
Temozolomide and the PARP Inhibitor Niraparib Enhance Expression of Natural Killer Group 2D Ligand ULBP1 and Gamma-Delta T Cell Cytotoxicity in Glioblastoma
 , , , , , , , and
, , , , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Cell Culture
2.3. Relative Viability
2.4. Transcriptional Profiling
| Human NKG2DL | Forward Primer Sequence | Reverse Primer Sequence |
| MICA | CCA CCA CGA TTT GCC AAG GAG A | CTG CCA ATG ACT CTG AAG CAC C |
| MICB | GGA ATG GAA CCT ACC AGA CCT G | CTG TCC GTT GAC TCT GAA GCA C |
| ULBP1 | CTG CTT GAC ATT CAA GTG GAG AAT | CTG TCC ATT GAA GAG GAA CTG CC |
| ULBP2 | GAG CAA CTG CGT GAC ATT CAG C | GCC CAT CGA AAC TGA ACT GCC A |
| ULBP3 | CAG ACT GGA ACT GGC TGA CACT | GGA GGA ACT TCC GTC CAT CGA A |
| ULBP4 | GGT ATA TGC CAC CAG CAC TTG G | TCT CGA CTT GCA GAG TGG AAG G |
| ULBP5 | CTG CTG AGA AGA GTC CTT TGG AG | GGT AAG GAG TGT GAG TCG TCT C |
| ULBP6 | AGA GCA ACT GCT TGA CAT TCA GC | GAG TAG GAA GGT CTG TCC ATC G |
| Murine NKG2DL | Forward Primer Sequence | Reverse Primer Sequence |
| Mult-1 | GTG CAG GAG ACT AAC ACA ACC G | TGC CAG TGC TTG TGT CAA CAC G |
2.5. Flow Cytometry
2.6. Cytotoxicity
2.7. Statistical Analysis
3. Results
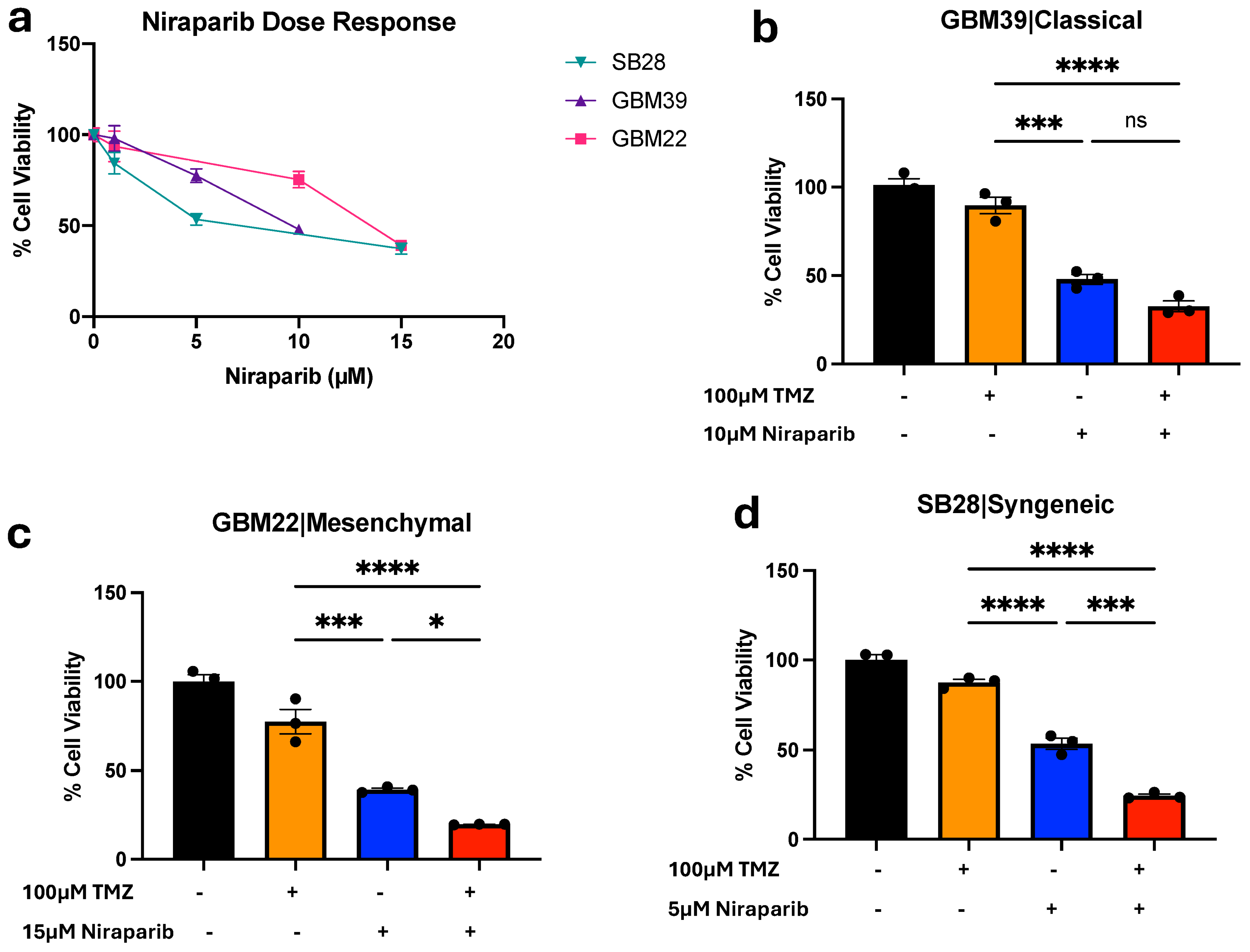
3.1. Temozolomide and Niraparib Decrease Human and Mouse Glioma Cell Growth
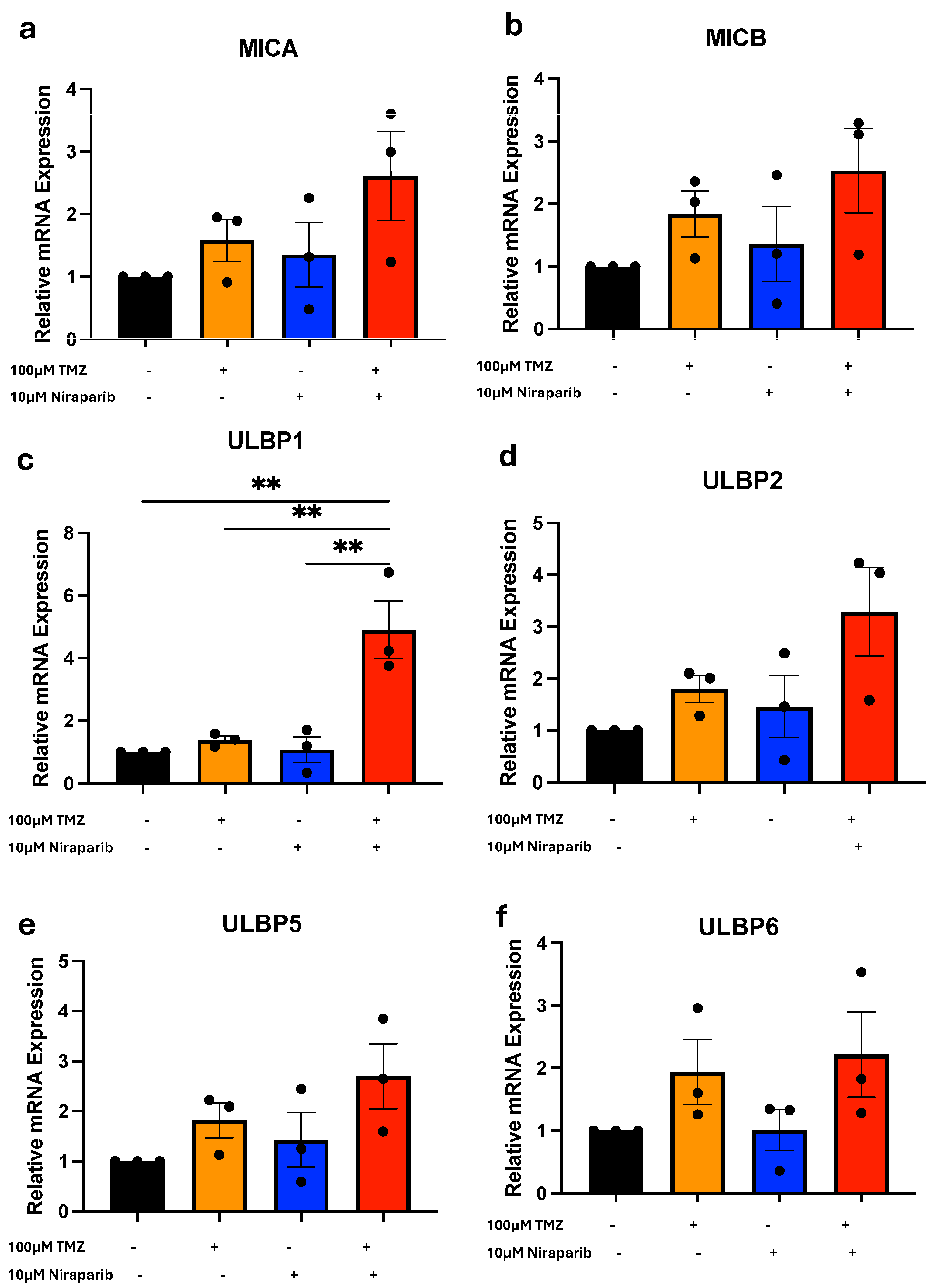
3.2. Temozolomide and Niraparib Increase ULBP1/Mult-1 mRNA
3.3. Temozolomide and Niraparib Increase ULBP1/Mult-1 Protein
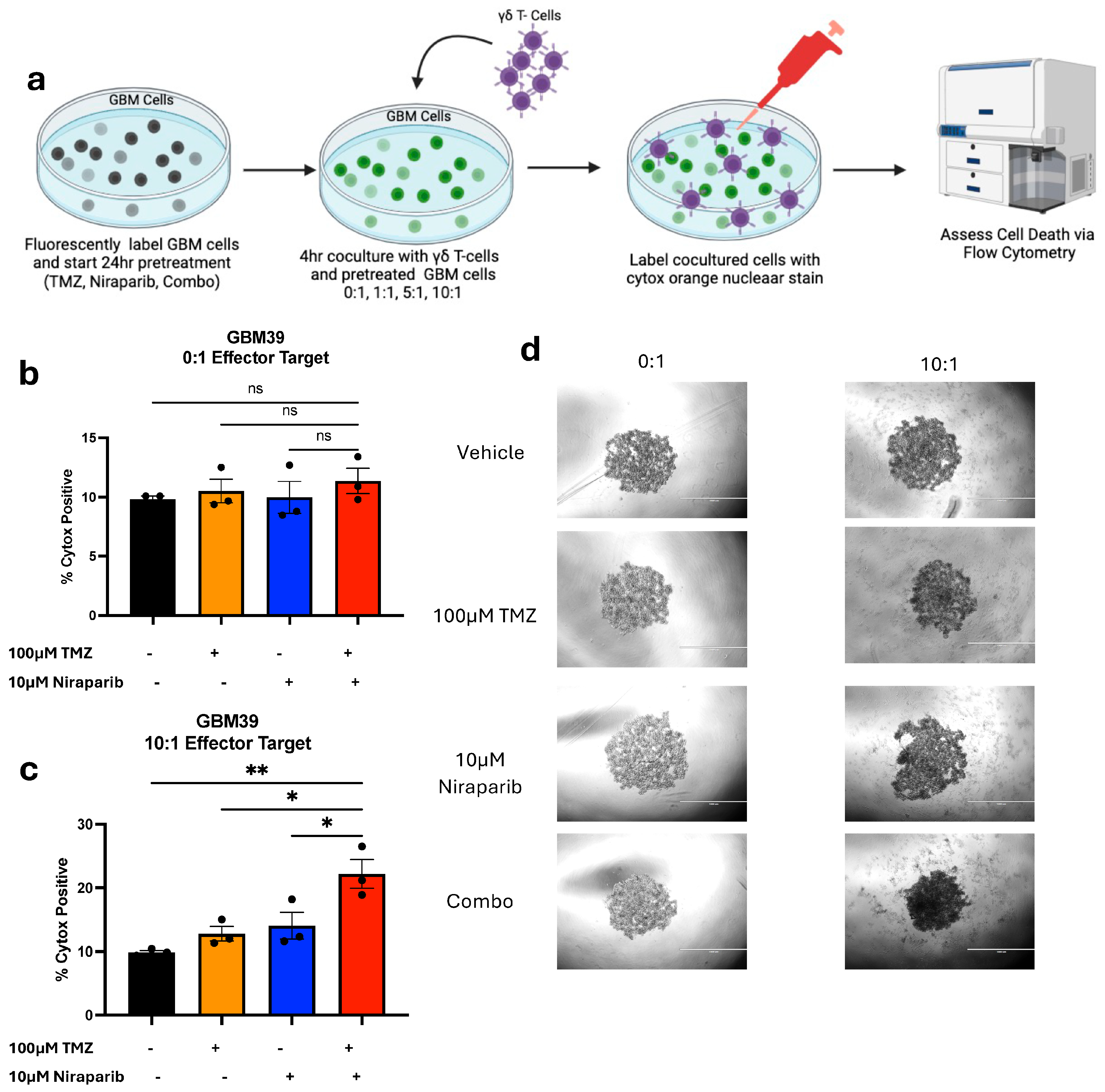
3.4. Pretreatment with Temozolomide and Niraparib Increases the Ability of Gamma Delta T Cells to Kill GBM39 Cells
4. Discussion
4.1. Overview of Findings and Clinical Implications
4.2. NKG2DL Regulation Including Specific Mechanisms for ULBP1 Induction Need Additional Investigation
4.3. ULBP1 Induction Can Be Distinct from Cell Stress Indicated by Decreased GBM Cell Growth
4.4. Limitations of the Study and Future Directions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Hwang, K.; Lee, J.H.; Kim, S.H.; Go, K.O.; Ji, S.Y.; Han, J.H.; Kim, C.Y. The Combination PARP Inhibitor Olaparib with Temozolomide in an Experimental Glioblastoma Model. In Vivo 2021, 35, 2015–2023. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.Y.; Tang, H.L.; Zheng, Y.H.; Feng, J.; Dong, B.X.; Chen, X.Q. The PARP1 Inhibitor Niraparib Represses DNA Damage Repair and Synergizes with Temozolomide for Antimyeloma Effects. J. Oncol. 2022, 2022, 2800488. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Li, X.; Gao, F.; de Groot, J.F.; Koul, D.; Yung, W.K.A. PARP-mediated PARylation of MGMT is critical to promote repair of temozolomide-induced O6-methylguanine DNA damage in glioblastoma. Neuro Oncol. 2021, 23, 920–931. [Google Scholar] [CrossRef] [PubMed]
- Cao, T.Q.; Wainwright, D.A.; Lee-Chang, C.; Miska, J.; Sonabend, A.M.; Heimberger, A.B.; Lukas, R.V. Next Steps for Immunotherapy in Glioblastoma. Cancers 2022, 14, 4023. [Google Scholar] [CrossRef] [PubMed]
- Woroniecka, K.I.; Rhodin, K.E.; Chongsathidkiet, P.; Keith, K.A.; Fecci, P.E. T-cell Dysfunction in Glioblastoma: Applying a New Framework. Clin. Cancer Res. 2018, 24, 3792–3802. [Google Scholar] [CrossRef] [PubMed]
- Zamora, A.E.; Crawford, J.C.; Thomas, P.G. Hitting the Target: How T Cells Detect and Eliminate Tumors. J. Immunol. 2018, 200, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Lohr, J.; Ratliff, T.; Huppertz, A.; Ge, Y.; Dictus, C.; Ahmadi, R.; Grau, S.; Hiraoka, N.; Eckstein, V.; Ecker, R.C.; et al. Effector T-cell infiltration positively impacts survival of glioblastoma patients and is impaired by tumor-derived TGF-beta. Clin. Cancer Res. 2011, 17, 4296–4308. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, R.; Sarkar, S.; Yong, V.W. T Cell Exhaustion in Glioblastoma: Intricacies of Immune Checkpoints. Trends Immunol. 2017, 38, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. The Microenvironmental Landscape of Brain Tumors. Cancer Cell 2017, 31, 326–341. [Google Scholar] [CrossRef] [PubMed]
- Lamb, L.S.; Pereboeva, L.; Youngblood, S.; Gillespie, G.Y.; Nabors, L.B.; Markert, J.M.; Dasgupta, A.; Langford, C.; Spencer, H.T. A combined treatment regimen of MGMT-modified gammadelta T cells and temozolomide chemotherapy is effective against primary high grade gliomas. Sci. Rep. 2021, 11, 21133. [Google Scholar] [CrossRef] [PubMed]
- Pressey, J.G.; Adams, J.; Harkins, L.; Kelly, D.; You, Z.; Lamb, L.S., Jr. In vivo expansion and activation of gammadelta T cells as immunotherapy for refractory neuroblastoma: A phase 1 study. Medicine 2016, 95, e4909. [Google Scholar] [CrossRef] [PubMed]
- Lamb, L.S., Jr.; Bowersock, J.; Dasgupta, A.; Gillespie, G.Y.; Su, Y.; Johnson, A.; Spencer, H.T. Engineered drug resistant gammadelta T cells kill glioblastoma cell lines during a chemotherapy challenge: A strategy for combining chemo- and immunotherapy. PLoS ONE 2013, 8, e51805. [Google Scholar] [CrossRef] [PubMed]
- Lamb, L.S., Jr. Gammadelta T cells as immune effectors against high-grade gliomas. Immunol. Res. 2009, 45, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Nussbaumer, O.; Koslowski, M. The emerging role of gammadelta T cells in cancer immunotherapy. Immunooncol. Technol. 2019, 1, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wang, S.; Xin, J.; Wang, J.; Yao, C.; Zhang, Z. Role of NKG2D and its ligands in cancer immunotherapy. Am. J. Cancer Res. 2019, 9, 2064–2078. [Google Scholar] [PubMed]
- Jelencic, V.; Lenartic, M.; Wensveen, F.M.; Polic, B. NKG2D: A versatile player in the immune system. Immunol. Lett. 2017, 189, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Galmarini, D.; Galmarini, C.M.; Galmarini, F.C. Cancer chemotherapy: A critical analysis of its 60 years of history. Crit. Rev. Oncol. Hematol. 2012, 84, 181–199. [Google Scholar] [CrossRef] [PubMed]
- Genoud, V.; Marinari, E.; Nikolaev, S.I.; Castle, J.C.; Bukur, V.; Dietrich, P.Y.; Okada, H.; Walker, P.R. Responsiveness to anti-PD-1 and anti-CTLA-4 immune checkpoint blockade in SB28 and GL261 mouse glioma models. Oncoimmunology 2018, 7, e1501137. [Google Scholar] [CrossRef] [PubMed]
- Eyler, C.E.; Foo, W.C.; LaFiura, K.M.; McLendon, R.E.; Hjelmeland, A.B.; Rich, J.N. Brain cancer stem cells display preferential sensitivity to Akt inhibition. Stem. Cells 2008, 26, 3027–3036. [Google Scholar] [CrossRef] [PubMed]
- Boyd, N.H.; Walker, K.; Fried, J.; Hackney, J.R.; McDonald, P.C.; Benavides, G.A.; Spina, R.; Audia, A.; Scott, S.E.; Landis, C.J.; et al. Addition of carbonic anhydrase 9 inhibitor SLC-0111 to temozolomide treatment delays glioblastoma growth in vivo. JCI Insight 2017, 2, 92928. [Google Scholar] [CrossRef] [PubMed]
- Bisht, P.; Kumar, V.U.; Pandey, R.; Velayutham, R.; Kumar, N. Role of PARP Inhibitors in Glioblastoma and Perceiving Challenges as Well as Strategies for Successful Clinical Development. Front. Pharmacol. 2022, 13, 939570. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, M.; Smetak, M.; Schaefer-Eckart, K.; Kimmel, B.; Birkmann, J.; Einsele, H.; Kunzmann, V. Successful adoptive transfer and in vivo expansion of haploidentical γδ T cells. J. Transl. Med. 2014, 12, 45. [Google Scholar] [CrossRef] [PubMed]
- Kierkels, G.J.J.; Scheper, W.; Meringa, A.D.; Johanna, I.; Beringer, D.X.; Janssen, A.; Schiffler, M.; Aarts-Riemens, T.; Kramer, L.; Straetemans, T.; et al. Identification of a tumor-specific allo-HLA-restricted γδTCR. Blood Adv. 2019, 3, 2870–2882. [Google Scholar] [CrossRef] [PubMed]
- Kabelitz, D.; Serrano, R.; Kouakanou, L.; Peters, C.; Kalyan, S. Cancer immunotherapy with γδ T cells: Many paths ahead of us. Cell Mol. Immunol. 2020, 17, 925–939. [Google Scholar] [CrossRef] [PubMed]
- Letchuman, V.; Ampie, L.; Shah, A.H.; Brown, D.A.; Heiss, J.D.; Chittiboina, P. Syngeneic murine glioblastoma models: Reactionary immune changes and immunotherapy intervention outcomes. Neurosurg. Focus. 2022, 52, E5. [Google Scholar] [CrossRef] [PubMed]
- Codo, P.; Weller, M.; Meister, G.; Szabo, E.; Steinle, A.; Wolter, M.; Reifenberger, G.; Roth, P. MicroRNA-mediated down-regulation of NKG2D ligands contributes to glioma immune escape. Oncotarget 2014, 5, 7651–7662. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.B.; Rocco, A.; Lamb, L.S.; Friedman, G.K.; Hjelmeland, A.B. Regulation of NKG2D Stress Ligands and Its Relevance in Cancer Progression. Cancers 2022, 14, 2339. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Bhat, K.P.L.; Balasubramaniyan, V.; Vaillant, B.; Ezhilarasan, R.; Hummelink, K.; Hollingsworth, F.; Wani, K.; Heathcock, L.; James, J.D.; Goodman, L.D.; et al. Mesenchymal differentiation mediated by NF-κB promotes radiation resistance in glioblastoma. Cancer Cell 2013, 24, 331–346. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lage, M.; Lynch, T.M.; Bi, Y.; Cocito, C.; Way, G.P.; Pal, S.; Haller, J.; Yan, R.E.; Ziober, A.; Nguyen, A.; et al. Immune landscapes associated with different glioblastoma molecular subtypes. Acta Neuropathol. Commun. 2019, 7, 203. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jones, A.B.; Tuy, K.; Hawkins, C.C.; Quinn, C.H.; Saad, J.; Gary, S.E.; Beierle, E.A.; Ding, L.; Rochlin, K.M.; Lamb, L.S.; et al. Temozolomide and the PARP Inhibitor Niraparib Enhance Expression of Natural Killer Group 2D Ligand ULBP1 and Gamma-Delta T Cell Cytotoxicity in Glioblastoma. Cancers 2024, 16, 2852. https://doi.org/10.3390/cancers16162852
Jones AB, Tuy K, Hawkins CC, Quinn CH, Saad J, Gary SE, Beierle EA, Ding L, Rochlin KM, Lamb LS, et al. Temozolomide and the PARP Inhibitor Niraparib Enhance Expression of Natural Killer Group 2D Ligand ULBP1 and Gamma-Delta T Cell Cytotoxicity in Glioblastoma. Cancers. 2024; 16(16):2852. https://doi.org/10.3390/cancers16162852
Chicago/Turabian StyleJones, Amber B., Kaysaw Tuy, Cyntanna C. Hawkins, Colin H. Quinn, Joelle Saad, Sam E. Gary, Elizabeth A. Beierle, Lei Ding, Kate M. Rochlin, Lawrence S. Lamb, and et al. 2024. "Temozolomide and the PARP Inhibitor Niraparib Enhance Expression of Natural Killer Group 2D Ligand ULBP1 and Gamma-Delta T Cell Cytotoxicity in Glioblastoma" Cancers 16, no. 16: 2852. https://doi.org/10.3390/cancers16162852
APA StyleJones, A. B., Tuy, K., Hawkins, C. C., Quinn, C. H., Saad, J., Gary, S. E., Beierle, E. A., Ding, L., Rochlin, K. M., Lamb, L. S., & Hjelmeland, A. B. (2024). Temozolomide and the PARP Inhibitor Niraparib Enhance Expression of Natural Killer Group 2D Ligand ULBP1 and Gamma-Delta T Cell Cytotoxicity in Glioblastoma. Cancers, 16(16), 2852. https://doi.org/10.3390/cancers16162852