


MERTK Is a Potential Therapeutic Target in Ewing Sarcoma
 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. CERES Dependency
2.2. RNA Expression Data
2.3. Cell Culture and Reagents
2.4. Small Molecule Inhibitors
2.5. Immunoblot Analysis
2.6. Immunoblot Analysis of MERTK
2.7. Downstream Signaling Assays
2.8. Cell Density Assay
2.9. Clonogenic Assay
2.10. Statistics
3. Results
3.1. Ewing Sarcoma Cell Lines Are Functionally Dependent on MERTK
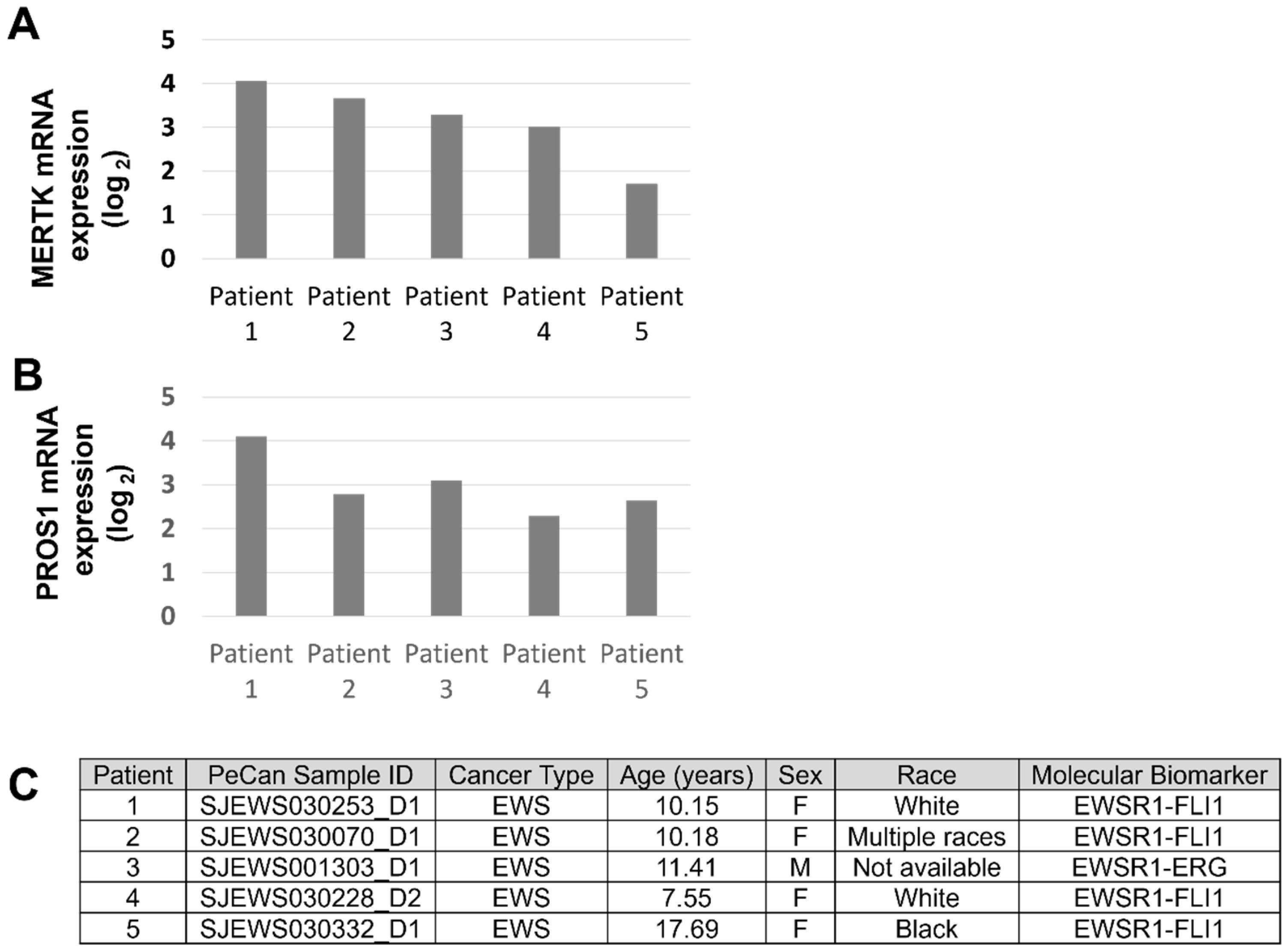
3.2. MERTK Is Expressed in Ewing Sarcoma
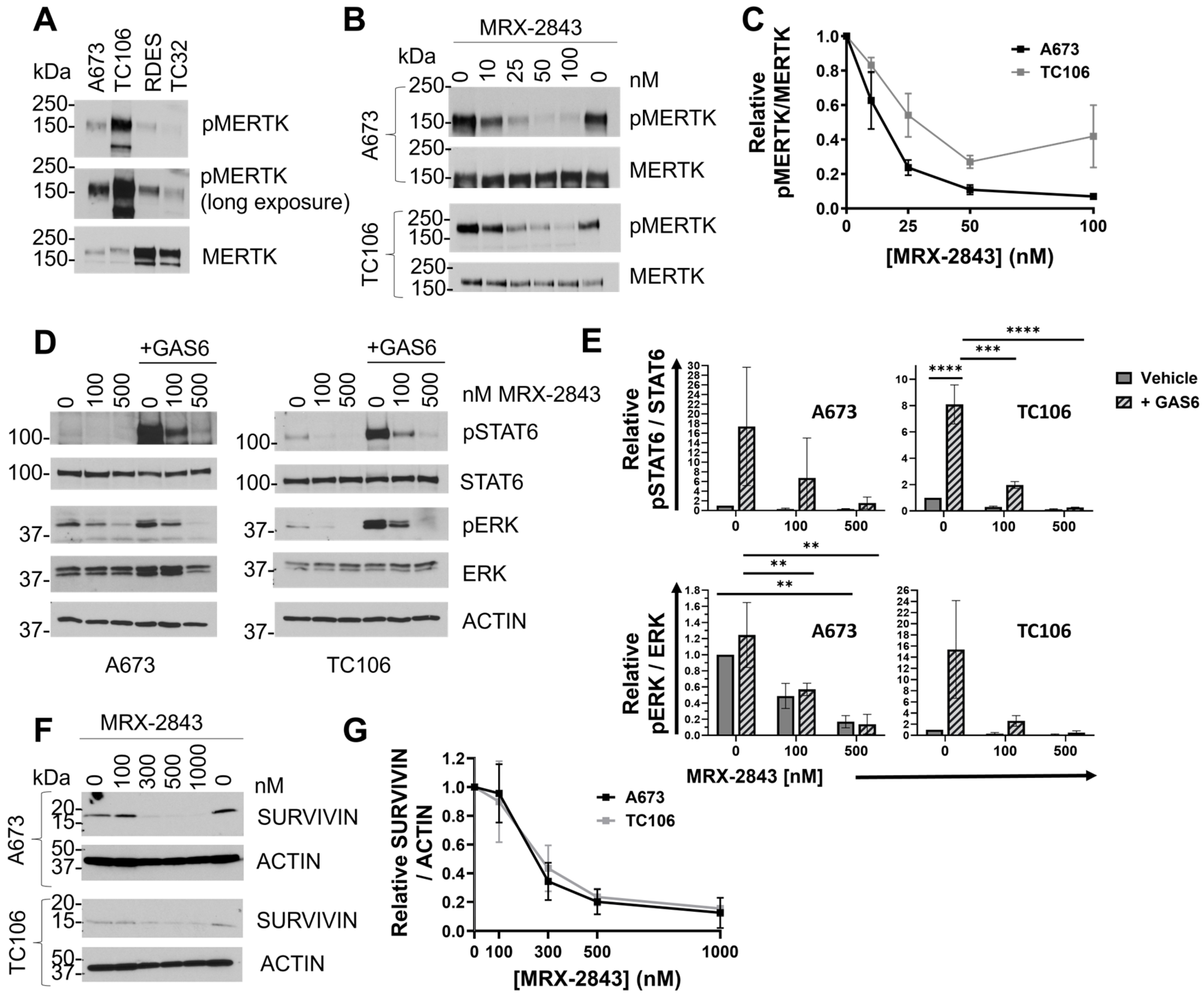
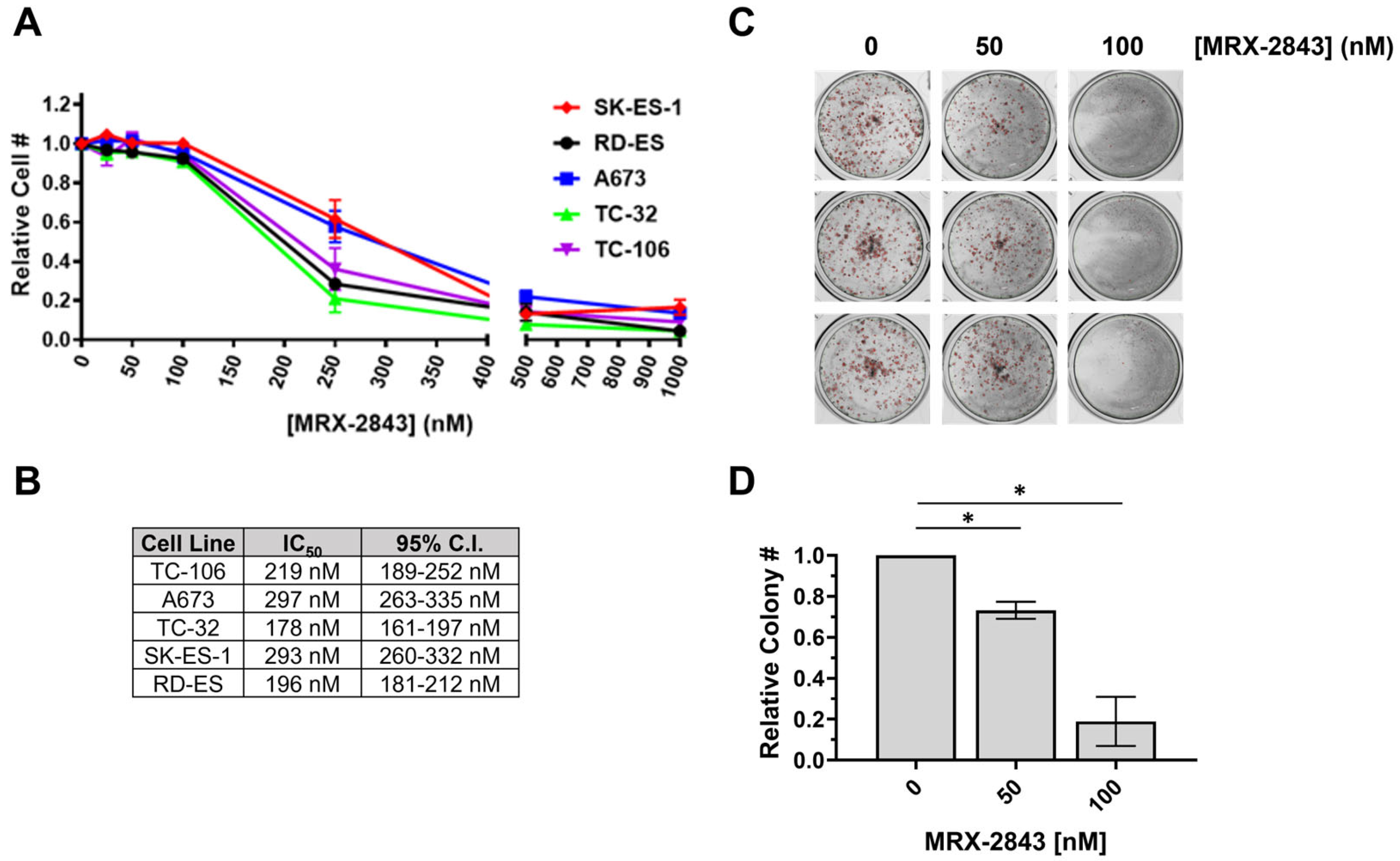
3.3. MRX-2843 Inhibits MERTK Kinase Activity and Has Potent Anti-Tumor Activity in EWS Cells
3.4. MRX-2843 Provides Enhanced Therapeutic Efficacy in Combination with BCL-2 Inhibitors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- PDQ Pediatric Treatment Editorial Board. Ewing Sarcoma and Undifferentiated Small Round Cell Sarcomas of Bone and Soft Tissue Treatment (PDQ®): Health Professional Version. In PDQ Cancer Information Summaries; National Cancer Institute (US): Bethesda, MD, USA, 2002. [Google Scholar]
- Durer, S.; Shaikh, H. Ewing Sarcoma. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2023. [Google Scholar]
- Tsuda, Y.; Dickson, B.C.; Swanson, D.; Sung, Y.S.; Zhang, L.; Meyers, P.; Healey, J.H.; Antonescu, C.R. Ewing sarcoma with FEV gene rearrangements is a rare subset with predilection for extraskeletal locations and aggressive behavior. Genes Chromosomes Cancer 2020, 59, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Allen-Rhoades, W.; Whittle, S.B.; Rainusso, N. Pediatric Solid Tumors in Children and Adolescents: An Overview. Pediatr. Rev. 2018, 39, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Zhan, H.; Mo, F.; Zhu, M.; Xu, X.; Zhang, B.; Liu, H.; Dai, M. A SEER-based nomogram accurately predicts prognosis in Ewing’s sarcoma. Sci. Rep. 2021, 11, 22723. [Google Scholar] [CrossRef] [PubMed]
- Bailey, K.; Cost, C.; Davis, I.; Glade-Bender, J.; Grohar, P.; Houghton, P.; Isakoff, M.; Stewart, E.; Laack, N.; Yustein, J.; et al. Emerging novel agents for patients with advanced Ewing sarcoma: A report from the Children’s Oncology Group (COG) New Agents for Ewing Sarcoma Task Force. F1000Research 2019, 8, 493. [Google Scholar] [CrossRef]
- Albarrán, V.; Villamayor, M.L.; Chamorro, J.; Rosero, D.I.; Pozas, J.; San Román, M.; Calvo, J.C.; Pérez De Aguado, P.; Moreno, J.; Guerrero, P.; et al. Receptor Tyrosine Kinase Inhibitors for the Treatment of Recurrent and Unresectable Bone Sarcomas. Int. J. Mol. Sci. 2022, 23, 13784. [Google Scholar] [CrossRef]
- Graham, D.K.; DeRyckere, D.; Davies, K.D.; Earp, H.S. The TAM family: Phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat. Rev. Cancer 2014, 14, 769–785. [Google Scholar] [CrossRef]
- Huelse, J.M.; Fridlyand, D.M.; Earp, S.; DeRyckere, D.; Graham, D.K. MERTK in cancer therapy: Targeting the receptor tyrosine kinase in tumor cells and the immune system. Pharmacol. Ther. 2020, 213, 107577. [Google Scholar] [CrossRef] [PubMed]
- Tsou, W.-I.; Nguyen, K.-Q.N.; Calarese, D.A.; Garforth, S.J.; Antes, A.L.; Smirnov, S.V.; Almo, S.C.; Birge, R.B.; Kotenko, S.V. Receptor Tyrosine Kinases, TYRO3, AXL, and MER, Demonstrate Distinct Patterns and Complex Regulation of Ligand-induced Activation. J. Biol. Chem. 2014, 289, 25750–25763. [Google Scholar] [CrossRef] [PubMed]
- Cummings, C.T.; Deryckere, D.; Earp, H.S.; Graham, D.K. Molecular pathways: MERTK signaling in cancer. Clin. Cancer Res. 2013, 19, 5275–5280. [Google Scholar] [CrossRef]
- Yan, D.; Parker, R.E.; Wang, X.; Frye, S.V.; Earp, H.S., 3rd; DeRyckere, D.; Graham, D.K. MERTK Promotes Resistance to Irreversible EGFR Tyrosine Kinase Inhibitors in Non-small Cell Lung Cancers Expressing Wild-type EGFR Family Members. Clin. Cancer Res. 2018, 24, 6523–6535. [Google Scholar] [CrossRef]
- Yan, D.; Huelse, J.M.; Kireev, D.; Tan, Z.; Chen, L.; Goyal, S.; Wang, X.; Frye, S.V.; Behera, M.; Schneider, F.; et al. MERTK activation drives osimertinib resistance in EGFR-mutant non–small cell lung cancer. J. Clin. Investig. 2022, 132, e150517. [Google Scholar] [CrossRef] [PubMed]
- Linger, R.M.A.; Cohen, R.A.; Cummings, C.T.; Sather, S.; Migdall-Wilson, J.; Middleton, D.H.G.; Lu, X.; Barón, A.E.; Franklin, W.A.; Merrick, D.T.; et al. Mer or Axl receptor tyrosine kinase inhibition promotes apoptosis, blocks growth and enhances chemosensitivity of human non-small cell lung cancer. Oncogene 2013, 32, 3420–3431. [Google Scholar] [CrossRef]
- Minson, K.A.; Smith, C.C.; Deryckere, D.; Libbrecht, C.; Lee-Sherick, A.B.; Huey, M.G.; Lasater, E.A.; Kirkpatrick, G.D.; Stashko, M.A.; Zhang, W.; et al. The MERTK/FLT3 inhibitor MRX-2843 overcomes resistance-conferring FLT3 mutations in acute myeloid leukemia. JCI Insight 2016, 1, e85630. [Google Scholar] [CrossRef]
- Zhang, W.; Deryckere, D.; Hunter, D.; Liu, J.; Stashko, M.A.; Minson, K.A.; Cummings, C.T.; Lee, M.; Glaros, T.G.; Newton, D.L.; et al. UNC2025, a Potent and Orally Bioavailable MER/FLT3 Dual Inhibitor. J. Med. Chem. 2014, 57, 7031–7041. [Google Scholar] [CrossRef] [PubMed]
- DeRyckere, D.; Lee-Sherick, A.B.; Huey, M.G.; Hill, A.A.; Tyner, J.W.; Jacobsen, K.M.; Page, L.S.; Kirkpatrick, G.G.; Eryildiz, F.; Montgomery, S.A.; et al. UNC2025, a MERTK Small-Molecule Inhibitor, Is Therapeutically Effective Alone and in Combination with Methotrexate in Leukemia Models. Clin. Cancer Res. 2017, 23, 1481–1492. [Google Scholar] [CrossRef]
- Cummings, C.T.; Zhang, W.; Davies, K.D.; Kirkpatrick, G.D.; Zhang, D.; DeRyckere, D.; Wang, X.; Frye, S.V.; Earp, H.S.; Graham, D.K. Small Molecule Inhibition of MERTK Is Efficacious in Non-Small Cell Lung Cancer Models Independent of Driver Oncogene Status. Mol. Cancer Ther. 2015, 14, 2014–2022. [Google Scholar] [CrossRef]
- Sufit, A.; Lee-Sherick, A.B.; DeRyckere, D.; Rupji, M.; Dwivedi, B.; Varella-Garcia, M.; Pierce, A.M.; Kowalski, J.; Wang, X.; Frye, S.V.; et al. MERTK Inhibition Induces Polyploidy and Promotes Cell Death and Cellular Senescence in Glioblastoma Multiforme. PLoS ONE 2016, 11, e0165107. [Google Scholar] [CrossRef]
- McDaniel, N.K.; Cummings, C.T.; Iida, M.; Hülse, J.; Pearson, H.E.; Vasileiadi, E.; Parker, R.E.; Orbuch, R.A.; Ondracek, O.J.; Welke, N.B.; et al. MERTK Mediates Intrinsic and Adaptive Resistance to AXL-targeting Agents. Mol. Cancer Ther. 2018, 17, 2297–2308. [Google Scholar] [CrossRef]
- Lee-Sherick, A.B.; Jacobsen, K.M.; Henry, C.J.; Huey, M.G.; Parker, R.E.; Page, L.S.; Hill, A.A.; Wang, X.; Frye, S.V.; Earp, H.S.; et al. MERTK inhibition alters the PD-1 axis and promotes anti-leukemia immunity. JCI Insight 2018, 3, e97941. [Google Scholar] [CrossRef]
- Sinik, L.; Minson, K.A.; Tentler, J.J.; Carrico, J.; Bagby, S.M.; Robinson, W.A.; Kami, R.; Burstyn-Cohen, T.; Eckhardt, S.G.; Wang, X.; et al. Inhibition of MERTK Promotes Suppression of Tumor Growth in BRAF Mutant and BRAF Wild-Type Melanoma. Mol. Cancer Ther. 2019, 18, 278–288. [Google Scholar] [CrossRef]
- Heisey, D.A.R.; Lochmann, T.L.; Floros, K.V.; Coon, C.M.; Powell, K.M.; Jacob, S.; Calbert, M.L.; Ghotra, M.S.; Stein, G.T.; Maves, Y.K.; et al. The Ewing Family of Tumors Relies on BCL-2 and BCL-XL to Escape PARP Inhibitor Toxicity. Clin. Cancer Res. 2019, 25, 1664–1675. [Google Scholar] [CrossRef] [PubMed]
- Hafezi, S.; Rahmani, M. Targeting BCL-2 in Cancer: Advances, Challenges, and Perspectives. Cancers 2021, 13, 1292. [Google Scholar] [CrossRef] [PubMed]
- Kaloni, D.; Diepstraten, S.T.; Strasser, A.; Kelly, G.L. BCL-2 protein family: Attractive targets for cancer therapy. Apoptosis 2023, 28, 20–38. [Google Scholar] [CrossRef] [PubMed]
- Kehr, S.; Vogler, M. It’s time to die: BH3 mimetics in solid tumors. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118987. [Google Scholar] [CrossRef] [PubMed]
- Fairlie, W.D.; Lee, E.F. Targeting the BCL-2-regulated apoptotic pathway for the treatment of solid cancers. Biochem. Soc. Trans. 2021, 49, 2397–2410. [Google Scholar] [CrossRef]
- D’Aguanno, S.; Del Bufalo, D. Inhibition of Anti-Apoptotic Bcl-2 Proteins in Preclinical and Clinical Studies: Current Overview in Cancer. Cells 2020, 9, 1287. [Google Scholar] [CrossRef] [PubMed]
- Kehr, S.; Haydn, T.; Bierbrauer, A.; Irmer, B.; Vogler, M.; Fulda, S. Targeting BCL-2 proteins in pediatric cancer: Dual inhibition of BCL-X(L) and MCL-1 leads to rapid induction of intrinsic apoptosis. Cancer Lett. 2020, 482, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Meyers, R.M.; Bryan, J.G.; McFarland, J.M.; Weir, B.A.; Sizemore, A.E.; Xu, H.; Dharia, N.V.; Montgomery, P.G.; Cowley, G.S.; Pantel, S.; et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet. 2017, 49, 1779–1784. [Google Scholar] [CrossRef] [PubMed]
- Puddu, A.; Ravera, S.; Panfoli, I.; Bertola, N.; Maggi, D. High Glucose Impairs Expression and Activation of MerTK in ARPE-19 Cells. Int. J. Mol. Sci. 2022, 23, 1144. [Google Scholar] [CrossRef]
- Vogler, M. Targeting BCL2-Proteins for the Treatment of Solid Tumours. Adv. Med. 2014, 2014, 943648. [Google Scholar] [CrossRef]
- Olsen, R.R.; Mary-Sinclair, M.N.; Yin, Z.; Freeman, K.W. Antagonizing Bcl-2 Family Members Sensitizes Neuroblastoma and Ewing’s Sarcoma to an Inhibitor of Glutamine Metabolism. PLoS ONE 2015, 10, e0116998. [Google Scholar] [CrossRef]
- Soldatenkov, V.; Notario, V.; Dritschilo, A. Expression of the human Bcl-2 increases resistance of Ewing’s sarcoma cells to apoptosis and inhibits poly(ADP-ribose) polymerase cleavage induced by radiation. Int. J. Oncol. 1996, 9, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Zhu, J.; Zeng, Y.; Liu, T.; Zhang, Y.; Cai, T.; Fu, Y.; Zhang, W.; Zhang, R.; Liu, Z.; et al. KPNB1-mediated nuclear translocation of PD-L1 promotes non-small cell lung cancer cell proliferation via the Gas6/MerTK signaling pathway. Cell Death Amp; Differ. 2021, 28, 1284–1300. [Google Scholar] [CrossRef] [PubMed]
- Pugazhenthi, S.; Nesterova, A.; Sable, C.; Heidenreich, K.A.; Boxer, L.M.; Heasley, L.E.; Reusch, J.E.B. Akt/Protein Kinase B Up-regulates Bcl-2 Expression through cAMP-response Element-binding Protein. J. Biol. Chem. 2000, 275, 10761–10766. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, K.C.; Verschuur, A.; Morgenstern, D.A.; van Eijkelenburg, N.; Federico, S.M.; Fraser, C.; Forlenza, C.J.; Ziegler, D.S.; Gerber, N.U.; Khaw, S.L.; et al. The first report of pediatric patients with solid tumors treated with venetoclax. J. Clin. Oncol. 2020, 38, 10524. [Google Scholar] [CrossRef]
- Nor Hisam, N.S.; Ugusman, A.; Rajab, N.F.; Ahmad, M.F.; Fenech, M.; Liew, S.L.; Mohamad Anuar, N.N. Combination Therapy of Navitoclax with Chemotherapeutic Agents in Solid Tumors and Blood Cancer: A Review of Current Evidence. Pharmaceutics 2021, 13, 1353. [Google Scholar] [CrossRef]
- Cleary, J.M.; Lima, C.M.; Hurwitz, H.I.; Montero, A.J.; Franklin, C.; Yang, J.; Graham, A.; Busman, T.; Mabry, M.; Holen, K.; et al. A phase I clinical trial of navitoclax, a targeted high-affinity Bcl-2 family inhibitor, in combination with gemcitabine in patients with solid tumors. Investig. New Drugs 2014, 32, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Pascual-Pasto, G.; Resa-Pares, C.; Castillo-Ecija, H.; Aschero, R.; Baulenas-Farres, M.; Vila-Ubach, M.; Burgueño, V.; Balaguer-Lluna, L.; Cuadrado-Vilanova, M.; Olaciregui, N.G.; et al. Low Bcl-2 is a robust biomarker of sensitivity to nab-paclitaxel in Ewing sarcoma. Biochem. Pharmacol. 2023, 208, 115408. [Google Scholar] [CrossRef]
- Tsafou, K.; Katschnig, A.M.; Radic-Sarikas, B.; Mutz, C.N.; Iljin, K.; Schwentner, R.; Kauer, M.O.; Mühlbacher, K.; Aryee, D.N.T.; Westergaard, D.; et al. Identifying the druggable interactome of EWS-FLI1 reveals MCL-1 dependent differential sensitivities of Ewing sarcoma cells to apoptosis inducers. Oncotarget 2018, 9, 31018–31031. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smart, S.K.; Yeung, T.Y.; Santos, M.O.; McSwain, L.F.; Wang, X.; Frye, S.V.; Earp, H.S., III; DeRyckere, D.; Graham, D.K. MERTK Is a Potential Therapeutic Target in Ewing Sarcoma. Cancers 2024, 16, 2831. https://doi.org/10.3390/cancers16162831
Smart SK, Yeung TY, Santos MO, McSwain LF, Wang X, Frye SV, Earp HS III, DeRyckere D, Graham DK. MERTK Is a Potential Therapeutic Target in Ewing Sarcoma. Cancers. 2024; 16(16):2831. https://doi.org/10.3390/cancers16162831
Chicago/Turabian StyleSmart, Sherri K., Tsz Y. Yeung, M. Olivia Santos, Leon F. McSwain, Xiaodong Wang, Stephen V. Frye, H. Shelton Earp, III, Deborah DeRyckere, and Douglas K. Graham. 2024. "MERTK Is a Potential Therapeutic Target in Ewing Sarcoma" Cancers 16, no. 16: 2831. https://doi.org/10.3390/cancers16162831
APA StyleSmart, S. K., Yeung, T. Y., Santos, M. O., McSwain, L. F., Wang, X., Frye, S. V., Earp, H. S., III, DeRyckere, D., & Graham, D. K. (2024). MERTK Is a Potential Therapeutic Target in Ewing Sarcoma. Cancers, 16(16), 2831. https://doi.org/10.3390/cancers16162831