J. Hepatol. 2021, 75, 1377–1386. [Google Scholar] [CrossRef]
- Corbella, E.; Fara, C.; Covarelli, F.; Porreca, V.; Palmisano, B.; Mignogna, G.; Corsi, A.; Riminucci, M.; Maras, B.; Mancone, C. THBS1 and THBS2 Enhance the In Vitro Proliferation, Adhesion, Migration and Invasion of Intrahepatic Cholangiocarcinoma Cells. Int. J. Mol. Sci. 2024, 25, 1782. [Google Scholar] [CrossRef] [PubMed]
- Lowery, M.A.; Ptashkin, R.; Jordan, E.; Berger, M.F.; Zehir, A.; Capanu, M.; Kemeny, N.E.; O’Reilly, E.M.; El-Dika, I.; Jarnagin, W.R.; et al. Comprehensive Molecular Profiling of Intrahepatic and Extrahepatic Cholangiocarcinomas: Potential Targets for Intervention. Clin. Cancer Res. 2018, 24, 4154–4161. [Google Scholar] [CrossRef] [PubMed]
- Silverman, I.M.; Hollebecque, A.; Friboulet, L.; Owens, S.; Newton, R.C.; Zhen, H.; Feliz, L.; Zecchetto, C.; Melisi, D.; Burn, T.C. Clinicogenomic Analysis of FGFR2-Rearranged Cholangiocarcinoma Identifies Correlates of Response and Mechanisms of Resistance to Pemigatinib. Cancer Discov. 2021, 11, 326–339. [Google Scholar] [CrossRef] [PubMed]
- Chung, T.; Park, Y.N. Up-to-Date Pathologic Classification and Molecular Characteristics of Intrahepatic Cholangiocarcinoma. Front. Med. 2022, 9, 857140. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, R.; Gupta, K.; Gupta, S. Cancer epigenetics: An introduction. Methods Mol. Biol. 2015, 1238, 3–25. [Google Scholar] [CrossRef]
- McGranahan, N.; Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef]
- Lundby, A.; Rossin, E.J.; Steffensen, A.B.; Acha, M.R.; Newton-Cheh, C.; Pfeufer, A.; Lynch, S.N.; QT Interval International GWAS Consortium (QT-IGC); Olesen, S.P.; Brunak, S.; et al. Annotation of loci from genome-wide association studies using tissue-specific quantitative interaction proteomics. Nat. Methods 2014, 11, 868–874. [Google Scholar] [CrossRef]
- Thomas, R.; Pontius, J.U.; Borst, L.B.; Breen, M. Development of a Genome-Wide Oligonucleotide Microarray Platform for Detection of DNA Copy Number Aberrations in Feline Cancers. Vet. Sci. 2020, 7, 88. [Google Scholar] [CrossRef]
- Chen, X.; Dong, Y.; Huang, Y.; Fan, J.; Yang, M.; Zhang, J. Whole-genome resequencing using next-generation and Nanopore sequencing for molecular characterization of T-DNA integration in transgenic poplar 741. BMC Genom. 2021, 22, 329. [Google Scholar] [CrossRef]
- Ou, Y.J.; Ren, Q.Q.; Fang, S.T.; Wu, J.G.; Jiang, Y.X.; Chen, Y.R.; Zhong, Y.; Wang, D.D.; Zhang, G.X. Complete Genome Insights into Lactococcus petauri CF11 Isolated from a Healthy Human Gut Using Second- and Third-Generation Sequencing. Front. Genet. 2020, 11, 119. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, C.; Xue, R.; Liu, M.; Bai, J.; Bao, J.; Wang, Y.; Jiang, N.; Li, Z.; Wang, W.; et al. Deep whole-genome analysis of 494 hepatocellular carcinomas. Nature 2024, 627, 586–593. [Google Scholar] [CrossRef] [PubMed]
- Rheinbay, E.; Nielsen, M.M.; Abascal, F.; Wala, J.A.; Shapira, O.; Tiao, G.; Hornshoj, H.; Hess, J.M.; Juul, R.I.; Lin, Z.; et al. Analyses of non-coding somatic drivers in 2,658 cancer whole genomes. Nature 2020, 578, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Dentro, S.C.; Leshchiner, I.; Haase, K.; Tarabichi, M.; Wintersinger, J.; Deshwar, A.G.; Yu, K.; Rubanova, Y.; Macintyre, G.; Demeulemeester, J.; et al. Characterizing genetic intra-tumor heterogeneity across 2658 human cancer genomes. Cell 2021, 184, 2239–2254.e2239. [Google Scholar] [CrossRef]
- Turner, K.M.; Deshpande, V.; Beyter, D.; Koga, T.; Rusert, J.; Lee, C.; Li, B.; Arden, K.; Ren, B.; Nathanson, D.A.; et al. Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature 2017, 543, 122–125. [Google Scholar] [CrossRef]
- Piunti, A.; Shilatifard, A. Epigenetic balance of gene expression by Polycomb and COMPASS families. Science 2016, 352, aad9780. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Chang, H.Y. Epigenomics: Technologies and Applications. Circ. Res. 2018, 122, 1191–1199. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Wan, H.; Tian, J.; Cheng, X.; Rao, M.; Li, J.; Zhang, H.; Zhang, M.; Cai, Y.; Lu, Z.; et al. CpG-methylation-based risk score predicts progression in colorectal cancer. Epigenomics 2020, 12, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Kuang, P.; Chen, P.; Wang, L.; Li, W.; Chen, B.; Liu, Y.; Xu, Y.; Wang, H.; Zhao, S.; Ye, L.; et al. RNA sequencing analysis of small cell lung cancer reveals candidate chemotherapy insensitivity long noncoding RNAs and microRNAs. Ann. Transl. Med. 2020, 8, 121. [Google Scholar] [CrossRef]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of transcription in human cells. Nature 2012, 489, 101–108. [Google Scholar] [CrossRef]
- Gilyazova, I.; Ivanova, E.; Gupta, H.; Mustafin, A.; Ishemgulov, R.; Izmailov, A.; Gilyazova, G.; Pudova, E.; Pavlov, V.; Khusnutdinova, E. miRNA Expression Patterns in Early- and Late-Stage Prostate Cancer Patients: High-Throughput Analysis. Biomedicines 2023, 11, 3073. [Google Scholar] [CrossRef] [PubMed]
- Yamada, A.; Yu, P.; Lin, W.; Okugawa, Y.; Boland, C.R.; Goel, A. A RNA-Sequencing approach for the identification of novel long non-coding RNA biomarkers in colorectal cancer. Sci. Rep. 2018, 8, 575. [Google Scholar] [CrossRef] [PubMed]
- Enroth, S.; Berggrund, M.; Lycke, M.; Broberg, J.; Lundberg, M.; Assarsson, E.; Olovsson, M.; Stalberg, K.; Sundfeldt, K.; Gyllensten, U. High throughput proteomics identifies a high-accuracy 11 plasma protein biomarker signature for ovarian cancer. Commun. Biol. 2019, 2, 221. [Google Scholar] [CrossRef]
- Zhou, Y.; Lih, T.M.; Pan, J.; Hoti, N.; Dong, M.; Cao, L.; Hu, Y.; Cho, K.C.; Chen, S.Y.; Eguez, R.V.; et al. Proteomic signatures of 16 major types of human cancer reveal universal and cancer-type-specific proteins for the identification of potential therapeutic targets. J. Hematol. Oncol. 2020, 13, 170. [Google Scholar] [CrossRef]
- Ziegler, Y.S.; Moresco, J.J.; Tu, P.G.; Yates, J.R., 3rd; Nardulli, A.M. Plasma membrane proteomics of human breast cancer cell lines identifies potential targets for breast cancer diagnosis and treatment. PLoS ONE 2014, 9, e102341. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, T.; Jung, A.; Reu, S.; Porzner, M.; Hlubek, F.; Kunz-Schughart, L.A.; Knuechel, R.; Kirchner, T. Variable beta-catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc. Natl. Acad. Sci. USA 2001, 98, 10356–10361. [Google Scholar] [CrossRef]
- Lee, S.I.; Kim, D.K.; Seo, E.J.; Choi, E.J.; Kwon, Y.W.; Jang, I.H.; Lee, J.C.; Kim, H.Y.; Shong, M.; Kim, J.H.; et al. Role of Kruppel-Like Factor 4 in the Maintenance of Chemoresistance of Anaplastic Thyroid Cancer. Thyroid 2017, 27, 1424–1432. [Google Scholar] [CrossRef]
- Selevsek, N.; Chang, C.Y.; Gillet, L.C.; Navarro, P.; Bernhardt, O.M.; Reiter, L.; Cheng, L.Y.; Vitek, O.; Aebersold, R. Reproducible and consistent quantification of the Saccharomyces cerevisiae proteome by SWATH-mass spectrometry. Mol. Cell. Proteom. 2015, 14, 739–749. [Google Scholar] [CrossRef]
- Choudhary, C.; Mann, M. Decoding signalling networks by mass spectrometry-based proteomics. Nat. Rev. Mol. Cell Biol. 2010, 11, 427–439. [Google Scholar] [CrossRef]
- Stack, E.C.; Wang, C.; Roman, K.A.; Hoyt, C.C. Multiplexed immunohistochemistry, imaging, and quantitation: A review, with an assessment of Tyramide signal amplification, multispectral imaging and multiplex analysis. Methods 2014, 70, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Tsujikawa, T.; Kumar, S.; Borkar, R.N.; Azimi, V.; Thibault, G.; Chang, Y.H.; Balter, A.; Kawashima, R.; Choe, G.; Sauer, D.; et al. Quantitative Multiplex Immunohistochemistry Reveals Myeloid-Inflamed Tumor-Immune Complexity Associated with Poor Prognosis. Cell Rep. 2017, 19, 203–217. [Google Scholar] [CrossRef] [PubMed]
- Govek, K.W.; Troisi, E.C.; Miao, Z.; Aubin, R.G.; Woodhouse, S.; Camara, P.G. Single-cell transcriptomic analysis of mIHC images via antigen mapping. Sci. Adv. 2021, 7, eabc5464. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.; Mahieu, N.G.; Johnson, S.L.; Patti, G.J. After the feature presentation: Technologies bridging untargeted metabolomics and biology. Curr. Opin. Biotechnol. 2014, 28, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Gauguier, D. Application of quantitative metabolomics in systems genetics in rodent models of complex phenotypes. Arch. Biochem. Biophys. 2016, 589, 158–167. [Google Scholar] [CrossRef]
- Dunn, W.B.; Broadhurst, D.I.; Atherton, H.J.; Goodacre, R.; Griffin, J.L. Systems level studies of mammalian metabolomes: The roles of mass spectrometry and nuclear magnetic resonance spectroscopy. Chem. Soc. Rev. 2011, 40, 387–426. [Google Scholar] [CrossRef] [PubMed]
- Schiliro, C.; Firestein, B.L. Mechanisms of Metabolic Reprogramming in Cancer Cells Supporting Enhanced Growth and Proliferation. Cells 2021, 10, 1056. [Google Scholar] [CrossRef]
- Kang, C.; Zhang, J.; Xue, M.; Li, X.; Ding, D.; Wang, Y.; Jiang, S.; Chu, F.F.; Gao, Q.; Zhang, M. Metabolomics analyses of cancer tissue from patients with colorectal cancer. Mol. Med. Rep. 2023, 28, 219. [Google Scholar] [CrossRef]
- Mei, L.; Zhang, Z.; Li, X.; Yang, Y.; Qi, R. Metabolomics profiling in prediction of chemo-immunotherapy efficiency in advanced non-small cell lung cancer. Front. Oncol. 2022, 12, 1025046. [Google Scholar] [CrossRef]
- Zhao, R.; Ren, S.; Li, C.; Guo, K.; Lu, Z.; Tian, L.; He, J.; Zhang, K.; Cao, Y.; Liu, S.; et al. Biomarkers for pancreatic cancer based on tissue and serum metabolomics analysis in a multicenter study. Cancer Med. 2023, 12, 5158–5171. [Google Scholar] [CrossRef]
- Subramanian, I.; Verma, S.; Kumar, S.; Jere, A.; Anamika, K. Multi-omics Data Integration, Interpretation, and Its Application. Bioinform. Biol. Insights 2020, 14, 1177932219899051. [Google Scholar] [CrossRef]
- Gambardella, V.; Tarazona, N.; Cejalvo, J.M.; Lombardi, P.; Huerta, M.; Rosello, S.; Fleitas, T.; Roda, D.; Cervantes, A. Personalized Medicine: Recent Progress in Cancer Therapy. Cancers 2020, 12, 1009. [Google Scholar] [CrossRef]
- Frohlich, H.; Balling, R.; Beerenwinkel, N.; Kohlbacher, O.; Kumar, S.; Lengauer, T.; Maathuis, M.H.; Moreau, Y.; Murphy, S.A.; Przytycka, T.M.; et al. From hype to reality: Data science enabling personalized medicine. BMC Med. 2018, 16, 150. [Google Scholar] [CrossRef]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M.; Cancer Genome Atlas Research Network. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Rudnick, P.A.; Markey, S.P.; Roth, J.; Mirokhin, Y.; Yan, X.; Tchekhovskoi, D.V.; Edwards, N.J.; Thangudu, R.R.; Ketchum, K.A.; Kinsinger, C.R.; et al. A Description of the Clinical Proteomic Tumor Analysis Consortium (CPTAC) Common Data Analysis Pipeline. J. Proteome Res. 2016, 15, 1023–1032. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xiao, C.; Yue, K.; Chen, M.; Zhou, H.; Yan, X. Identification of multi-omics biomarkers and construction of the novel prognostic model for hepatocellular carcinoma. Sci. Rep. 2022, 12, 12084. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, N.; Roy, S.; Vengadesan, K.; Gupta, D. Multi-omics approach for identifying CNV-associated lncRNA signatures with prognostic value in prostate cancer. Noncoding RNA Res. 2024, 9, 66–75. [Google Scholar] [CrossRef] [PubMed]
- Kafita, D.; Nkhoma, P.; Zulu, M.; Sinkala, M. Proteogenomic analysis of pancreatic cancer subtypes. PLoS ONE 2021, 16, e0257084. [Google Scholar] [CrossRef]
- Farshidfar, F.; Zheng, S.; Gingras, M.C.; Newton, Y.; Shih, J.; Robertson, A.G.; Hinoue, T.; Hoadley, K.A.; Gibb, E.A.; Roszik, J.; et al. Integrative Genomic Analysis of Cholangiocarcinoma Identifies Distinct IDH-Mutant Molecular Profiles. Cell Rep. 2017, 19, 2878–2880. [Google Scholar] [CrossRef] [PubMed]
- Sia, D.; Losic, B.; Moeini, A.; Cabellos, L.; Hao, K.; Revill, K.; Bonal, D.; Miltiadous, O.; Zhang, Z.; Hoshida, Y.; et al. Massive parallel sequencing uncovers actionable FGFR2-PPHLN1 fusion and ARAF mutations in intrahepatic cholangiocarcinoma. Nat. Commun. 2015, 6, 6087. [Google Scholar] [CrossRef]
- Arai, Y.; Totoki, Y.; Hosoda, F.; Shirota, T.; Hama, N.; Nakamura, H.; Ojima, H.; Furuta, K.; Shimada, K.; Okusaka, T.; et al. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology 2014, 59, 1427–1434. [Google Scholar] [CrossRef]
- Simbolo, M.; Fassan, M.; Ruzzenente, A.; Mafficini, A.; Wood, L.D.; Corbo, V.; Melisi, D.; Malleo, G.; Vicentini, C.; Malpeli, G.; et al. Multigene mutational profiling of cholangiocarcinomas identifies actionable molecular subgroups. Oncotarget 2014, 5, 2839–2852. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Pawlik, T.M.; Anders, R.A.; Selaru, F.M.; Streppel, M.M.; Lucas, D.J.; Niknafs, N.; Guthrie, V.B.; Maitra, A.; Argani, P.; et al. Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinomas. Nat. Genet. 2013, 45, 1470–1473. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Y.; Zhu, W.W.; Wang, Z.; Huang, J.B.; Wang, S.H.; Bai, F.M.; Li, T.E.; Zhu, Y.; Zhao, J.; Yang, X.; et al. Driver mutations of intrahepatic cholangiocarcinoma shape clinically relevant genomic clusters with distinct molecular features and therapeutic vulnerabilities. Theranostics 2022, 12, 260–276. [Google Scholar] [CrossRef]
- Song, H.; Huang, Y.; Jiang, X. Mutation spectrum associated with metastasis of advanced cholangiocarcinoma. J. Int. Med. Res. 2022, 50, 3000605221102080. [Google Scholar] [CrossRef]
- Sia, D.; Hoshida, Y.; Villanueva, A.; Roayaie, S.; Ferrer, J.; Tabak, B.; Peix, J.; Sole, M.; Tovar, V.; Alsinet, C.; et al. Integrative molecular analysis of intrahepatic cholangiocarcinoma reveals 2 classes that have different outcomes. Gastroenterology 2013, 144, 829–840. [Google Scholar] [CrossRef]
- Dalmasso, C.; Carpentier, W.; Guettier, C.; Camilleri-Broet, S.; Borelli, W.V.; Campos Dos Santos, C.R.; Castaing, D.; Duclos-Vallee, J.C.; Broet, P. Patterns of chromosomal copy-number alterations in intrahepatic cholangiocarcinoma. BMC Cancer 2015, 15, 126. [Google Scholar] [CrossRef]
- Goeppert, B.; Toth, R.; Singer, S.; Albrecht, T.; Lipka, D.B.; Lutsik, P.; Brocks, D.; Baehr, M.; Muecke, O.; Assenov, Y.; et al. Integrative Analysis Defines Distinct Prognostic Subgroups of Intrahepatic Cholangiocarcinoma. Hepatology 2019, 69, 2091–2106. [Google Scholar] [CrossRef]
- Peng, Y.; Meng, G.; Sheng, X.; Gao, H. Transcriptome and DNA methylation analysis reveals molecular mechanisms underlying intrahepatic cholangiocarcinoma progression. J. Cell. Mol. Med. 2021, 25, 6373–6387. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Feng, Y.; An, S.; Liu, F.; Xiang, G. Integrative epigenomic profiling reveal AP-1 is a key regulator in intrahepatich cholangiocarcinoma. Genomics 2022, 114, 241–252. [Google Scholar] [CrossRef]
- Dragomir, M.P.; Calina, T.G.; Perez, E.; Schallenberg, S.; Chen, M.; Albrecht, T.; Koch, I.; Wolkenstein, P.; Goeppert, B.; Roessler, S.; et al. DNA methylation-based classifier differentiates intrahepatic pancreato-biliary tumours. EBioMedicine 2023, 93, 104657. [Google Scholar] [CrossRef]
- Chen, X.; Dong, L.; Chen, L.; Wang, Y.; Du, J.; Ma, L.; Yan, X.; Huang, J.; Liao, M.; Chen, X.; et al. Epigenome-wide development and validation of a prognostic methylation score in intrahepatic cholangiocarcinoma based on machine learning strategies. Hepatobiliary Surg. Nutr. 2023, 12, 478–494. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.; Chen, X.; Wang, H.; Lin, Y.; Chen, L.; Yuan, K.; Liao, M.; Jiang, H.; Peng, J.; Wu, Z.; et al. Whole-Genome DNA Methylation Profiling of Intrahepatic Cholangiocarcinoma Reveals Prognostic Subtypes with Distinct Biological Drivers. Cancer Res. 2024, 84, 1747–1763. [Google Scholar] [CrossRef]
- Job, S.; Rapoud, D.; Dos Santos, A.; Gonzalez, P.; Desterke, C.; Pascal, G.; Elarouci, N.; Ayadi, M.; Adam, R.; Azoulay, D.; et al. Identification of Four Immune Subtypes Characterized by Distinct Composition and Functions of Tumor Microenvironment in Intrahepatic Cholangiocarcinoma. Hepatology 2020, 72, 965–981. [Google Scholar] [CrossRef]
- Ahn, K.S.; O’Brien, D.; Kang, Y.N.; Mounajjed, T.; Kim, Y.H.; Kim, T.S.; Kocher, J.A.; Allotey, L.K.; Borad, M.J.; Roberts, L.R.; et al. Prognostic subclass of intrahepatic cholangiocarcinoma by integrative molecular-clinical analysis and potential targeted approach. Hepatol. Int. 2019, 13, 490–500. [Google Scholar] [CrossRef]
- Silvestri, M.; Nghia Vu, T.; Nichetti, F.; Niger, M.; Di Cosimo, S.; De Braud, F.; Pruneri, G.; Pawitan, Y.; Calza, S.; Cappelletti, V. Comprehensive transcriptomic analysis to identify biological and clinical differences in cholangiocarcinoma. Cancer Med. 2023, 12, 10156–10168. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, D.; Liu, P.; Chen, Y.; Yu, H.; Zhang, Q. Identification of biomarkers of intrahepatic cholangiocarcinoma via integrated analysis of mRNA and miRNA microarray data. Mol. Med. Rep. 2017, 15, 1051–1056. [Google Scholar] [CrossRef]
- Yang, W.; Li, Y.; Song, X.; Xu, J.; Xie, J. Genome-wide analysis of long noncoding RNA and mRNA co-expression profile in intrahepatic cholangiocarcinoma tissue by RNA sequencing. Oncotarget 2017, 8, 26591–26599. [Google Scholar] [CrossRef] [PubMed]
- Peraldo-Neia, C.; Ostano, P.; Cavalloni, G.; Pignochino, Y.; Sangiolo, D.; De Cecco, L.; Marchesi, E.; Ribero, D.; Scarpa, A.; De Rose, A.M.; et al. Transcriptomic analysis and mutational status of IDH1 in paired primary-recurrent intrahepatic cholangiocarcinoma. BMC Genom. 2018, 19, 440. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Sun, L.; Li, J.; Zhou, C.; Cheng, L.; Chen, K.; Yan, B.; Qian, W.; Ma, Q.; Duan, W. A novel three-miRNA signature predicts survival in cholangiocarcinoma based on RNA-Seq data. Oncol. Rep. 2018, 40, 1422–1434. [Google Scholar] [CrossRef]
- Ye, Z.; Zeng, Z.; Wang, D.; Lei, S.; Shen, Y.; Chen, Z. Identification of key genes associated with the progression of intrahepatic cholangiocarcinoma using weighted gene co-expression network analysis. Oncol. Lett. 2020, 20, 483–494. [Google Scholar] [CrossRef]
- Xia, L.; Chen, X.; Yang, J.; Zhu, S.; Zhang, L.; Yin, Q.; Hong, Y.; Chen, H.; Chen, G.; Li, H. Long Non-Coding RNA-PAICC Promotes the Tumorigenesis of Human Intrahepatic Cholangiocarcinoma by Increasing YAP1 Transcription. Front. Oncol. 2020, 10, 595533. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Qu, L.; Zhang, H.; Liu, J.; Zhang, X. A comprehensive transcriptomic landscape of cholangiocarcinoma based on bioinformatics analysis from large cohort of patients. Sci. Rep. 2021, 11, 13713. [Google Scholar] [CrossRef] [PubMed]
- Rhee, H.; Ko, J.E.; Chung, T.; Jee, B.A.; Kwon, S.M.; Nahm, J.H.; Seok, J.Y.; Yoo, J.E.; Choi, J.S.; Thorgeirsson, S.S.; et al. Transcriptomic and histopathological analysis of cholangiolocellular differentiation trait in intrahepatic cholangiocarcinoma. Liver Int. 2018, 38, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Yang, H.; Wan, L.; Wang, Z.; Wang, H.; Ge, C.; Liu, Y.; Hao, Y.; Zhang, D.; Shi, G.; et al. Single-cell transcriptomic architecture and intercellular crosstalk of human intrahepatic cholangiocarcinoma. J. Hepatol. 2020, 73, 1118–1130. [Google Scholar] [CrossRef]
- Xiang, X.; Liu, Z.; Zhang, C.; Li, Z.; Gao, J.; Zhang, C.; Cao, Q.; Cheng, J.; Liu, H.; Chen, D.; et al. IDH Mutation Subgroup Status Associates with Intratumor Heterogeneity and the Tumor Microenvironment in Intrahepatic Cholangiocarcinoma. Adv. Sci. 2021, 8, e2101230. [Google Scholar] [CrossRef]
- Zhou, Q.; Ji, L.; Shi, X.; Deng, D.; Guo, F.; Wang, Z.; Liu, W.; Zhang, J.; Xia, S.; Shang, D. INTS8 is a therapeutic target for intrahepatic cholangiocarcinoma via the integration of bioinformatics analysis and experimental validation. Sci. Rep. 2021, 11, 23649. [Google Scholar] [CrossRef]
- Liao, W.; Du, J.; Li, L.; Wu, X.; Chen, X.; Feng, Q.; Xu, L.; Chen, X.; Liao, M.; Huang, J.; et al. CircZNF215 promotes tumor growth and metastasis through inactivation of the PTEN/AKT pathway in intrahepatic cholangiocarcinoma. J. Exp. Clin. Cancer Res. 2023, 42, 125. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.T.; Yang, Y.F.; Chen, X.M.; Wang, S.B.; Tong, G.L. IL23R as an indicator of immune infiltration and poor prognosis in intrahepatic cholangiocarcinoma: A bioinformatics analysis. Transl. Cancer Res. 2023, 12, 2461–2476. [Google Scholar] [CrossRef] [PubMed]
- Correa-Gallego, C.; Maddalo, D.; Doussot, A.; Kemeny, N.; Kingham, T.P.; Allen, P.J.; D’Angelica, M.I.; DeMatteo, R.P.; Betel, D.; Klimstra, D.; et al. Circulating Plasma Levels of MicroRNA-21 and MicroRNA-221 Are Potential Diagnostic Markers for Primary Intrahepatic Cholangiocarcinoma. PLoS ONE 2016, 11, e0163699. [Google Scholar] [CrossRef]
- Zhang, Y.; Ma, Z.; Li, C.; Wang, C.; Jiang, W.; Chang, J.; Han, S.; Lu, Z.; Shao, Z.; Wang, Y.; et al. The genomic landscape of cholangiocarcinoma reveals the disruption of post-transcriptional modifiers. Nat. Commun. 2022, 13, 3061. [Google Scholar] [CrossRef]
- Carpino, G.; Overi, D.; Melandro, F.; Grimaldi, A.; Cardinale, V.; Di Matteo, S.; Mennini, G.; Rossi, M.; Alvaro, D.; Barnaba, V.; et al. Matrisome analysis of intrahepatic cholangiocarcinoma unveils a peculiar cancer-associated extracellular matrix structure. Clin. Proteom. 2019, 16, 37. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, A.; Court, M.; Thiers, V.; Sar, S.; Guettier, C.; Samuel, D.; Brechot, C.; Garin, J.; Demaugre, F.; Masselon, C.D. Identification of cellular targets in human intrahepatic cholangiocarcinoma using laser microdissection and accurate mass and time tag proteomics. Mol. Cell. Proteom. 2010, 9, 1991–2004. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Xu, S.; Ye, C.; Li, Q.; Chen, R.; Wu, W.; Jiang, Q.; Jia, Y.; Zhang, X.; Fan, L.; et al. Proteomic and single-cell landscape reveals novel pathogenic mechanisms of HBV-infected intrahepatic cholangiocarcinoma. iScience 2023, 26, 106003. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.; Zhu, J.; Liu, W.; Peng, L.; Lu, C.; Sun, P.; Huang, L.; Nie, X.; Huang, S.; Guo, T.; et al. Proteome Landscapes of Human Hepatocellular Carcinoma and Intrahepatic Cholangiocarcinoma. Mol. Cell. Proteom. 2023, 22, 100604. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Li, Q.; Ren, W.; Wu, H.; Wang, C.; Li, X.; Xue, B.; Qiu, Y.; Zhang, J.; Chen, J.; et al. Quantitative Proteomics Reveals Down-Regulated Glycolysis/Gluconeogenesis in the Large-Duct Type Intrahepatic Cholangiocarcinoma. J. Proteome Res. 2022, 21, 2504–2514. [Google Scholar] [CrossRef] [PubMed]
- Polidoro, M.A.; Franceschini, B.; Milana, F.; Soldani, C.; Carriero, R.; Aghemo, A.; Donadon, M.; Torzilli, G.; Pastorelli, R.; Brunelli, L.; et al. Decoding human Intrahepatic Cholangiocarcinoma Metabolism: Unveiling the Impact of SLC2A3 on Aggressiveness and Prognosis. Dig. Liver Dis. 2024, 56, S6–S7. [Google Scholar] [CrossRef]
- Cavalloni, G.; Peraldo-Neia, C.; Massa, A.; Bergamini, C.; Trentini, A.; De Rosa, G.; Daniele, L.; Ciccosanti, F.; Cervellati, C.; Leone, F.; et al. Proteomic analysis identifies deregulated metabolic and oxidative-associated proteins in Italian intrahepatic cholangiocarcinoma patients. BMC Cancer 2021, 21, 865. [Google Scholar] [CrossRef]
- Li, J.; Lu, J.; Lv, S.; Sun, S.; Liu, C.; Xu, F.; Sun, H.; Yang, J.; Wang, X.; Zhong, X.; et al. Linoleic acid pathway disturbance contributing to potential cancerization of intrahepatic bile duct stones into intrahepatic cholangiocarcinoma. BMC Gastroenterol. 2022, 22, 269. [Google Scholar] [CrossRef]
- Haznadar, M.; Diehl, C.M.; Parker, A.L.; Krausz, K.W.; Bowman, E.D.; Rabibhadana, S.; Forgues, M.; Bhudhisawasdi, V.; Gonzalez, F.J.; Mahidol, C.; et al. Urinary Metabolites Diagnostic and Prognostic of Intrahepatic Cholangiocarcinoma. Cancer Epidemiol. Biomark. Prev. 2019, 28, 1704–1711. [Google Scholar] [CrossRef]
- Javle, M.M.; Murugesan, K.; Shroff, R.T.; Borad, M.J.; Abdel-Wahab, R.; Schrock, A.B.; Chung, J.; Goyal, L.; Frampton, G.M.; Kelley, R.K.; et al. Profiling of 3,634 cholangiocarcinomas (CCA) to identify genomic alterations (GA), tumor mutational burden (TMB), and genomic loss of heterozygosity (gLOH). J. Clin. Oncol. 2019, 37, 4087. [Google Scholar] [CrossRef]
- Romanidou, O.; Kotoula, V.; Fountzilas, G. Bridging Cancer Biology with the Clinic: Comprehending and Exploiting IDH Gene Mutations in Gliomas. Cancer Genom. Proteom. 2018, 15, 421–436. [Google Scholar] [CrossRef]
- Wang, P.; Dong, Q.; Zhang, C.; Kuan, P.F.; Liu, Y.; Jeck, W.R.; Andersen, J.B.; Jiang, W.; Savich, G.L.; Tan, T.X.; et al. Mutations in isocitrate dehydrogenase 1 and 2 occur frequently in intrahepatic cholangiocarcinomas and share hypermethylation targets with glioblastomas. Oncogene 2013, 32, 3091–3100. [Google Scholar] [CrossRef] [PubMed]
- Grassian, A.R.; Pagliarini, R.; Chiang, D.Y. Mutations of isocitrate dehydrogenase 1 and 2 in intrahepatic cholangiocarcinoma. Curr. Opin. Gastroenterol. 2014, 30, 295–302. [Google Scholar] [CrossRef]
- Nakagawa, M.; Yamaguchi, M.; Endo, M.; Machida, Y.; Hattori, A.; Tanzawa, F.; Tsutsumi, S.; Kitabayashi, I.; Kawai, A.; Nakatani, F. Clinical usefulness of 2-hydroxyglutarate as a biomarker in IDH-mutant chondrosarcoma. J. Bone Oncol. 2022, 34, 100430. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef]
- Grassian, A.R.; Lin, F.; Barrett, R.; Liu, Y.; Jiang, W.; Korpal, M.; Astley, H.; Gitterman, D.; Henley, T.; Howes, R.; et al. Isocitrate dehydrogenase (IDH) mutations promote a reversible ZEB1/microRNA (miR)-200-dependent epithelial-mesenchymal transition (EMT). J. Biol. Chem. 2012, 287, 42180–42194. [Google Scholar] [CrossRef] [PubMed]
- Chan-On, W.; Nairismagi, M.L.; Ong, C.K.; Lim, W.K.; Dima, S.; Pairojkul, C.; Lim, K.H.; McPherson, J.R.; Cutcutache, I.; Heng, H.L.; et al. Exome sequencing identifies distinct mutational patterns in liver fluke-related and non-infection-related bile duct cancers. Nat. Genet. 2013, 45, 1474–1478. [Google Scholar] [CrossRef]
- Javle, M.; Bekaii-Saab, T.; Jain, A.; Wang, Y.; Kelley, R.K.; Wang, K.; Kang, H.C.; Catenacci, D.; Ali, S.; Krishnan, S.; et al. Biliary cancer: Utility of next-generation sequencing for clinical management. Cancer 2016, 122, 3838–3847. [Google Scholar] [CrossRef] [PubMed]
- Mitelman, F.; Johansson, B.; Mertens, F. The impact of translocations and gene fusions on cancer causation. Nat. Rev. Cancer 2007, 7, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.S.; Wang, K.; Gay, L.; Al-Rohil, R.; Rand, J.V.; Jones, D.M.; Lee, H.J.; Sheehan, C.E.; Otto, G.A.; Palmer, G.; et al. New routes to targeted therapy of intrahepatic cholangiocarcinomas revealed by next-generation sequencing. Oncologist 2014, 19, 235–242. [Google Scholar] [CrossRef]
- Lamarca, A.; Barriuso, J.; McNamara, M.G.; Valle, J.W. Molecular targeted therapies: Ready for “prime time” in biliary tract cancer. J. Hepatol. 2020, 73, 170–185. [Google Scholar] [CrossRef]
- Graham, R.P.; Barr Fritcher, E.G.; Pestova, E.; Schulz, J.; Sitailo, L.A.; Vasmatzis, G.; Murphy, S.J.; McWilliams, R.R.; Hart, S.N.; Halling, K.C.; et al. Fibroblast growth factor receptor 2 translocations in intrahepatic cholangiocarcinoma. Hum. Pathol. 2014, 45, 1630–1638. [Google Scholar] [CrossRef]
- Goyal, L.; Meric-Bernstam, F.; Hollebecque, A.; Valle, J.W.; Morizane, C.; Karasic, T.B.; Abrams, T.A.; Furuse, J.; Kelley, R.K.; Cassier, P.A.; et al. Futibatinib for FGFR2-Rearranged Intrahepatic Cholangiocarcinoma. N. Engl. J. Med. 2023, 388, 228–239. [Google Scholar] [CrossRef]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef]
- Sirica, A.E. Role of ErbB family receptor tyrosine kinases in intrahepatic cholangiocarcinoma. World J. Gastroenterol. 2008, 14, 7033–7058. [Google Scholar] [CrossRef] [PubMed]
- Endo, K.; Yoon, B.I.; Pairojkul, C.; Demetris, A.J.; Sirica, A.E. ERBB-2 overexpression and cyclooxygenase-2 up-regulation in human cholangiocarcinoma and risk conditions. Hepatology 2002, 36, 439–450. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, D.; Ojima, H.; Iwasaki, M.; Hiraoka, N.; Kosuge, T.; Kasai, S.; Hirohashi, S.; Shibata, T. Clinicopathological and prognostic significance of EGFR, VEGF, and HER2 expression in cholangiocarcinoma. Br. J. Cancer 2008, 98, 418–425. [Google Scholar] [CrossRef]
- Tsutaho, A.; Hashimoto, A.; Hashimoto, S.; Hata, S.; Kachi, S.; Hirano, S.; Sabe, H. High expression of AMAP1, an ARF6 effector, is associated with elevated levels of PD-L1 and fibrosis of pancreatic cancer. Cell Commun. Signal 2020, 18, 101. [Google Scholar] [CrossRef] [PubMed]
- Frost, N.; Kollmeier, J.; Vollbrecht, C.; Grah, C.; Matthes, B.; Pultermann, D.; von Laffert, M.; Luders, H.; Olive, E.; Raspe, M.; et al. KRAS(G12C)/TP53 co-mutations identify long-term responders to first line palliative treatment with pembrolizumab monotherapy in PD-L1 high (>/=50%) lung adenocarcinoma. Transl. Lung Cancer Res. 2021, 10, 737–752. [Google Scholar] [CrossRef]
- Tran, C.G.; Goffredo, P.; Mott, S.L.; Hart, A.; You, Y.N.; Vauthey, J.N.; Weigel, R.J.; Hassan, I. The impact of KRAS mutation, microsatellite instability, and tumor laterality on the prognosis of nonmetastatic colon cancer. Surgery 2022, 171, 657–665. [Google Scholar] [CrossRef]
- Shi, D.; Jiang, P. A Different Facet of p53 Function: Regulation of Immunity and Inflammation during Tumor Development. Front. Cell Dev. Biol. 2021, 9, 762651. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Liu, Z.; Yu, Y.; Chen, Y.; Liu, H.; Guo, Y.; Peng, Z.; Cai, G.; Hua, Z.; Han, X.; et al. TP53/KRAS Co-Mutations Create Divergent Prognosis Signatures in Intrahepatic Cholangiocarcinoma. Front. Genet. 2022, 13, 844800. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.F.; Sun, J.P.; Zhang, S.R.; Zhu, G.S.; Li, L.B.; Liao, Y.L.; Xie, J.M.; Liao, W.J. KRAS and PIK3CA but not BRAF genes are frequently mutated in Chinese cholangiocarcinoma patients. Biomed. Pharmacother. 2011, 65, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.C.; Jan, Y.Y.; Yeh, T.S. K-ras mutation is strongly associated with perineural invasion and represents an independent prognostic factor of intrahepatic cholangiocarcinoma after hepatectomy. Ann. Surg. Oncol. 2012, 19 (Suppl. S3), S675–S681. [Google Scholar] [CrossRef] [PubMed]
- Furubo, S.; Harada, K.; Shimonishi, T.; Katayanagi, K.; Tsui, W.; Nakanuma, Y. Protein expression and genetic alterations of p53 and ras in intrahepatic cholangiocarcinoma. Histopathology 1999, 35, 230–240. [Google Scholar] [CrossRef]
- Tannapfel, A.; Weinans, L.; Geissler, F.; Schutz, A.; Katalinic, A.; Kockerling, F.; Hauss, J.; Wittekind, C. Mutations of p53 tumor suppressor gene, apoptosis, and proliferation in intrahepatic cholangiocellular carcinoma of the liver. Dig. Dis. Sci. 2000, 45, 317–324. [Google Scholar] [CrossRef]
- Xiaofang, L.; Kun, T.; Shaoping, Y.; Zaiqiu, W.; Hailong, S. Correlation between promoter methylation of p14(ARF), TMS1/ASC, and DAPK, and p53 mutation with prognosis in cholangiocarcinoma. World J. Surg. Oncol. 2012, 10, 5. [Google Scholar] [CrossRef] [PubMed]
- Goeppert, B.; Frauenschuh, L.; Renner, M.; Roessler, S.; Stenzinger, A.; Klauschen, F.; Warth, A.; Vogel, M.N.; Mehrabi, A.; Hafezi, M.; et al. BRAF V600E-specific immunohistochemistry reveals low mutation rates in biliary tract cancer and restriction to intrahepatic cholangiocarcinoma. Mod. Pathol. 2014, 27, 1028–1034. [Google Scholar] [CrossRef]
- Tannapfel, A.; Sommerer, F.; Benicke, M.; Katalinic, A.; Uhlmann, D.; Witzigmann, H.; Hauss, J.; Wittekind, C. Mutations of the BRAF gene in cholangiocarcinoma but not in hepatocellular carcinoma. Gut 2003, 52, 706–712. [Google Scholar] [CrossRef] [PubMed]
- Robertson, S.; Hyder, O.; Dodson, R.; Nayar, S.K.; Poling, J.; Beierl, K.; Eshleman, J.R.; Lin, M.T.; Pawlik, T.M.; Anders, R.A. The frequency of KRAS and BRAF mutations in intrahepatic cholangiocarcinomas and their correlation with clinical outcome. Hum. Pathol. 2013, 44, 2768–2773. [Google Scholar] [CrossRef]
- Andrici, J.; Goeppert, B.; Sioson, L.; Clarkson, A.; Renner, M.; Stenzinger, A.; Tayao, M.; Watson, N.; Farzin, M.; Toon, C.W.; et al. Loss of BAP1 Expression Occurs Frequently in Intrahepatic Cholangiocarcinoma. Medicine 2016, 95, e2491. [Google Scholar] [CrossRef]
- Rizzo, A.; Carloni, R.; Ricci, A.D.; Di Federico, A.; Guven, D.C.; Yalcin, S.; Brandi, G. Molecular Profile and Prognostic Value of BAP1 Mutations in Intrahepatic Cholangiocarcinoma: A Genomic Database Analysis. J. Pers. Med. 2022, 12, 1247. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.K.; Kim, W.H.; Jang, J.J. Expression of G1-S modulators (p53, p16, p27, cyclin D1, Rb) and Smad4/Dpc4 in intrahepatic cholangiocarcinoma. Hum. Pathol. 2002, 33, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Tavolari, S.; Brandi, G. Mutational Landscape of Cholangiocarcinoma According to Different Etiologies: A Review. Cells 2023, 12, 1216. [Google Scholar] [CrossRef]
- Yan, X.Q.; Zhang, W.; Zhang, B.X.; Liang, H.F.; Zhang, W.G.; Chen, X.P. Inactivation of Smad4 is a prognostic factor in intrahepatic cholangiocarcinoma. Chin. Med. J. 2013, 126, 3039–3043. [Google Scholar] [CrossRef] [PubMed]
- Hohmann, A.F.; Vakoc, C.R. A rationale to target the SWI/SNF complex for cancer therapy. Trends Genet. 2014, 30, 356–363. [Google Scholar] [CrossRef]
- Wiegand, K.C.; Shah, S.P.; Al-Agha, O.M.; Zhao, Y.; Tse, K.; Zeng, T.; Senz, J.; McConechy, M.K.; Anglesio, M.S.; Kalloger, S.E.; et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N. Engl. J. Med. 2010, 363, 1532–1543. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Sato, Y.; Nakanuma, Y. Cholangiolocellular Carcinoma with “Ductal Plate Malformation” Pattern May Be Characterized by ARID1A Genetic Alterations. Am. J. Surg. Pathol. 2019, 43, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Shiao, M.S.; Chiablaem, K.; Charoensawan, V.; Ngamphaiboon, N.; Jinawath, N. Emergence of Intrahepatic Cholangiocarcinoma: How High-Throughput Technologies Expedite the Solutions for a Rare Cancer Type. Front. Genet. 2018, 9, 309. [Google Scholar] [CrossRef]
- Sasaki, M.; Nitta, T.; Sato, Y.; Nakanuma, Y. Loss of ARID1A Expression Presents a Novel Pathway of Carcinogenesis in Biliary Carcinomas. Am. J. Clin. Pathol. 2016, 145, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Xu, Y.; Wu, W.; Wang, P.; Wang, Y.; Jiang, H.; Zhu, J. ARID1A Variations in Cholangiocarcinoma: Clinical Significances and Molecular Mechanisms. Front. Oncol. 2021, 11, 693295. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.Z.; Wang, A.Q.; Du, J.; Wang, J.T.; Yu, W.W.; Liu, Q.; Wu, Y.F.; Chen, S.G. Low expression of ARID1A correlates with poor prognosis in intrahepatic cholangiocarcinoma. World J. Gastroenterol. 2016, 22, 5814–5821. [Google Scholar] [CrossRef]
- Bi, C.; Liu, M.; Rong, W.; Wu, F.; Zhang, Y.; Lin, S.; Liu, Y.; Wu, J.; Wang, L. High Beclin-1 and ARID1A expression corelates with poor survival and high recurrence in intrahepatic cholangiocarcinoma: A histopathological retrospective study. BMC Cancer 2019, 19, 213. [Google Scholar] [CrossRef]
- Cong, W.M.; Bakker, A.; Swalsky, P.A.; Raja, S.; Woods, J.; Thomas, S.; Demetris, A.J.; Finkelstein, S.D. Multiple genetic alterations involved in the tumorigenesis of human cholangiocarcinoma: A molecular genetic and clinicopathological study. J. Cancer Res. Clin. Oncol. 2001, 127, 187–192. [Google Scholar] [CrossRef]
- Kim, H.; Kim, J.Y.; Park, K.U. Clinical Implications of BRCA Mutations in Advanced Biliary Tract Cancer. Oncology 2023, 101, 41–48. [Google Scholar] [CrossRef]
- Demols, A.; Perez-Casanova, L.; Rocq, L.; Charry, M.; De Nève, N.; Verrellen, A.; Ramadhan, A.; Van Campenhout, C.; De Clercq, S.; Maris, C.; et al. NTRK gene fusions in bilio-pancreatic cancers. J. Clin. Oncol. 2020, 38, e16664. [Google Scholar] [CrossRef]
- Andersen, J.B.; Thorgeirsson, S.S. Genetic profiling of intrahepatic cholangiocarcinoma. Curr. Opin. Gastroenterol. 2012, 28, 266–272. [Google Scholar] [CrossRef]
- Li, S.; Chai, Y.; Ding, Y.; Yuan, T.; Wu, C.; Huang, C. CHD1L is associated with poor survival and promotes the proliferation and metastasis of intrahepatic cholangiocarcinoma. Oncol. Rep. 2019, 42, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Rimini, M.; Fabregat-Franco, C.; Burgio, V.; Lonardi, S.; Niger, M.; Scartozzi, M.; Rapposelli, I.G.; Aprile, G.; Ratti, F.; Pedica, F.; et al. Molecular profile and its clinical impact of IDH1 mutated versus IDH1 wild type intrahepatic cholangiocarcinoma. Sci. Rep. 2022, 12, 18775. [Google Scholar] [CrossRef] [PubMed]
- Deenonpoe, R.; Sa-Ngiamwibool, P.; Watcharadetwittaya, S.; Thanee, M.; Intuyod, K.; Kongpan, T.; Padthaisong, S.; Nutalai, R.; Chamgramol, Y.; Pairojkul, C. Fluorescence in situ hybridization detection of chromosome 7 and/or 17 polysomy as a prognostic marker for cholangiocarcinoma. Sci. Rep. 2022, 12, 8441. [Google Scholar] [CrossRef]
- Yang, X.; Wang, W.; Wang, C.; Wang, L.; Yang, M.; Qi, M.; Su, H.; Sun, X.; Liu, Z.; Zhang, J.; et al. Characterization of EGFR family gene aberrations in cholangiocarcinoma. Oncol. Rep. 2014, 32, 700–708. [Google Scholar] [CrossRef] [PubMed]
- Lavingia, V.; Fakih, M. Impressive response to dual BRAF and MEK inhibition in patients with BRAF mutant intrahepatic cholangiocarcinoma-2 case reports and a brief review. J. Gastrointest. Oncol. 2016, 7, E98–E102. [Google Scholar] [CrossRef]
- Miyamoto, M.; Ojima, H.; Iwasaki, M.; Shimizu, H.; Kokubu, A.; Hiraoka, N.; Kosuge, T.; Yoshikawa, D.; Kono, T.; Furukawa, H.; et al. Prognostic significance of overexpression of c-Met oncoprotein in cholangiocarcinoma. Br. J. Cancer 2011, 105, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Li, B.; Duan, C.; Cheng, Y.; Xiao, B.; Yao, F.; Wei, M.; Tao, Q.; Feng, C.; Xia, X.; et al. c-Myc promotes cholangiocarcinoma cells to overcome contact inhibition via the mTOR pathway. Oncol. Rep. 2017, 38, 2498–2506. [Google Scholar] [CrossRef]
- Li, Z.R.; Wu, Y.F.; Ma, C.Y.; Nie, S.D.; Mao, X.H.; Shi, Y.Z. Down-regulation of c-Myc expression inhibits the invasion of bile duct carcinoma cells. Cell Biol. Int. 2011, 35, 799–802. [Google Scholar] [CrossRef]
- O’Rourke, C.J.; Lafuente-Barquero, J.; Andersen, J.B. Epigenome Remodeling in Cholangiocarcinoma. Trends Cancer 2019, 5, 335–350. [Google Scholar] [CrossRef]
- O’Rourke, C.J.; Munoz-Garrido, P.; Aguayo, E.L.; Andersen, J.B. Epigenome dysregulation in cholangiocarcinoma. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1423–1434. [Google Scholar] [CrossRef] [PubMed]
- Merino-Azpitarte, M.; Lozano, E.; Perugorria, M.J.; Esparza-Baquer, A.; Erice, O.; Santos-Laso, A.; O’Rourke, C.J.; Andersen, J.B.; Jimenez-Aguero, R.; Lacasta, A.; et al. SOX17 regulates cholangiocyte differentiation and acts as a tumor suppressor in cholangiocarcinoma. J. Hepatol. 2017, 67, 72–83. [Google Scholar] [CrossRef]
- Jusakul, A.; Cutcutache, I.; Yong, C.H.; Lim, J.Q.; Huang, M.N.; Padmanabhan, N.; Nellore, V.; Kongpetch, S.; Ng, A.W.T.; Ng, L.M.; et al. Whole-Genome and Epigenomic Landscapes of Etiologically Distinct Subtypes of Cholangiocarcinoma. Cancer Discov. 2017, 7, 1116–1135. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203.e113. [Google Scholar] [CrossRef]
- Endo, Y.; Fujimoto, M.; Ito, N.; Takahashi, Y.; Kitago, M.; Gotoh, M.; Hiraoka, N.; Yoshida, T.; Kitagawa, Y.; Kanai, Y.; et al. Clinicopathological impacts of DNA methylation alterations on pancreatic ductal adenocarcinoma: Prediction of early recurrence based on genome-wide DNA methylation profiling. J. Cancer Res. Clin. Oncol. 2021, 147, 1341–1354. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Goeppert, B.; Stichel, D.; Toth, R.; Fritzsche, S.; Loeffler, M.A.; Schlitter, A.M.; Neumann, O.; Assenov, Y.; Vogel, M.N.; Mehrabi, A.; et al. Integrative analysis reveals early and distinct genetic and epigenetic changes in intraductal papillary and tubulopapillary cholangiocarcinogenesis. Gut 2022, 71, 391–401. [Google Scholar] [CrossRef]
- Leader, Y.; Lev Maor, G.; Sorek, M.; Shayevitch, R.; Hussein, M.; Hameiri, O.; Tammer, L.; Zonszain, J.; Keydar, I.; Hollander, D.; et al. The upstream 5′ splice site remains associated to the transcription machinery during intron synthesis. Nat. Commun. 2021, 12, 4545. [Google Scholar] [CrossRef]
- Zhao, Z.; Shilatifard, A. Epigenetic modifications of histones in cancer. Genome Biol. 2019, 20, 245. [Google Scholar] [CrossRef] [PubMed]
- Iannone, C.; Pohl, A.; Papasaikas, P.; Soronellas, D.; Vicent, G.P.; Beato, M.; ValcaRcel, J. Relationship between nucleosome positioning and progesterone-induced alternative splicing in breast cancer cells. RNA 2015, 21, 360–374. [Google Scholar] [CrossRef]
- Kremer, A.; Holdenrieder, S.; Stieber, P.; Wilkowski, R.; Nagel, D.; Seidel, D. Nucleosomes in colorectal cancer patients during radiochemotherapy. Tumour Biol. 2006, 27, 235–242. [Google Scholar] [CrossRef]
- Andersen, J.B.; Spee, B.; Blechacz, B.R.; Avital, I.; Komuta, M.; Barbour, A.; Conner, E.A.; Gillen, M.C.; Roskams, T.; Roberts, L.R.; et al. Genomic and genetic characterization of cholangiocarcinoma identifies therapeutic targets for tyrosine kinase inhibitors. Gastroenterology 2012, 142, 1021–1031.e1015. [Google Scholar] [CrossRef]
- Oishi, N.; Kumar, M.R.; Roessler, S.; Ji, J.; Forgues, M.; Budhu, A.; Zhao, X.; Andersen, J.B.; Ye, Q.H.; Jia, H.L.; et al. Transcriptomic profiling reveals hepatic stem-like gene signatures and interplay of miR-200c and epithelial-mesenchymal transition in intrahepatic cholangiocarcinoma. Hepatology 2012, 56, 1792–1803. [Google Scholar] [CrossRef]
- Montal, R.; Sia, D.; Montironi, C.; Leow, W.Q.; Esteban-Fabro, R.; Pinyol, R.; Torres-Martin, M.; Bassaganyas, L.; Moeini, A.; Peix, J.; et al. Molecular classification and therapeutic targets in extrahepatic cholangiocarcinoma. J. Hepatol. 2020, 73, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Kubo, S.; Tamori, A.; Itami, S.; Kawamura, E.; Iwaisako, K.; Ikeda, K.; Kawada, N.; Ochiya, T.; Taguchi, Y.H. Comprehensive analysis of transcriptome and metabolome analysis in Intrahepatic Cholangiocarcinoma and Hepatocellular Carcinoma. Sci. Rep. 2015, 5, 16294. [Google Scholar] [CrossRef]
- Ahn, K.S.; Kang, K.J.; Kim, Y.H.; Kim, T.S.; Song, B.I.; Kim, H.W.; O’Brien, D.; Roberts, L.R.; Lee, J.W.; Won, K.S. Genetic features associated with (18)F-FDG uptake in intrahepatic cholangiocarcinoma. Ann. Surg. Treat. Res. 2019, 96, 153–161. [Google Scholar] [CrossRef]
- Gao, C.; Zhou, G.; Shi, J.; Shi, P.; Jin, L.; Li, Y.; Wang, X.; Liao, S.; Yan, H.; Wu, J.; et al. The A-to-I editing of KPC1 promotes intrahepatic cholangiocarcinoma by attenuating proteasomal processing of NF-kappaB1 p105 to p50. J. Exp. Clin. Cancer Res. 2022, 41, 338. [Google Scholar] [CrossRef]
- Kang, Z.; Guo, L.; Zhu, Z.; Qu, R. Identification of prognostic factors for intrahepatic cholangiocarcinoma using long non-coding RNAs-associated ceRNA network. Cancer Cell Int. 2020, 20, 315. [Google Scholar] [CrossRef]
- Chaisaingmongkol, J.; Budhu, A.; Dang, H.; Rabibhadana, S.; Pupacdi, B.; Kwon, S.M.; Forgues, M.; Pomyen, Y.; Bhudhisawasdi, V.; Lertprasertsuke, N.; et al. Common Molecular Subtypes Among Asian Hepatocellular Carcinoma and Cholangiocarcinoma. Cancer Cell 2017, 32, 57–70.e53. [Google Scholar] [CrossRef] [PubMed]
- Perez-Mancera, P.A.; Rust, A.G.; van der Weyden, L.; Kristiansen, G.; Li, A.; Sarver, A.L.; Silverstein, K.A.; Grutzmann, R.; Aust, D.; Rummele, P.; et al. The deubiquitinase USP9X suppresses pancreatic ductal adenocarcinoma. Nature 2012, 486, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Wang, H.; Li, Z.; Li, F.; Liang, L.; Zou, Y.; Shen, H.; Li, J.; Xia, Y.; Cheng, Z.; et al. Circular RNA ACTN4 promotes intrahepatic cholangiocarcinoma progression by recruiting YBX1 to initiate FZD7 transcription. J. Hepatol. 2022, 76, 135–147. [Google Scholar] [CrossRef]
- Zhao, Z.; Meng, J.; Su, R.; Zhang, J.; Chen, J.; Ma, X.; Xia, Q. Epitranscriptomics in liver disease: Basic concepts and therapeutic potential. J. Hepatol. 2020, 73, 664–679. [Google Scholar] [CrossRef]
- Dai, X.; Shen, L. Advances and Trends in Omics Technology Development. Front. Med. 2022, 9, 911861. [Google Scholar] [CrossRef]
- Frohlich, K.; Brombacher, E.; Fahrner, M.; Vogele, D.; Kook, L.; Pinter, N.; Bronsert, P.; Timme-Bronsert, S.; Schmidt, A.; Barenfaller, K.; et al. Benchmarking of analysis strategies for data-independent acquisition proteomics using a large-scale dataset comprising inter-patient heterogeneity. Nat. Commun. 2022, 13, 2622. [Google Scholar] [CrossRef] [PubMed]
- Seeree, P.; Pearngam, P.; Kumkate, S.; Janvilisri, T. An Omics Perspective on Molecular Biomarkers for Diagnosis, Prognosis, and Therapeutics of Cholangiocarcinoma. Int. J. Genomics 2015, 2015, 179528. [Google Scholar] [CrossRef]
- Cao, X.L.; Li, H.; Yu, X.L.; Liang, P.; Dong, B.W.; Fan, J.; Li, M.; Liu, F.Y. Predicting early intrahepatic recurrence of hepatocellular carcinoma after microwave ablation using SELDI-TOF proteomic signature. PLoS ONE 2013, 8, e82448. [Google Scholar] [CrossRef]
- Sirica, A.E.; Gores, G.J. Desmoplastic stroma and cholangiocarcinoma: Clinical implications and therapeutic targeting. Hepatology 2014, 59, 2397–2402. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.H.; Yang, J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes. Dev. 2013, 27, 2192–2206. [Google Scholar] [CrossRef]
- Vaquero, J.; Guedj, N.; Claperon, A.; Nguyen Ho-Bouldoires, T.H.; Paradis, V.; Fouassier, L. Epithelial-mesenchymal transition in cholangiocarcinoma: From clinical evidence to regulatory networks. J. Hepatol. 2017, 66, 424–441. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.D.; Hainaut, P.; Gores, G.J.; Amadou, A.; Plymoth, A.; Roberts, L.R. A global view of hepatocellular carcinoma: Trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 589–604. [Google Scholar] [CrossRef]
- Lee, Y.T.; Wang, J.J.; Luu, M.; Noureddin, M.; Nissen, N.N.; Patel, T.C.; Roberts, L.R.; Singal, A.G.; Gores, G.J.; Yang, J.D. Comparison of Clinical Features and Outcomes between Intrahepatic Cholangiocarcinoma and Hepatocellular Carcinoma in the United States. Hepatology 2021, 74, 2622–2632. [Google Scholar] [CrossRef]
- Thelen, A.; Scholz, A.; Weichert, W.; Wiedenmann, B.; Neuhaus, P.; Gessner, R.; Benckert, C.; Jonas, S. Tumor-associated angiogenesis and lymphangiogenesis correlate with progression of intrahepatic cholangiocarcinoma. Am. J. Gastroenterol. 2010, 105, 1123–1132. [Google Scholar] [CrossRef]
- Petrov, V.A.; Fernandez-Peralbo, M.A.; Derks, R.; Knyazeva, E.M.; Merzlikin, N.V.; Sazonov, A.E.; Mayboroda, O.A.; Saltykova, I.V. Biliary Microbiota and Bile Acid Composition in Cholelithiasis. Biomed. Res. Int. 2020, 2020, 1242364. [Google Scholar] [CrossRef]
- Pant, K.; Richard, S.; Peixoto, E.; Gradilone, S.A. Role of Glucose Metabolism Reprogramming in the Pathogenesis of Cholangiocarcinoma. Front. Med. 2020, 7, 113. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, J.; Lei, Z.; Wu, D.; Si, A.; Wang, K.; Wang, Y.; Wan, X.; Lau, W.Y.; Shen, F. Prognosis of Intrahepatic Cholangiocarcinomas with HBV Infection is Better than Those with Hepatolithiasis after R0 Liver Resection: A Propensity Score Matching Analysis. Ann. Surg. Oncol. 2017, 24, 1579–1587. [Google Scholar] [CrossRef] [PubMed]
- Liau, J.Y.; Tsai, J.H.; Yuan, R.H.; Chang, C.N.; Lee, H.J.; Jeng, Y.M. Morphological subclassification of intrahepatic cholangiocarcinoma: Etiological, clinicopathological, and molecular features. Mod. Pathol. 2014, 27, 1163–1173. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Li, Q.; Chen, J.; Chen, D.; Ye, C.; Dai, X.; Wang, Y.; Li, X.; Rong, X.; Cheng, F.; et al. Molecular Subgroups of Intrahepatic Cholangiocarcinoma Discovered by Single-Cell RNA Sequencing-Assisted Multiomics Analysis. Cancer Immunol. Res. 2022, 10, 811–828. [Google Scholar] [CrossRef]
- Ding, G.Y.; Ma, J.Q.; Yun, J.P.; Chen, X.; Ling, Y.; Zhang, S.; Shi, J.Y.; Chang, Y.Q.; Ji, Y.; Wang, X.Y.; et al. Distribution and density of tertiary lymphoid structures predict clinical outcome in intrahepatic cholangiocarcinoma. J. Hepatol. 2022, 76, 608–618. [Google Scholar] [CrossRef]
- Lin, J.; Dai, Y.; Sang, C.; Song, G.; Xiang, B.; Zhang, M.; Dong, L.; Xia, X.; Ma, J.; Shen, X.; et al. Multimodule characterization of immune subgroups in intrahepatic cholangiocarcinoma reveals distinct therapeutic vulnerabilities. J. Immunother. Cancer 2022, 10, e004892. [Google Scholar] [CrossRef]
- Lin, Y.; Peng, L.; Dong, L.; Liu, D.; Ma, J.; Lin, J.; Chen, X.; Lin, P.; Song, G.; Zhang, M.; et al. Geospatial Immune Heterogeneity Reflects the Diverse Tumor-Immune Interactions in Intrahepatic Cholangiocarcinoma. Cancer Discov. 2022, 12, 2350–2371. [Google Scholar] [CrossRef]
- Zhu, C.; Ma, J.; Zhu, K.; Yu, L.; Zheng, B.; Rao, D.; Zhang, S.; Dong, L.; Gao, Q.; Zhang, X.; et al. Spatial immunophenotypes predict clinical outcome in intrahepatic cholangiocarcinoma. JHEP Rep. 2023, 5, 100762. [Google Scholar] [CrossRef]
- Martin-Serrano, M.A.; Kepecs, B.; Torres-Martin, M.; Bramel, E.R.; Haber, P.K.; Merritt, E.; Rialdi, A.; Param, N.J.; Maeda, M.; Lindblad, K.E.; et al. Novel microenvironment-based classification of intrahepatic cholangiocarcinoma with therapeutic implications. Gut 2023, 72, 736–748. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Xie, Y.; Cai, Y.; Hu, H.; He, M.; Liu, L.; Liao, C.; Wang, Y.; Wang, J.; Ren, X.; et al. Multiomic Analysis Reveals Comprehensive Tumor Heterogeneity and Distinct Immune Subtypes in Multifocal Intrahepatic Cholangiocarcinoma. Clin. Cancer Res. 2022, 28, 1896–1910. [Google Scholar] [CrossRef]
- Dong, L.; Lu, D.; Chen, R.; Lin, Y.; Zhu, H.; Zhang, Z.; Cai, S.; Cui, P.; Song, G.; Rao, D.; et al. Proteogenomic characterization identifies clinically relevant subgroups of intrahepatic cholangiocarcinoma. Cancer Cell 2022, 40, 70–87.e15. [Google Scholar] [CrossRef]
- Cho, S.Y.; Hwang, H.; Kim, Y.H.; Yoo, B.C.; Han, N.; Kong, S.Y.; Baek, M.J.; Kim, K.H.; Lee, M.R.; Park, J.G.; et al. Refining Classification of Cholangiocarcinoma Subtypes via Proteogenomic Integration Reveals New Therapeutic Prospects. Gastroenterology 2023, 164, 1293–1309. [Google Scholar] [CrossRef]
- Liu, W.; Wang, H.; Zhao, Q.; Tao, C.; Qu, W.; Hou, Y.; Huang, R.; Sun, Z.; Zhu, G.; Jiang, X.; et al. Multiomics analysis reveals metabolic subtypes and identifies diacylglycerol kinase α (DGKA) as a potential therapeutic target for intrahepatic cholangiocarcinoma. Cancer Commun. 2023, 44, 226–250. [Google Scholar] [CrossRef]
- Calderaro, J.; Petitprez, F.; Becht, E.; Laurent, A.; Hirsch, T.Z.; Rousseau, B.; Luciani, A.; Amaddeo, G.; Derman, J.; Charpy, C.; et al. Intra-tumoral tertiary lymphoid structures are associated with a low risk of early recurrence of hepatocellular carcinoma. J. Hepatol. 2019, 70, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Sautes-Fridman, C.; Petitprez, F.; Calderaro, J.; Fridman, W.H. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat. Rev. Cancer 2019, 19, 307–325. [Google Scholar] [CrossRef] [PubMed]
- Kurebayashi, Y.; Ojima, H.; Tsujikawa, H.; Kubota, N.; Maehara, J.; Abe, Y.; Kitago, M.; Shinoda, M.; Kitagawa, Y.; Sakamoto, M. Landscape of immune microenvironment in hepatocellular carcinoma and its additional impact on histological and molecular classification. Hepatology 2018, 68, 1025–1041. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Sprengers, D.; Mancham, S.; Erkens, R.; Boor, P.P.C.; van Beek, A.A.; Doukas, M.; Noordam, L.; Campos Carrascosa, L.; de Ruiter, V.; et al. Reduction of immunosuppressive tumor microenvironment in cholangiocarcinoma by ex vivo targeting immune checkpoint molecules. J. Hepatol. 2019, 71, 753–762. [Google Scholar] [CrossRef]
- Kumar, M.; Zhao, X.; Wang, X.W. Molecular carcinogenesis of hepatocellular carcinoma and intrahepatic cholangiocarcinoma: One step closer to personalized medicine? Cell Biosci. 2011, 1, 5. [Google Scholar] [CrossRef]
- Hasita, H.; Komohara, Y.; Okabe, H.; Masuda, T.; Ohnishi, K.; Lei, X.F.; Beppu, T.; Baba, H.; Takeya, M. Significance of alternatively activated macrophages in patients with intrahepatic cholangiocarcinoma. Cancer Sci. 2010, 101, 1913–1919. [Google Scholar] [CrossRef]
- Sia, D.; Jiao, Y.; Martinez-Quetglas, I.; Kuchuk, O.; Villacorta-Martin, C.; Castro de Moura, M.; Putra, J.; Camprecios, G.; Bassaganyas, L.; Akers, N.; et al. Identification of an Immune-specific Class of Hepatocellular Carcinoma, Based on Molecular Features. Gastroenterology 2017, 153, 812–826. [Google Scholar] [CrossRef]
- Kang, N.; Gores, G.J.; Shah, V.H. Hepatic stellate cells: Partners in crime for liver metastases? Hepatology 2011, 54, 707–713. [Google Scholar] [CrossRef]
- Helmink, B.A.; Reddy, S.M.; Gao, J.; Zhang, S.; Basar, R.; Thakur, R.; Yizhak, K.; Sade-Feldman, M.; Blando, J.; Han, G.; et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 2020, 577, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Milardi, G.; Franceschini, B.; Camisaschi, C.; Costa, G.; Soldani, C.; Uva, P.; Cangelosi, D.; Carriero, R.; Lodetti-Zangrandi, G.; Malenica, I.; et al. Immunosuppressive contribution of tumor-infiltrating B cell subsets in human intrahepatic cholangiocarcinoma and their role in immunotherapy response. Dig. Liver Dis. 2024, 56, S94–S95. [Google Scholar] [CrossRef]
- Amendola, C.R.; Mahaffey, J.P.; Parker, S.J.; Ahearn, I.M.; Chen, W.C.; Zhou, M.; Court, H.; Shi, J.; Mendoza, S.L.; Morten, M.J.; et al. KRAS4A directly regulates hexokinase 1. Nature 2019, 576, 482–486. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Zeng, Y. Artificial intelligence and multi-omics driven models would be the future of intrahepatic cholangiocarcinoma prediction research. Hepatobiliary Surg. Nutr. 2024, 13, 560–561. [Google Scholar] [CrossRef]
- Sha, M.; Jeong, S.; Qiu, B.J.; Tong, Y.; Xia, L.; Xu, N.; Zhang, J.J.; Xia, Q. Isolation of cancer-associated fibroblasts and its promotion to the progression of intrahepatic cholangiocarcinoma. Cancer Med. 2018, 7, 4665–4677. [Google Scholar] [CrossRef]
- Kim, J.; Han, D.H.; Choi, G.H.; Kim, K.S.; Choi, J.S.; Kim, S.H. The prognostic value of the number of metastatic lymph nodes on the long-term survival of intrahepatic cholangiocarcinoma using the SEER database. J. Gastrointest. Oncol. 2023, 14, 2511–2520. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Single-Omics | Sample Type | Methods | Results | Refs. |
|---|---|---|---|---|
| Genomics | Ex vivo | RNA-seq; FISH; WES; Gene-array | Identification of 13 novel inter- and intrachromosomal fusion events; Top ranked fusion event: fusion of FGFR2 and PPHLN1 | [89] |
| Ex vivo | NGS | 5547 genomic alterations in 335 genes: 3424 short variants, 1676 copy number variations | [43] | |
| Ex vivo | WES | Detection of fusion transcripts FGFR2-AHCYL1, FGFR2- BICC1, AHCYL1-FGFR2, and BICC1-FGFR2 | [90] | |
| Ex vivo | NGS | Most commonly mutated genes: KRAS (28.1%), TP53 (18.3%), ARID1A (11.8%), IDH1/IDH2 (9.2%), PBRM1 (9.2%), BAP1 (7.2%), and PIK3CA (7.2%) | [91] | |
| Ex vivo | NGS | 1259 somatic mutations in 1128 genes | [92] | |
| Ex vivo | Gene-array | 775 genetic alterations and 38 structural alterations | [42] | |
| Ex vivo | NGS, WES, and WGS | Most recurrently mutated genes: TP53, KRAS, IDH1/2, ARID1A, BAP1, PBRM1, FGFR2 fusion, CDKN2A, SMAD4, PIK3CA, EPHA2 (mutation frequency > 5%) | [93] | |
| Ex vivo | NGS | Most commonly mutated genes: TP53, ARID1A, KRAS, CDKN2A, SMAD4, and PBRM1 | [94] | |
| Ex vivo | SNP-array | Copy number gain: chromosome 1q (32%) and 7p (25%); Copy number loss: 6q (52%), 9q (45%), 9p (42%), 3p (41%), 13q (38%), 14q (36%), 8p (30%), 21q (20%), 17p (21%), 4q (18%), 1p (16%), 18q (15%), and 11p (13%) | [95] | |
| Ex vivo | Exome-array | Exclusively deleted cytobands: 1p36.33–1p35.1, 3p26.3–3p14.25, and 14q24.1–14q32.33; Exclusively amplified cytobands: 1p11.2–1p41.1, 1q21.1–1q44, 2q23.1–2q35, 7p22.3–7p11.1, 7q11.1–7q36.3, and 8q23.2–8q24.3 | [96] | |
| Epigenomics | Ex vivo | Methylation-array | Methylation data used to classify tumors; general hypermethylation in iCCA samples | [97] |
| Ex vivo | Methylation-array | cg15026696 hypomethylation and cg06972969 hypermethylation | [98] | |
| In vitro | ChIP-sequencing | Reduced levels of H3K4me1, H3K4me3, and H3K27ac in tumor cells compared to normal cells | [99] | |
| Ex vivo | Methylation-array | 2048 most variable CpG sites used to classify tumors | [100] | |
| Ex vivo | WGBS | Identification of prognostically methylated regions (PMRs) as biomarkers | [101] | |
| Ex vivo | WGBS | DNA methylation data used to classify tumors | [102] | |
| Transcriptomics | Ex vivo | Transcriptome-array | Gene expression profiles used to classify iCCA tumors | [103] |
| Ex vivo | RNA-sequencing | Gene expression profiles used to classify iCCA tumors | [104] | |
| Ex vivo | RNA-sequencing and Transcriptome-array | C19orf33, S100P, CEACAM6, KRT17, GPRC5A, MSLN, SERPINB5, TCN1, OLFM4, VTN, ADH1C, ALDOB, FABP1, ADH1B, UGT2B7, APOC4, AQP9, CDO1, GSTA2 as biomarkers of specific iCCA subgroups | [105] | |
| Ex vivo | mRNA and microRNA-array | 2327 DE mRNAs (1113 up- and 1214 downregulated) and 70 DE miRNAs (65 up- and 5 downregulated) in iCCA vs. paired non-cancerous liver tissue samples | [106] | |
| Ex vivo | RNA-seq | 230 DE lncRNAs (97 up- and 133 downregulated) and 2220 DE mRNAs (640 down- and 1580 upregulated) in iCCA vs. paired unaffected tissues; top downregulated genes: RP11-328K4.1, LINC01093, LINC00844, RP11-372E1.4, ITIH4-AS1; top upregulated genes: RP11-532F12.5, AC016735.1, RP11-284F21.7, LINC01123, AC013275.2 | [107] | |
| Ex vivo | GEP; Transcriptome-array; NGS | 315 DE genes (65 down- and 250 upregulated) in recurrent compared with primary tumors | [108] | |
| Ex vivo | miRNA-seq | miR-10b, miR-22, and miR-551b significantly associated with OS in iCCA patients; Overexpression of miR-551b decreased HuCCT-1 proliferation and promoted apoptosis | [109] | |
| Ex vivo | RNA-sequencing | 1643 DE transcripts (1098 up- and 545 downregulated) | [110] | |
| Ex vivo | microRNA-array, and RNA-sequencing | Pathway enrichment analysis performed on DE lncRNAs and mRNAs revealed their involvement in the Hippo pathway | [111] | |
| Ex vivo | Transcriptome-array | Gene expression profiles used to classify iCCA tumors; identification of ICCA-specific DE genes (395 up- and 5889 downregulated); Upregulated: ARHGAP21; Downregulated: SCP2, UBIAD1, TJP2, RAP1A, HDAC9, FKBP2, MRPL2, and MRPL27 | [112] | |
| Ex vivo | Transcriptome-array | Classification of iCCA tumors according to 794 DE transcripts (486 up- and 308 downregulated) specific for cholangiolocellular differentiation (CD); specific of CD-tumors: CRP; specific of non-CD-tumors: S100P, TFF1, AGR2, CLDN18, KRT17, and CTSE | [113] | |
| Ex vivo | single-cell RNA-seq | Gene expression profiles used to classify cells | [114] | |
| Ex vivo | RNA-sequencing | Gene expression profiles used to classify iCCA tumors | [115] | |
| Ex vivo | Transcriptome-array | 2611 (1142 up- and 1469 downregulated) | [116] | |
| Ex vivo | STS; WES; WGS; NGS; Methylation-array | Gene expression profiles used to classify iCCA tumors; 52 genes showed differential expression between Cluster1A and Cluster2 | [93] | |
| Ex vivo | circRNA-sequencing | 4 circRNAs upregulated in metastatic vs. non-metastatic tumors (circZNF215, circBNIP3L, circCD109, and circPLOD2) | [117] | |
| Ex vivo | Transcriptome-array | 173 DE circRNAs (69 up- and 104 downregulated), 58 DE miRNAs (30 up- and 28 downregulated), and 4437 DE mRNAs (2234 up- and 2203 downregulated) | [118] | |
| Ex vivo | RNA-sequencing | 262 dysregulated miRNAs (128 up- and 134 downregulated); top upregulated miRNAs: miR-21, miR-34c, miR-200b, and miR-221 | [119] | |
| Epitransctriptomics | Ex vivo | Methylated RNA immunoprecipitation sequencing | METTL14 p.R298H mutation affected m6A modifications on the target MACF1 | [120] |
| Proteomics | Ex vivo | Nano-LC-MALDI-TOF/TOF | Overexpression of COL3A1 and low levels of reticular and elastic fibers; Basement membrane dismantling; Reduced angiogenesis; COL3A1 promotes iCCA cell migration | [121] |
| Ex vivo | AMT Tag | Altered protein network involved in cytoskeleton organization and cell motility (increase in vimentin, cofilin-1, profilin-1, and transgelin-2, S100A11, reduction in annexins) | [122] | |
| Ex vivo | TMT | Decreased cell–cell junction level in HBV-iCCA; Inverse relationship between cell–cell junction level and EMT program: reduced abundance of epithelial cells is correlated with low expression levels of E-CAD in HBV-iCCA | [123] | |
| Ex vivo | DIA-MS | Enrichment of proteins related to lipid metabolism (i.e., AMACR, SREBP, FASN) in HCC tissues; High levels of proteins involved in ECM-related pathways (S100A6, laminin interactions, assembly of collagen fibrils, PTK2 signaling) in iCCA tissues; S100A6 potential diagnosis biomarker to discriminate iCCA from HCC. | [124] | |
| Ex vivo | LC-MS/MS | Overexpression of angio-inhibitory proteins in iCCA-ECF; Upregulation of THBS1, THBS2, and PEDF inhibits angiogenesis on behalf of lymphangiogenesis promoting the trans-differentiation of vascular endothelial cells toward a lymphatic phenotype | [40] | |
| Metabolomics | Ex vivo | iTRAQ labeling and LC-MS/MS | Downregulation of enzymes (TPI1, GAPDH, and PGK1) involved in glycolysis and gluconeogenesis pathways in LDT compared to SDT | [125] |
| Ex vivo | MS | High mitochondrial activity, with an enrichment of glutamine and glucose uptake; upregulation of glucose transporter SLC2A3; positive correlation between SLC2A3 and EMT markers (i.e., N-CAD) | [126] | |
| Ex vivo | MALDI-MS and MALDI-MS/MS | Upregulation of enzymes (i.e., CAT, PRDX2/6, and SODM) involved in redox homeostasis causing accumulation of free radicals in iCCA patients | [127] | |
| Ex vivo | GC–MS | Overactivation of linoleic acid metabolism pathway in IBDS rather than in iCCA tissues; 9,12-octadecadienoic acid potential biomarker for long-term monitoring, and treatment of IBDS to prevent the risk of iCCA development | [128] | |
| Ex vivo | UPLC-MS/MS | 4 urinary metabolites (CR, NANA, CS, and 561+) are upregulated in iCCA compared to HCC patients; Potential model of iCCA diagnosis based on the combination of the 4 urinary metabolites and CA19-9 | [129] |
| Multi-Omics | Sample Type | No of iCCA Patients | Methods | Results | Refs. |
|---|---|---|---|---|---|
| Genomics + Epigenomics | Ex vivo and In vitro | 71 | WGS; MiGS | 4 clusters based on strict correlation between phenotype and etiological agents | [189] |
| Genomics + Epigenomics + Transcriptomics | Ex vivo | 38 | NGS; WES; RNA-seq; miRNA-seq; SNP-array | 4 clusters according to genomics, epigenomics, and transcriptomics profile | [88] |
| Genomics + Epigenomics | Ex vivo | 52 | WGS; WES | 4 subgroups (IDH, L, M, H) based on genetic and epigenetic features | [97] |
| Genomics + Epigenomics integrated with WES, RNA-seq, and proteome datasets | Ex vivo and Invitro | 331 | WGBS; TMA; WES | 4 clusters (S1–S4) according to hyper-differentially methylated regions (DMRs) | [102] |
| Genomics + Epigenomics + Transcriptomics | Ex vivo | 149 | bs-DNA; Methylation-array; RNA-seq | 2 iCCA classes (inflammation and proliferation) based on genomic and mutational profiles | [95] |
| Epigenomics + Transcriptomics | Ex vivo | 116 | Methylation-Array; Transcriptome-Array | 4 iCCA subtypes are identified according to different escape mechanisms in TME (immune desert, immunogenic, myeloid, and mesenchymal | [103] |
| Proteomics + Genomics + Epigenomics + Transcriptomics | Ex vivo | 110 | TMT; NGS; WES; Single-cell RNA-seq | 3 molecular iCCA subtypes (chromatin remodeling, metabolism, and chronic inflammation) correlated to OS | [223] |
| Proteomics + Epigenomics | Ex vivo | 962 | mIHC; WES | 4 iCCA classes based on the spatial distribution, abundance, and functional orientation of TLSs (immune excluded, altered, active) | [224] |
| Genomics + Transcriptomics + Proteomics | Ex vivo | 255 | WES; Single-cell RNA-seq; MS; mIHC + TMA; | 3 iCCA subgroups (IG1-3) established by immune landscape | [225] |
| Genomics + Transcriptomics + Proteomics | Ex vivo | 45 | WGS, RNA-seq; mIHC | 3 iCCA subgroups (sparsely, heterogeneously, and highly infiltrated) correlated to immunogenomics profiles | [226] |
| Genomics + Transcriptomics + Proteomics | Ex vivo | 192 | mIHC; Molecular Signatures Database ((MSigDB) | 3 iCCA spatial immunophenotypes (inflamed, excluded, and ignored) based on immune infiltration | [227] |
| Genomics + Transcriptomics | Ex vivo | 961 | NMF; Transcriptomic tools (i.e., EnrichR); Molecular Signatures Database (MSigDB) | 5 iCCA classes (STIM classification) related to the stroma, tumor, and immune microenvironment | [228] |
| Genomics + Epigenomics + Transcriptomics + Proteomics | Ex vivo | 16 | WES; RNA-seq; Methylation-Array; mIHC; scRNA-seq; Single-cell RNA-seq | 2 iCCA groups according to ITH index (-high and -low) | [229] |
| Genomics + Transcriptomics + Proteomics | Ex vivo | 262 | RNA-seq; TCR-seq; FISH; Fusion-derived peptides; peptide-array | 4 iCCA subgroups (S1–S4) with specific features in terms of genetic alterations, microenvironment dysregulation, tumor microbiota composition, and OS | [230] |
| Genomics + Transcriptomics + Proteomics | Ex vivo | 102 | WES; RNA-seq; LC-MS/MS | 3 iCCA clinical subtypes (stem-like, poorly immunogenic, and metabolism) | [231] |
| Genomics + Transcriptomics + Proteomics | Ex vivo | 116 | WES; RNA-seq; MS | 3 iCCA subgroups (S1–S3) according to metabolomics profile | [232] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Porreca, V.; Barbagallo, C.; Corbella, E.; Peres, M.; Stella, M.; Mignogna, G.; Maras, B.; Ragusa, M.; Mancone, C. Unveil Intrahepatic Cholangiocarcinoma Heterogeneity through the Lens of Omics and Multi-Omics Approaches. Cancers 2024, 16, 2889. https://doi.org/10.3390/cancers16162889
Porreca V, Barbagallo C, Corbella E, Peres M, Stella M, Mignogna G, Maras B, Ragusa M, Mancone C. Unveil Intrahepatic Cholangiocarcinoma Heterogeneity through the Lens of Omics and Multi-Omics Approaches. Cancers. 2024; 16(16):2889. https://doi.org/10.3390/cancers16162889
Chicago/Turabian StylePorreca, Veronica, Cristina Barbagallo, Eleonora Corbella, Marco Peres, Michele Stella, Giuseppina Mignogna, Bruno Maras, Marco Ragusa, and Carmine Mancone. 2024. "Unveil Intrahepatic Cholangiocarcinoma Heterogeneity through the Lens of Omics and Multi-Omics Approaches" Cancers 16, no. 16: 2889. https://doi.org/10.3390/cancers16162889
APA StylePorreca, V., Barbagallo, C., Corbella, E., Peres, M., Stella, M., Mignogna, G., Maras, B., Ragusa, M., & Mancone, C. (2024). Unveil Intrahepatic Cholangiocarcinoma Heterogeneity through the Lens of Omics and Multi-Omics Approaches. Cancers, 16(16), 2889. https://doi.org/10.3390/cancers16162889