
Breaking Boundaries: Immunotherapy for Myeloid Malignancies


{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
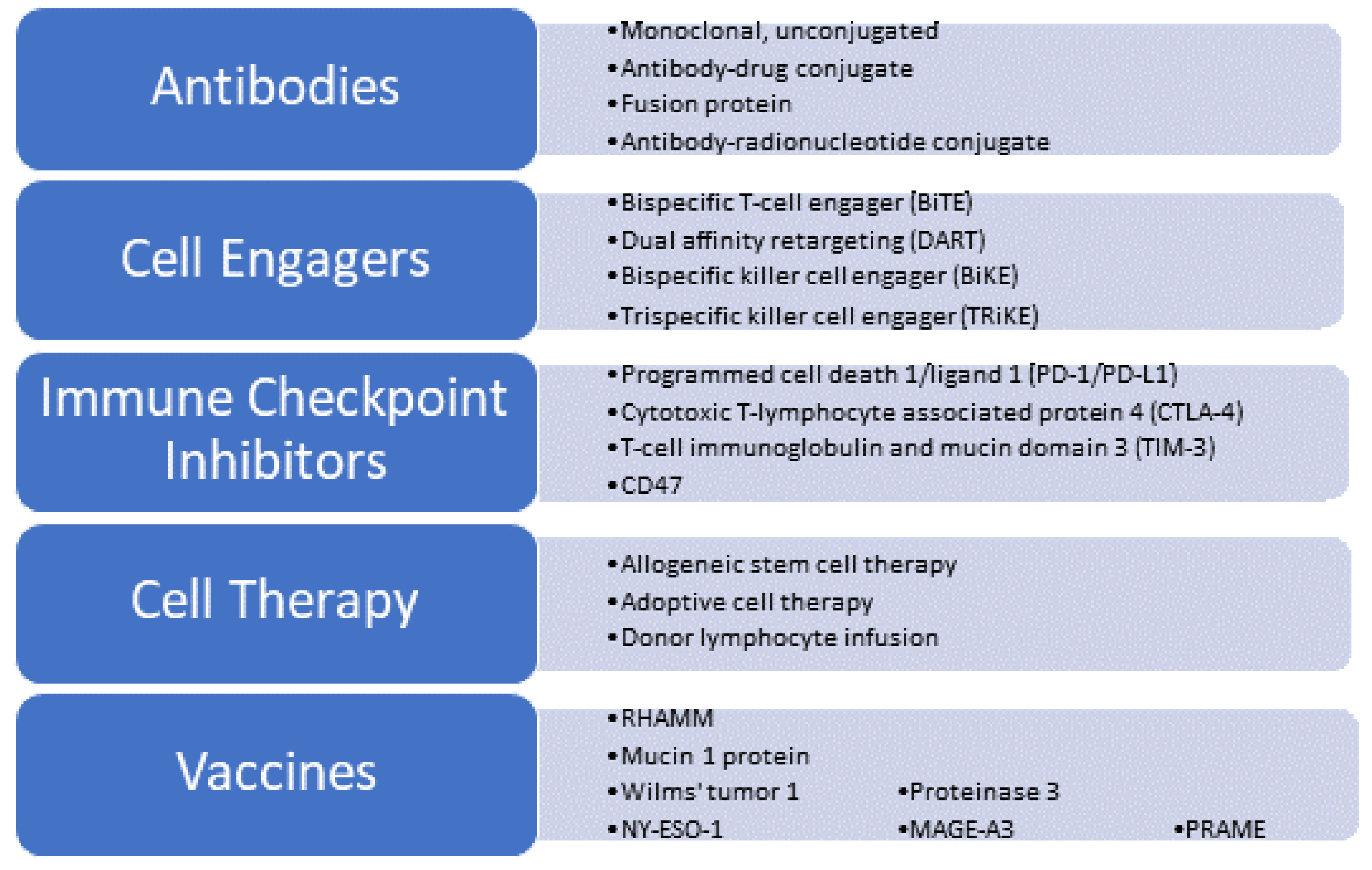
2. Immunotherapy for Myeloid Malignancies: Finding the Right Target
2.1. Antibodies
2.1.1. CD33
2.1.2. CD123
2.2. Cell Engagers: BiTEs and DARTs
2.3. Immune Checkpoint Inhibitors
2.3.1. CD47
2.3.2. T-Cell Immunoglobin and Mucin Domain 3 (TIM-3) Inhibitor
2.4. Adoptive Cell Therapy (ACT)
2.5. Allogeneic Stem Cell Transplantation
2.6. DLI
2.7. Vaccines
2.8. NK Cells
3. Future Directions
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sekeres, M.A.; Taylor, J. Diagnosis and Treatment of Myelodysplastic Syndromes: A Review. JAMA 2022, 328, 872–880. [Google Scholar] [CrossRef] [PubMed]
- Niederwieser, C.; Kroger, N. Hematopoietic cell transplantation (HCT) in MDS patients of older age. Leuk Lymphoma 2024, 65, 570–584. [Google Scholar] [CrossRef] [PubMed]
- Jentzsch, M.; Bischof, L.; Ussmann, J.; Backhaus, D.; Brauer, D.; Metzeler, K.H.; Schwind, S. Prognostic Impact of the 2022 European Leukemia Net Risk Classification in Patients with Acute Myeloid Leukemia Undergoing Allogeneic Stem Cell Transplantation. Blood 2022, 140 (Suppl. S1), 10601–10602. [Google Scholar] [CrossRef]
- Greenberg, P.; Cox, C.; LeBeau, M.M.; Fenaux, P.; Morel, P.; Sanz, G.; Sanz, M.; Vallespi, T.; Hamblin, T.; Oscier, D.; et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood 1997, 89, 2079–2088. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, P.L.; Tuechler, H.; Schanz, J.; Sanz, G.; Garcia-Manero, G.; Solé, F.; Bennett, J.M.; Bowen, D.; Fenaux, P.; Dreyfus, F.; et al. Revised International Prognostic Scoring System for Myelodysplastic Syndromes. Blood 2012, 120, 2454–2465. [Google Scholar] [CrossRef] [PubMed]
- Zeidan, A.M.; Platzbecker, U.; Bewersdorf, J.P.; Stahl, M.; Adès, L.; Borate, U.; Bowen, D.T.; Buckstein, R.J.; Brunner, A.M.; E Carraway, H.; et al. Consensus proposal for revised International Working Group response criteria for higher risk myelodysplastic syndromes. Blood 2023, 141, 2047–2061. [Google Scholar] [CrossRef] [PubMed]
- Bernard, E.; Tuechler, H.; Greenberg, P.L.; Hasserjian, R.P.; Ossa, J.E.A.; Nannya, Y.; Devlin, S.M.; Creignou, M.; Pinel, P.; Monnier, L.; et al. Molecular International Prognostic Scoring System for Myelodysplastic Syndromes. NEJM Évid. 2022, 1, EVIDoa2200008. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.-M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating morphologic, clinical, and genomic data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef]
- Awada, H.; Gurnari, C.; Xie, Z.; Bewersdorf, J.P.; Zeidan, A.M. What’s Next after Hypomethylating Agents Failure in Myeloid Neoplasms? A Rational Approach. Cancers 2023, 15, 2248. [Google Scholar] [CrossRef] [PubMed]
- Santini, V. How I treat MDS after hypomethylating agent failure. Blood 2019, 133, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Cherry, E.M.; Abbott, D.; Amaya, M.; McMahon, C.; Schwartz, M.; Rosser, J.; Sato, A.; Schowinsky, J.T.; Inguva, A.; Minhajuddin, M.; et al. Venetoclax and azacitidine compared with induction chemotherapy for newly diagnosed patients with acute myeloid leukemia. Blood Adv. 2021, 5, 5565–5573. [Google Scholar] [CrossRef] [PubMed]
- Vago, L.; Gojo, I. Immune escape and immunotherapy of acute myeloid leukemia. J. Clin. Investig. 2020, 130, 1552–1564. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Yu, J.; Li, Y.; Wang, K. Neoantigen-specific TCR-T cell-based immunotherapy for acute myeloid leukemia. Exp. Hematol. Oncol. 2022, 11, 100. [Google Scholar] [CrossRef] [PubMed]
- Roerden, M.; Nelde, A.; Walz, J.S. Neoantigens in Hematological Malignancies-Ultimate Targets for Immunotherapy? Front. Immunol. 2019, 10, 3004. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.; Alotaibi, A.S.; Bucklein, V.; Subklewe, M. T-cell-based immunotherapy of acute myeloid leukemia: Current concepts and future developments. Leukemia 2021, 35, 1843–1863. [Google Scholar] [CrossRef] [PubMed]
- Lichtenegger, F.S.; Krupka, C.; Haubner, S.; Kohnke, T.; Subklewe, M. Recent developments in immunotherapy of acute myeloid leukemia. J. Hematol. Oncol. 2017, 10, 142. [Google Scholar] [CrossRef] [PubMed]
- Kirkey, D.C.; Loeb, A.M.; Castro, S.; McKay, C.N.; Perkins, L.; Pardo, L.; Leonti, A.R.; Tang, T.T.; Loken, M.R.; Brodersen, L.E.; et al. Therapeutic targeting of PRAME with mTCRCAR T cells in acute myeloid leukemia. Blood Adv. 2023, 7, 1178–1189. [Google Scholar] [CrossRef] [PubMed]
- Schorr, C.; Perna, F. Targets for chimeric antigen receptor T-cell therapy of acute myeloid leukemia. Front. Immunol. 2022, 13, 1085978. [Google Scholar] [CrossRef]
- de Visser, K.E.; Joyce, J.A. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell 2023, 41, 374–403. [Google Scholar] [CrossRef]
- Dysthe, M.; Parihar, R. Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1224, 117–140. [Google Scholar]
- Mantovani, A.; Allavena, P.; Marchesi, F.; Garlanda, C. Macrophages as tools and targets in cancer therapy. Nat. Rev. Drug Discov. 2022, 21, 799–820. [Google Scholar] [CrossRef] [PubMed]
- Ephraim, R.; Fraser, S.; Nurgali, K.; Apostolopoulos, V. Checkpoint Markers and Tumor Microenvironment: What Do We Know? Cancers 2022, 14, 3788. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.K.; Cho, S.W. The Evasion Mechanisms of Cancer Immunity and Drug Intervention in the Tumor Microenvironment. Front. Pharmacol. 2022, 13, 868695. [Google Scholar] [CrossRef] [PubMed]
- Fathi, E.; Farahzadi, R.; Sheervalilou, R.; Sanaat, Z.; Vietor, I. A general view of CD33(+) leukemic stem cells and CAR-T cells as interesting targets in acute myeloblatsic leukemia therapy. Blood Res. 2020, 55, 10–16. [Google Scholar] [CrossRef]
- Liu, J.; Tong, J.; Yang, H. Targeting CD33 for acute myeloid leukemia therapy. BMC Cancer 2022, 22, 24. [Google Scholar] [CrossRef] [PubMed]
- Feldman, E.J.; Brandwein, J.; Stone, R.; Kalaycio, M.; Moore, J.; O’Connor, J.; Wedel, N.; Roboz, G.J.; Miller, C.; Chopra, R.; et al. Phase III randomized multicenter study of a humanized anti-CD33 monoclonal antibody, lintuzumab, in combination with chemotherapy, versus chemotherapy alone in patients with refractory or first-relapsed acute myeloid leukemia. J. Clin. Oncol. 2005, 23, 4110–4116. [Google Scholar] [CrossRef] [PubMed]
- Petersdorf, S.H.; Kopecky, K.J.; Slovak, M.; Willman, C.; Nevill, T.; Brandwein, J.; Larson, R.A.; Erba, H.P.; Stiff, P.J.; Stuart, R.K.; et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood 2013, 121, 4854–4860. [Google Scholar] [CrossRef] [PubMed]
- Burnett, A.K.; Hills, R.K.; Milligan, D.; Kjeldsen, L.; Kell, J.; Russell, N.H.; Yin, J.A.; Hunter, A.; Goldstone, A.H.; Wheatley, K. Identification of Patients with Acute Myeloblastic Leukemia Who Benefit from the Addition of Gemtuzumab Ozogamicin: Results of the MRC AML15 Trial. J. Clin. Oncol. 2011, 29, 369–377. [Google Scholar] [CrossRef]
- Freeman, S.D.; Thomas, A.; Thomas, I.; Hills, R.K.; Vyas, P.; Gilkes, A.; Metzner, M.; Jakobsen, N.A.; Kennedy, A.; Moore, R.; et al. Fractionated vs single-dose gemtuzumab ozogamicin with determinants of benefit in older patients with AML: The UK NCRI AML18 trial. Blood 2023, 142, 1697–1707. [Google Scholar] [CrossRef]
- Hills, R.K.; Castaigne, S.; Appelbaum, F.R.; Delaunay, J.; Petersdorf, S.; Othus, M.; Estey, E.H.; Dombret, H.; Chevret, S.; Ifrah, N.; et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: A meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014, 15, 986–996. [Google Scholar] [CrossRef]
- Castaigne, S.; Pautas, C.; Terré, C.; Raffoux, E.; Bordessoule, D.; Bastie, J.-N.; Legrand, O.; Thomas, X.; Turlure, P.; Reman, O.; et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): A randomised, open-label, phase 3 study. Lancet 2012, 379, 1508–1516. [Google Scholar] [CrossRef] [PubMed]
- Norsworthy, K.J.; Ko, C.W.; Lee, J.E.; Liu, J.; John, C.S.; Przepiorka, D. FDA Approval Summary: Mylotarg for Treatment of Patients with Relapsed or Refractory CD33-Positive Acute Myeloid Leukemia. Oncologist 2018, 23, 1103–1108. [Google Scholar] [CrossRef]
- Gbadamosi, M.; Meshinchi, S.; Lamba, J.K. Gemtuzumab ozogamicin for treatment of newly diagnosed CD33-positive acute myeloid leukemia. Future Oncol. 2018, 14, 3199–3213. [Google Scholar] [CrossRef] [PubMed]
- Ravandi, F.; Estey, E.; Jones, D.; Faderl, S.; O’Brien, S.; Fiorentino, J.; Pierce, S.; Blamble, D.; Estrov, Z.; Wierda, W.; et al. Effective treatment of acute promyelocytic leukemia with all-trans-retinoic acid, arsenic trioxide, and gemtuzumab ozogamicin. J. Clin. Oncol. 2009, 27, 504–510. [Google Scholar] [CrossRef]
- Abedin, S.M.; Murthy, G.S.G.; Desai, A.; Chen, M.; Atallah, E.L. Sequential salvage chemotherapy and lintuzumab-Ac225 results in deep responses and prolonged survival in adverse risk relapsed/refractory AML and in AML patients that received prior venetoclax therapy. J. Clin. Oncol. 2023, 41 (Suppl. S16), e19030. [Google Scholar] [CrossRef]
- El Achi, H.; Dupont, E.; Paul, S.; Khoury, J.D. CD123 as a Biomarker in Hematolymphoid Malignancies: Principles of Detection and Targeted Therapies. Cancers 2020, 12, 3087. [Google Scholar] [CrossRef]
- E Hogge, D.; Yalcintepe, L.; Wong, S.-H.; Gerhard, B.; E Frankel, A. Variant diphtheria toxin-interleukin-3 fusion proteins with increased receptor affinity have enhanced cytotoxicity against acute myeloid leukemia progenitors. Clin. Cancer Res. 2006, 12, 1284–1291. [Google Scholar] [CrossRef]
- Pemmaraju, N.; Konopleva, M. Approval of tagraxofusp-erzs for blastic plasmacytoid dendritic cell neoplasm. Blood Adv. 2020, 4, 4020–4027. [Google Scholar] [CrossRef] [PubMed]
- Mani, R.; Goswami, S.; Gopalakrishnan, B.; Ramaswamy, R.; Wasmuth, R.; Tran, M.; Mo, X.K.; Gordon, A.; Gordon, D.; Lucas, D.M.; et al. The interleukin-3 receptor CD123 targeted SL-401 mediates potent cytotoxic activity against CD34(+)CD123(+) cells from acute myeloid leukemia/myelodysplastic syndrome patients and healthy donors. Haematologica 2018, 103, 1288–1297. [Google Scholar] [CrossRef]
- Lane, A.A.; Stein, A.S.; Garcia, J.S.; Garzon, J.L.; Galinsky, I.; Luskin, M.R.; Stone, R.M.; Winer, E.S.; Leonard, R.; Mughal, T.I.; et al. Safety and Efficacy of Combining Tagraxofusp (SL-401) with Azacitidine or Azacitidine and Venetoclax in a Phase 1b Study for CD123 Positive AML, MDS, or BPDCN. Blood 2021, 138 (Suppl. S1), 2346. [Google Scholar] [CrossRef]
- Minetto, P.; Rosellini, S.; Guolo, F.; Tedone, E.; Audisio, E.; Cattaneo, C.; Bocchia, M.; Fracchiolla, N.; Martelli, M.P.; Crea, E.; et al. Single Agent Tagraxofusp in Relapsed/Refractory CD123-Positive Acute Myeloid Leukemia: A Preliminary Analysis of Italian Gimema AML2020 Trial. Blood 2023, 142 (Suppl. S1), 2918. [Google Scholar] [CrossRef]
- Mouhayar, E.N.; Hammond, D.; Lopez-Mattei, J.; Banchs, J.; Konopleva, M.; Pemmaraju, N. Reversible Myocardial Edema Secondary to Tagraxofusp-Induced Capillary Leak Syndrome. JACC Cardio Oncol. 2021, 3, 752–755. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.G.; Montesinos, P.; DeAngelo, D.J.; Wang, E.S.; Papadantonakis, N.; Todisco, E.; Sweet, K.L.; Pemmaraju, N.; Lane, A.A.; Torres-Miñana, L.; et al. Pivekimab sunirine (IMGN632), a novel CD123-targeting antibody-drug conjugate, in relapsed or refractory acute myeloid leukaemia: A phase 1/2 study. Lancet Oncol. 2024, 25, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Kuruvilla, V.M.; Zhang, Q.; Daver, N.; Watkins, K.; Sloss, C.M.; Zweidler-McKay, P.A.; Romanelli, A.; Konopleva, M. Combining IMGN632, a Novel CD123-Targeting Antibody Drug Conjugate with Azacitidine and Venetoclax Facilitates Apoptosis In Vitro and Prolongs Survival In Vivo in AML Models. Blood 2020, 136 (Suppl. S1), 32–33. [Google Scholar] [CrossRef]
- Daver, N.; Montesinos, P.; Aribi, A.; Altman, J.K.; Wang, E.S.; Roboz, G.J.; Burke, P.W.; Gaidano, G.; Walter, R.B.; Thomas, X.; et al. Broad Activity for the Pivekimab Sunirine (PVEK, IMGN632), Azacitidine, and Venetoclax Triplet in High-Risk Patients with Relapsed/Refractory Acute Myeloid Leukemia (AML). Blood 2022, 140 (Suppl. S1), 145–149. [Google Scholar] [CrossRef]
- Tapia-Galisteo, A.; Alvarez-Vallina, L.; Sanz, L. Bi- and trispecific immune cell engagers for immunotherapy of hematological malignancies. J. Hematol. Oncol. 2023, 16, 83. [Google Scholar] [CrossRef]
- Smits, N.C.; Sentman, C.L. Bispecific T-Cell Engagers (BiTEs) as Treatment of B-Cell Lymphoma. J. Clin. Oncol. 2016, 34, 1131–1133. [Google Scholar] [CrossRef]
- Przepiorka, D.; Ko, C.W.; Deisseroth, A.; Yancey, C.L.; Candau-Chacon, R.; Chiu, H.J.; Gehrke, B.J.; Gomez-Broughton, C.; Kane, R.C.; Kirshner, S.; et al. FDA Approval: Blinatumomab. Clin. Cancer Res. 2015, 21, 4035–4039. [Google Scholar] [CrossRef]
- Kontermann, R.E. Strategies for extended serum half-life of protein therapeutics. Curr. Opin. Biotechnol. 2011, 22, 868–876. [Google Scholar] [CrossRef] [PubMed]
- Gogesch, P.; Dudek, S.; van Zandbergen, G.; Waibler, Z.; Anzaghe, M. The Role of Fc Receptors on the Effectiveness of Therapeutic Monoclonal Antibodies. Int. J. Mol. Sci. 2021, 22, 8947. [Google Scholar] [CrossRef]
- Balendran, S.; Tam, C.; Ku, M. T-Cell Engaging Antibodies in Diffuse Large B Cell Lymphoma—An Update. J. Clin. Med. 2023, 12, 6737. [Google Scholar] [CrossRef]
- Krupka, C.; Kufer, P.; Kischel, R.; Zugmaier, G.; Bögeholz, J.; Köhnke, T.; Lichtenegger, F.S.; Schneider, S.; Metzeler, K.; Fiegl, M.; et al. CD33 target validation and sustained depletion of AML blasts in long-term cultures by the bispecific T-cell–engaging antibody AMG 330. Blood 2014, 123, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Sidori, A.; Cerchione, C.; Daver, N.; DiNardo, C.; Garcia-Manero, G.; Konopleva, M.; Jabbour, E.; Ravandi, F.; Kadia, T.; Burguera, A.d.l.F.; et al. Immunotherapy in Acute Myeloid Leukemia: Where We Stand. Front. Oncol. 2021, 11, 656218. [Google Scholar]
- Westervelt, P.; Cortes, J.E.; Altman, J.K.; Long, M.; Oehler, V.G.; Gojo, I.; Guenot, J.; Chun, P.; Roboz, G.J. Phase 1 First-in-Human Trial of AMV564, a Bivalent Bispecific (2:2) CD33/CD3 T-Cell Engager, in Patients with Relapsed/Refractory Acute Myeloid Leukemia (AML). Blood 2019, 134 (Suppl. S1), 834. [Google Scholar] [CrossRef]
- Ravandi, F.; Bashey, A.; Foran, J.; Stock, W.; Mawad, R.; Short, N.; Yilmaz, M.; Kantarjian, H.; Odenike, O.; Patel, A.; et al. Phase 1 study of vibecotamab identifies an optimized dose for treatment of relapsed/refractory acute myeloid leukemia. Blood Adv. 2023, 7, 6492–6505. [Google Scholar] [CrossRef]
- Uy, G.L.; Aldoss, I.; Foster, M.C.; Sayre, P.H.; Wieduwilt, M.J.; Advani, A.S.; Godwin, J.E.; Arellano, M.L.; Sweet, K.L.; Emadi, A.; et al. Flotetuzumab as salvage immunotherapy for refractory acute myeloid leukemia. Blood 2021, 137, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Riva, C.; Vernarecci, C.; Minetto, P.; Goda, R.; Greppi, M.; Pesce, S.; Chies, M.; Zecchetti, G.; Ferro, B.; Maio, E.; et al. Harnessing Immune Response in Acute Myeloid Leukemia. J. Clin. Med. 2023, 12, 582. [Google Scholar] [CrossRef]
- Braciak, T.A.; Roskopf, C.C.; Wildenhain, S.; Fenn, N.C.; Schiller, C.B.; Schubert, I.A.; Jacob, U.; Honegger, A.; Krupka, C.; Subklewe, M.; et al. Dual-targeting triplebody 33-16-123 (SPM-2) mediates effective redirected lysis of primary blasts from patients with a broad range of AML subtypes in combination with natural killer cells. OncoImmunology 2018, 7, e1472195. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Bueso-Ramos, C.; Dinardo, C.; Estecio, M.R.; Davanlou, M.; Geng, Q.-R.; Fang, Z.; Nguyen, M.; Pierce, S.; Wei, Y.; et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia 2014, 28, 1280–1288. [Google Scholar] [CrossRef]
- Stahl, M.; Goldberg, A.D. Immune Checkpoint Inhibitors in Acute Myeloid Leukemia: Novel Combinations and Therapeutic Targets. Curr. Oncol. Rep. 2019, 21, 37. [Google Scholar] [CrossRef] [PubMed]
- Perna, F.; Espinoza-Gutarra, M.R.; Bombaci, G.; Farag, S.S.; Schwartz, J.E. Immune-Based Therapeutic Interventions for Acute Myeloid Leukemia. Cancer Treat Res. 2022, 183, 225–254. [Google Scholar] [PubMed]
- Chan, T.; Yarchoan, M.; Jaffee, E.; Swanton, C.; Quezada, S.; Stenzinger, A.; Peters, S. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann. Oncol. 2019, 30, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Salik, B.; Smyth, M.J.; Nakamura, K. Targeting immune checkpoints in hematological malignancies. J. Hematol. Oncol. 2020, 13, 111. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Ma, L.; Zhang, X.; Huang, L.; Wei, J. Targeting PD-1/PD-L1 pathway in myelodysplastic syndromes and acute myeloid leukemia. Exp. Hematol. Oncol. 2022, 11, 11. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.P.; Takimoto, C.H.; Feng, D.D.; McKenna, K.; Gip, P.; Liu, J.; Volkmer, J.-P.; Weissman, I.L.; Majeti, R.; Gibb, P. Therapeutic Targeting of the Macrophage Immune Checkpoint CD47 in Myeloid Malignancies. Front. Oncol. 2019, 9, 1380. [Google Scholar] [CrossRef] [PubMed]
- Kikushige, Y. Clinical roles of TIM-3 in myeloid malignancies and its importance in cellular therapy. Blood Cell Ther. Off. J. APBMT 2022, 5, S1–S5. [Google Scholar]
- Ricciuti, B.; Wang, X.; Alessi, J.V.; Rizvi, H.; Mahadevan, N.R.; Li, Y.Y.; Polio, A.; Lindsay, J.; Umeton, R.; Sinha, R.; et al. Association of High Tumor Mutation Burden in Non–Small Cell Lung Cancers with Increased Immune Infiltration and Improved Clinical Outcomes of PD-L1 Blockade Across PD-L1 Expression Levels. JAMA Oncol. 2022, 8, 1160–1168. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Chen, Y.; Wang, C. Beyond Tumor Mutation Burden: Tumor Neoantigen Burden as a Biomarker for Immunotherapy and Other Types of Therapy. Front. Oncol. 2021, 11, 672677. [Google Scholar] [CrossRef]
- Daver, N.; Konopleva, M.; Maiti, A.; Kadia, T.M.; DiNardo, C.D.; Loghavi, S.; Pemmaraju, N.; Jabbour, E.; Montalban-Bravo, G.; Tang, G.L.; et al. Phase I/II Study of Azacitidine (AZA) with Venetoclax (VEN) and Magrolimab (Magro) in Patients (pts) with Newly Diagnosed Older/Unfit or High-Risk Acute Myeloid Leukemia (AML) and Relapsed/Refractory (R/R) AML. Blood 2021, 138 (Suppl. S1), 141–144. [Google Scholar] [CrossRef]
- Zeidan, A.M.; Boss, I.W.; Beach, C.L.; Copeland, W.B.; Thompson, E.G.; Fox, B.A.; Hasle, V.E.; Ogasawara, K.; Cavenagh, J.; Silverman, L.R.; et al. A randomized phase 2 trial of azacitidine with or without durvalumab as first-line therapy for higher-risk myelodysplastic syndromes. Blood Adv. 2022, 6, 2207–2218. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Sasaki, K.; Montalban-Bravo, G.; Daver, N.G.; Jabbour, E.J.; Alvarado, Y.; DiNardo, C.D.; Ravandi, F.; Borthakur, G.; Bose, P.; et al. A Phase II Study of Nivolumab or Ipilimumab with or without Azacitidine for Patients with Myelodysplastic Syndrome (MDS). Blood 2018, 132 (Suppl. S1), 465. [Google Scholar] [CrossRef]
- Willingham, S.B.; Volkmer, J.-P.; Gentles, A.J.; Sahoo, D.; Dalerba, P.; Mitra, S.S.; Wang, J.; Contreras-Trujillo, H.; Martin, R.; Cohen, J.D.; et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 6662–6667. [Google Scholar] [CrossRef] [PubMed]
- Linder, K.; Lulla, P. Myelodysplastic syndrome and immunotherapy novel to next in-line treatments. Hum. Vaccines Immunother. 2021, 17, 2602–2616. [Google Scholar] [CrossRef] [PubMed]
- Sallman, D.A.; Al Malki, M.M.; Asch, A.S.; Wang, E.S.; Jurcic, J.G.; Bradley, T.J.; Flinn, I.W.; Pollyea, D.A.; Kambhampati, S.; Tanaka, T.N.; et al. Magrolimab in Combination with Azacitidine in Patients with Higher-Risk Myelodysplastic Syndromes: Final Results of a Phase Ib Study. J. Clin. Oncol. 2023, 41, 2815–2826. [Google Scholar] [CrossRef] [PubMed]
- A Sallman, D.; Asch, A.S.; Al Malki, M.M.; Lee, D.J.; Donnellan, W.B.; Marcucci, G.; Kambhampati, S.; Daver, N.G.; Garcia-Manero, G.; Komrokji, R.S.; et al. The First-in-Class Anti-CD47 Antibody Magrolimab (5F9) in Combination with Azacitidine Is Effective in MDS and AML Patients: Ongoing Phase 1b Results. Blood 2019, 134 (Suppl. S1), 569. [Google Scholar] [CrossRef]
- Garcia-Manero, G. Current status of phase 3 clinical trials in high-risk myelodysplastic syndromes: Pitfalls and recommendations. Lancet Haematol. 2023, 10, e71–e78. [Google Scholar] [CrossRef] [PubMed]
- Stirling, E.R.; Terabe, M.; Wilson, A.S.; Kooshki, M.; Yamaleyeva, L.M.; Alexander-Miller, M.A.; Zhang, W.; Miller, L.D.; Triozzi, P.L.; Soto-Pantoja, D.R. Targeting the CD47/thrombospondin-1 signaling axis regulates immune cell bioenergetics in the tumor microenvironment to potentiate antitumor immune response. J. Immunother. Cancer 2022, 10, e004712. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Guo, X.; Ma, W. Opportunities and challenges of CD47-targeted therapy in cancer immunotherapy. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 2023, 32, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Yamada-Hunter, S.A.; Theruvath, J.; McIntosh, B.J.; Freitas, K.A.; Lin, F.; Radosevich, M.T.; Leruste, A.; Dhingra, S.; Martinez-Velez, N.; Xu, P.; et al. Engineered CD47 protects T cells for enhanced antitumour immunity. Nature 2024, 630, 457–465. [Google Scholar] [CrossRef]
- Tahk, S.; Vick, B.; Hiller, B.; Schmitt, S.; Marcinek, A.; Perini, E.D.; Leutbecher, A.; Leutbecher, C.; Leutbecher, A.; Tast, B.; et al. SIRPalpha-alphaCD123 fusion antibodies targeting CD123 in conjunction with CD47 blockade enhance the clearance of AML-initiating cells. J. Hematol. Oncol. 2021, 14, 155. [Google Scholar] [CrossRef]
- Das, M.; Zhu, C.; Kuchroo, V.K. Tim-3 and its role in regulating anti-tumor immunity. Immunol. Rev. 2017, 276, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Tan, H.; Li, Y. Targeting TIM-3 for hematological malignancy: Latest updates from the 2022 ASH annual meeting. Exp. Hematol. Oncol. 2023, 12, 62. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Zhang, N.; Rinne, M.L.; Sun, H.; Stein, A.M. Sabatolimab (MBG453) model-informed drug development for dose selection in patients with myelodysplastic syndrome/acute myeloid leukemia and solid tumors. CPT Pharmacometrics Syst. Pharmacol. 2023, 12, 1653–1665. [Google Scholar] [CrossRef] [PubMed]
- Borate, U.; Esteve, J.; Porkka, K.; Knapper, S.; Vey, N.; Scholl, S.; Garcia-Manero, G.; Wermke, M.; Janssen, J.; Traer, E.; et al. Phase Ib Study of the Anti-TIM-3 Antibody MBG453 in Combination with Decitabine in Patients with High-Risk Myelodysplastic Syndrome (MDS) and Acute Myeloid Leukemia (AML). Blood 2019, 134 (Suppl. S1), 570. [Google Scholar] [CrossRef]
- Dao, T.; Xiong, G.; Mun, S.S.; Meyerberg, J.; Korontsvit, T.; Xiang, J.; Cui, Z.; Chang, A.Y.; Jarvis, C.; Cai, W.; et al. A dual-receptor T-cell platform with Ab-TCR and costimulatory receptor achieves specificity and potency against AML. Blood 2024, 143, 507–521. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.N.; Azzi, J.; Cooper, B.W.; Deol, A.; DiPersio, J.; Koura, D.; McClune, B.; Muffly, L.S.; Mushtaq, M.U.; Narayan, R.; et al. Phase 1/2 Study of Donor-Derived Anti-CD33 Chimeric Antigen Receptor Expressing T Cells (VCAR33) in Patients with Relapsed or Refractory Acute Myeloid Leukemia after Allogeneic Hematopoietic Cell Transplantation. Blood 2023, 142, 4862. [Google Scholar] [CrossRef]
- Tambaro, F.P.; Singh, H.; Jones, E.; Rytting, M.; Mahadeo, K.M.; Thompson, P.; Daver, N.; DiNardo, C.; Kadia, T.; Garcia-Manero, G.; et al. Autologous CD33-CAR-T cells for treatment of relapsed/refractory acute myelogenous leukemia. Leukemia 2021, 35, 3282–3286. [Google Scholar] [CrossRef] [PubMed]
- Budde, L.; Song, J.Y.; Kim, Y.; Blanchard, S.; Wagner, J.; Stein, A.S.; Weng, L.; Del Real, M.; Hernandez, R.; Marcucci, E.; et al. Remissions of Acute Myeloid Leukemia and Blastic Plasmacytoid Dendritic Cell Neoplasm Following Treatment with CD123-Specific CAR T Cells: A First-in-Human Clinical Trial. Blood 2017, 130 (Suppl. S1), 811. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Chen, H.; Li, W.; Huang, T.; Zhang, W.; Ling, W.; Lai, P.; Wang, Y.; Geng, S.; et al. C-Type Lectin-Like Molecule-1 as a Biomarker for Diagnosis and Prognosis in Acute Myeloid Leukemia: A Preliminary Study. BioMed. Res. Int. 2021, 2021, 6643948. [Google Scholar] [CrossRef]
- Zhang, H.; Bu, C.; Peng, Z.; Li, G.; Zhou, Z.; Ding, W.; Zheng, Y.; He, Y.; Hu, Z.; Pei, K.; et al. Characteristics of anti-CLL1 based CAR-T therapy for children with relapsed or refractory acute myeloid leukemia: The multi-center efficacy and safety interim analysis. Leukemia 2022, 36, 2596–2604. [Google Scholar] [CrossRef]
- Mardiros, A.; Dos Santos, C.; McDonald, T.; Brown, C.E.; Wang, X.; Budde, L.E.; Hoffman, L.; Aguilar, B.; Chang, W.-C.; Bretzlaff, W.; et al. T cells expressing CD123-specific chimeric antigen receptors exhibit specific cytolytic effector functions and antitumor effects against human acute myeloid leukemia. Blood 2013, 122, 3138–3148. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.; Tasian, S.K.; Ruella, M.; Shestova, O.; Li, Y.; Porter, D.L.; Carroll, M.; Danet-Desnoyers, G.; Scholler, J.; Grupp, S.A.; et al. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood 2014, 123, 2343–2354. [Google Scholar] [CrossRef] [PubMed]
- Stevens, B.M.; Zhang, W.; Pollyea, D.A.; Winters, A.; Gutman, J.; Smith, C.; Budde, E.; Forman, S.J.; Jordan, C.T.; Purev, E. CD123 CAR T cells for the treatment of myelodysplastic syndrome. Exp. Hematol. 2019, 74, 52–63.e3. [Google Scholar] [CrossRef] [PubMed]
- Xie, D.; Jin, X.; Sun, R.; Zhang, M.; Lu, W.; Cao, X.; Guo, R.; Zhang, Y.; Zhao, M. Bicistronic CAR-T cells targeting CD123 and CLL1 for AML to reduce the risk of antigen escape. Transl. Oncol. 2023, 34, 101695. [Google Scholar] [CrossRef]
- Martínez, D.S.; Tirado, N.; Mensurado, S.; Martínez-Moreno, A.; Romecín, P.; Agüera, F.G.; Correia, D.V.; Silva-Santos, B.; Menéndez, P. Generation and proof-of-concept for allogeneic CD123 CAR-Delta One T (DOT) cells in acute myeloid leukemia. J. Immunother. Cancer 2022, 10, e005400. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.; Venugopal, S.; Ravandi, F. FLT3 mutated acute myeloid leukemia: 2021 treatment algorithm. Blood Cancer J. 2021, 11, 104. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Mao, H.; Zhang, J.; Chu, J.; Devine, S.; A Caligiuri, M.; Yu, J. Targeting FLT3 by chimeric antigen receptor T cells for the treatment of acute myeloid leukemia. Leukemia 2017, 31, 1830–1834. [Google Scholar] [CrossRef] [PubMed]
- Karbowski, C.; Goldstein, R.; Frank, B.; Kim, K.; Li, C.M.; Homann, O.; Hensley, K.; Brooks, B.; Wang, X.; Yan, Q.; et al. Nonclinical Safety Assessment of AMG 553, an Investigational Chimeric Antigen Receptor T-Cell Therapy for the Treatment of Acute Myeloid Leukemia. Toxicol. Sci. 2020, 177, 94–107. [Google Scholar] [CrossRef] [PubMed]
- Jetani, H.; Garcia-Cadenas, I.; Nerreter, T.; Thomas, S.; Rydzek, J.; Meijide, J.B.; Bonig, H.; Herr, W.; Sierra, J.; Einsele, H.; et al. CAR T-cells targeting FLT3 have potent activity against FLT3−ITD+ AML and act synergistically with the FLT3-inhibitor crenolanib. Leukemia 2018, 32, 1168–1179. [Google Scholar] [CrossRef]
- Cornelissen, J.J.; Breems, D.; van Putten, W.L.; Gratwohl, A.A.; Passweg, J.R.; Pabst, T.; Maertens, J.; Beverloo, H.B.; Kooy, M.v.M.; Wijermans, P.W.; et al. Comparative Analysis of the Value of Allogeneic Hematopoietic Stem-Cell Transplantation in Acute Myeloid Leukemia with Monosomal Karyotype Versus Other Cytogenetic Risk Categories. J. Clin. Oncol. 2012, 30, 2140–2146. [Google Scholar] [CrossRef]
- Hu, Y.; Zhang, M.; Yang, T.; Mo, Z.; Wei, G.; Jing, R.; Zhao, H.; Chen, R.; Zu, C.; Gu, T.; et al. Sequential CD7 CAR T-Cell Therapy and Allogeneic HSCT without GVHD Prophylaxis. N. Engl. J. Med. 2024, 390, 1467–1480. [Google Scholar] [CrossRef]
- Krakow, E.F.; Brault, M.; Summers, C.; Cunningham, T.M.; A Biernacki, M.; Black, R.G.; Woodward, K.B.; Vartanian, N.; Kanaan, S.B.; Yeh, A.C.; et al. HA-1-targeted T cell receptor (TCR) T cell therapy for recurrent leukemia after hematopoietic stem cell transplantation. Blood 2024, in press. [CrossRef] [PubMed]
- Döhner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and management of AML in adults: 2022 recommendations from an international expert panel on behalf of the ELN. Blood 2022, 140, 1345–1377. [Google Scholar] [CrossRef] [PubMed]
- Cornelissen, J.J.; Blaise, D. Hematopoietic stem cell transplantation for patients with AML in first complete remission. Blood 2016, 127, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Robin, M.; Porcher, R.; Ruggeri, A.; Blaise, D.; Wolschke, C.; Koster, L.; Angelucci, E.; Stölzel, F.; Potter, V.; Yakoub-Agha, I.; et al. HLA-Mismatched Donors in Patients with Myelodysplastic Syndrome: An EBMT Registry Analysis. Biol. Blood Marrow Transplant. 2019, 25, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Kröger, N.; Iacobelli, S.; Franke, G.-N.; Platzbecker, U.; Uddin, R.; Hübel, K.; Scheid, C.; Weber, T.; Robin, M.; Stelljes, M.; et al. Dose-Reduced versus Standard Conditioning Followed by Allogeneic Stem-Cell Transplantation for Patients with Myelodysplastic Syndrome: A Prospective Randomized Phase III Study of the EBMT (RICMAC Trial). J. Clin. Oncol. 2017, 35, 2157–2164. [Google Scholar] [CrossRef]
- Mina, A.; Greenberg, P.L.; Deeg, H.J. How I reduce and treat posttransplant relapse of MDS. Blood 2024, 143, 1344–1354. [Google Scholar] [CrossRef]
- Tentori, C.A.; Gregorio, C.; Robin, M.; Gagelmann, N.; Gurnari, C.; Ball, S.; Caballero Berrocal, J.C.; Lanino, L.; D’Amico, S.; Spreafico, M.; et al. Clinical and Genomic-Based Decision Support System to Define the Optimal Timing of Allogeneic Hematopoietic Stem-Cell Transplantation in Patients with Myelodysplastic Syndromes. J. Clin. Oncol. 2024, JCO2302175. [Google Scholar] [CrossRef]
- Maurer, K.; Antin, J.H. The graft versus leukemia effect: Donor lymphocyte infusions and cellular therapy. Front. Immunol. 2024, 15, 1328858. [Google Scholar] [CrossRef]
- Bar, M.; Sandmaier, B.M.; Inamoto, Y.; Bruno, B.; Hari, P.; Chauncey, T.; Martin, P.J.; Storb, R.; Maloney, D.G.; Storer, B.; et al. Donor Lymphocyte Infusion for Relapsed Hematological Malignancies after Allogeneic Hematopoietic Cell Transplantation: Prognostic Relevance of the Initial CD3+ T Cell Dose. Biol. Blood Marrow Transplant. 2013, 19, 949–957. [Google Scholar] [CrossRef]
- Giralt, S.; Hester, J.; Huh, Y.; Hirsch-Ginsberg, C.; Rondón, G.; Seong, D.; Lee, M.; Gajewski, J.; Van Besien, K.; Khouri, I.; et al. CD8-depleted donor lymphocyte infusion as treatment for relapsed chronic myelogenous leukemia after allogeneic bone marrow transplantation. Blood 1995, 86, 4337–4343. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.S.; Weisdorf, D.J.; Burns, L.J.; Slungaard, A.; Wagner, J.E.; Verneris, M.R.; Cooley, S.; Wangen, R.; Fautsch, S.K.; Nicklow, R.; et al. Lymphodepletion followed by donor lymphocyte infusion (DLI) causes significantly more acute graft-versus-host disease than DLI alone. Blood 2007, 110, 2761–2763. [Google Scholar] [CrossRef]
- Ye, Y.; Yang, L.; Yuan, X.; Huang, H.; Luo, Y. Optimization of Donor Lymphocyte Infusion for AML Relapse After Allo-HCT in the Era of New Drugs and Cell Engineering. Front. Oncol. 2021, 11, 790299. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H.J.; Mittermüller, J.; Clemm, C.; Holler, E.; Ledderose, G.; Brehm, G.; Heim, M.; Wilmanns, W. Donor leukocyte transfusions for treatment of recurrent chronic myelogenous leukemia in marrow transplant patients. Blood 1990, 76, 2462–2465. [Google Scholar] [CrossRef] [PubMed]
- Deol, A.; Lum, L.G. Role of donor lymphocyte infusions in relapsed hematological malignancies after stem cell transplantation revisited. Cancer Treat. Rev. 2010, 36, 528–538. [Google Scholar] [CrossRef]
- Schmid, C.; Labopin, M.; Nagler, A.; Bornhäuser, M.; Finke, J.; Fassas, A.; Volin, L.; Gürman, G.; Maertens, J.; Bordigoni, P.; et al. Donor Lymphocyte Infusion in the Treatment of First Hematological Relapse after Allogeneic Stem-Cell Transplantation in Adults with Acute Myeloid Leukemia: A Retrospective Risk Factors Analysis and Comparison with Other Strategies by the EBMT Acute Leukemia Working Party. J. Clin. Oncol. 2007, 25, 4938–4945. [Google Scholar]
- Minculescu, L.; Reekie, J.; Petersen, S.L.; Kornblit, B.T.; Schjoedt, I.; Andersen, N.S.; Andersen, L.P.; Fischer-Nielsen, A.; Haastrup, E.K.; Friis, L.S.; et al. Donor Lymphocyte Infusion Is a Feasible Way to Improve Survival in Patients with Acute Myeloid Leukemia and Myelodysplastic Syndromes Who Relapse after Allogeneic Stem Cell Transplantation. Acta Haematol. 2023, 147, 325–332. [Google Scholar] [CrossRef]
- Krishnamurthy, P.; Potter, V.T.; Barber, L.D.; Kulasekararaj, A.G.; Lim, Z.Y.; Pearce, R.M.; de Lavallade, H.; Kenyon, M.; Ireland, R.M.; Marsh, J.C.; et al. Outcome of Donor Lymphocyte Infusion after T Cell–depleted Allogeneic Hematopoietic Stem Cell Transplantation for Acute Myelogenous Leukemia and Myelodysplastic Syndromes. Biol. Blood Marrow Transplant. 2013, 19, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Schmid, C.; Labopin, M.; Schaap, N.; Veelken, H.; Schleuning, M.; Stadler, M.; Finke, J.; Hurst, E.; Baron, F.; Ringden, O.; et al. Prophylactic donor lymphocyte infusion after allogeneic stem cell transplantation in acute leukaemia—A matched pair analysis by the Acute Leukaemia Working Party of EBMT. Br. J. Haematol. 2019, 184, 782–787. [Google Scholar] [CrossRef]
- Akatsuka, Y. TCR-Like CAR-T Cells Targeting MHC-Bound Minor Histocompatibility Antigens. Front. Immunol. 2020, 11, 257. [Google Scholar] [CrossRef]
- Olsen, K.S.; Jadi, O.; Dexheimer, S.; Bortone, D.S.; Vensko, S.P.; Bennett, S.; Tang, H.; Diiorio, M.; Saran, T.; Dingfelder, D.; et al. Shared graft-versus-leukemia minor histocompatibility antigens in DISCOVeRY-BMT. Blood Adv. 2023, 7, 1635–1649. [Google Scholar] [CrossRef] [PubMed]
- Al Malki, M.M.; Keyzner, A.; Suh, H.C.; Matin, A.; Buonomo, E.; Wang, Y.; Abelowitz, N.; Murray, J.; Macbeath, G.; Barton, D.; et al. Initial Results of a Phase 1 Trial of TSC-100 and TSC-101, Engineered T Cell Therapies That Target Minor Histocompatibility Antigens to Prevent Relapse after Allogeneic Hematopoietic Cell Transplantation. Blood 2023, 142, 2090. [Google Scholar] [CrossRef]
- Kreutmair, S.; Pfeifer, D.; Waterhouse, M.; Takács, F.; Graessel, L.; Döhner, K.; Duyster, J.; Illert, A.L.; Frey, A.-V.; Schmitt, M.; et al. First-in-human study of WT1 recombinant protein vaccination in elderly patients with AML in remission: A single-center experience. Cancer Immunol. Immunother. 2022, 71, 2913–2928. [Google Scholar] [CrossRef] [PubMed]
- Scheibenbogen, C.; Letsch, A.; Thiel, E.; Schmittel, A.; Mailaender, V.; Baerwolf, S.; Nagorsen, D.; Keilholz, U. CD8 T-cell responses to Wilms tumor gene product WT1 and proteinase 3 in patients with acute myeloid leukemia. Blood 2002, 100, 2132–2137. [Google Scholar] [CrossRef] [PubMed]
- Kuball, J.; de Boer, K.; Wagner, E.; Wattad, M.; Antunes, E.; Weeratna, R.D.; Vicari, A.P.; Lotz, C.; van Dorp, S.; Hol, S.; et al. Pitfalls of vaccinations with WT1-, Proteinase3- and MUC1-derived peptides in combination with MontanideISA51 and CpG7909. Cancer Immunol. Immunother. 2011, 60, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, H. Wilms’ Tumor GeneWT1: Its Oncogenic Function and Clinical Application. Int. J. Hematol. 2001, 73, 177–187. [Google Scholar] [CrossRef]
- Brayer, J.; Lancet, J.E.; Powers, J.; List, A.; Balducci, L.; Komrokji, R.; Pinilla-Ibarz, J. WT1 vaccination in AML and MDS: A pilot trial with synthetic analog peptides. Am. J. Hematol. 2015, 90, 602–607. [Google Scholar] [CrossRef]
- Di Stasi, A.; Jimenez, A.M.; Eminagawa, K.; Eal-Obaidi, M.; Erezvani, K. Review of the Results of WT1 Peptide Vaccination Strategies for Myelodysplastic Syndromes and Acute Myeloid Leukemia from Nine Different Studies. Front. Immunol. 2015, 6, 36. [Google Scholar] [CrossRef]
- Alatrash, G.; Molldrem, J.J.; Qazilbas, M.H. Targeting PR1 in myeloid leukemia. Oncotarget 2018, 9, 4280–4281. [Google Scholar] [CrossRef]
- Rezvani, K.; Yong, A.S.M.; Mielke, S.; Savani, B.N.; Musse, L.; Superata, J.; Jafarpour, B.; Boss, C.; Barrett, A.J. Leukemia-associated antigen-specific T-cell responses following combined PR1 and WT1 peptide vaccination in patients with myeloid malignancies. Blood 2008, 111, 236–242. [Google Scholar] [CrossRef]
- Hinneh, J.A.; Gillis, J.L.; Moore, N.L.; Butler, L.M.; Centenera, M.M. The role of RHAMM in cancer: Exposing novel therapeutic vulnerabilities. Front. Oncol. 2022, 12, 982231. [Google Scholar] [CrossRef] [PubMed]
- Greiner, J.; Schmitt, A.; Giannopoulos, K.; Rojewski, M.T.; Götz, M.; Funk, I.; Ringhoffer, M.; Bunjes, D.; Hofmann, S.; Ritter, G.; et al. High-dose RHAMM-R3 peptide vaccination for patients with acute myeloid leukemia, myelodysplastic syndrome and multiple myeloma. Haematologica 2010, 95, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Rausch, J.; Ullrich, E.; Kühn, M.W. Epigenetic targeting to enhance acute myeloid leukemia-directed immunotherapy. Front. Immunol. 2023, 14, 1269012. [Google Scholar] [CrossRef] [PubMed]
- Holmberg-Thydén, S.; Dufva, I.H.; Gang, A.O.; Breinholt, M.F.; Schejbel, L.; Andersen, D.M.K.; Kadivar, M.; Svane, I.M.; Grønbæk, K.; Hadrup, S.R.; et al. Therapeutic Cancer Vaccination Targeting Shared Tumor Associated Antigens in Combination with Azacitidine for High Risk Myelodysplastic Syndrome—A Phase I Clinical Trial. Blood 2020, 136 (Suppl. S1), 23–24. [Google Scholar] [CrossRef]
- Keilholz, U.; Letsch, A.; Busse, A.; Asemissen, A.M.; Bauer, S.; Blau, I.W.; Hofmann, W.-K.; Uharek, L.; Thiel, E.; Scheibenbogen, C. A clinical and immunologic phase 2 trial of Wilms tumor gene product 1 (WT1) peptide vaccination in patients with AML and MDS. Blood 2009, 113, 6541–6548. [Google Scholar] [CrossRef] [PubMed]
- Naoe, T.; Saito, A.; Hosono, N.; Kasahara, S.; Muto, H.; Hatano, K.; Ogura, M.; Masunari, T.; Tanaka, M.; Usuki, K.; et al. Immunoreactivity to WT1 peptide vaccine is associated with prognosis in elderly patients with acute myeloid leukemia: Follow-up study of randomized phase II trial of OCV-501, an HLA class II-binding WT1 polypeptide. Cancer Immunol. Immunother. 2023, 72, 2865–2871. [Google Scholar] [CrossRef]
- Schmitt, M.; Schmitt, A.; Rojewski, M.T.; Chen, J.; Giannopoulos, K.; Fei, F.; Yu, Y.; Götz, M.; Heyduk, M.; Ritter, G.; et al. RHAMM-R3 peptide vaccination in patients with acute myeloid leukemia, myelodysplastic syndrome, and multiple myeloma elicits immunologic and clinical responses. Blood 2008, 111, 1357–1365. [Google Scholar] [CrossRef]
- Griffiths, E.A.; Srivastava, P.; Matsuzaki, J.; Brumberger, Z.; Wang, E.S.; Kocent, J.; Miller, A.; Roloff, G.W.; Wong, H.Y.; Paluch, B.E.; et al. NY-ESO-1 Vaccination in Combination with Decitabine Induces Antigen-Specific T-lymphocyte Responses in Patients with Myelodysplastic Syndrome. Clin. Cancer Res. 2018, 24, 1019–1029. [Google Scholar] [CrossRef]
- Avigan, D.; Rosenblatt, J. Vaccine therapy in hematologic malignancies. Blood 2018, 131, 2640–2650. [Google Scholar] [CrossRef]
- Cooley, S.; Parham, P.; Miller, J.S. Strategies to activate NK cells to prevent relapse and induce remission following hematopoietic stem cell transplantation. Blood 2018, 131, 1053–1062. [Google Scholar] [CrossRef]
- Guzman, L.G.M.; Keating, N.; Nicholson, S.E. Natural Killer Cells: Tumor Surveillance and Signaling. Cancers 2020, 12, 952. [Google Scholar] [CrossRef] [PubMed]
- Bednarski, J.J.; Zimmerman, C.; Berrien-Elliott, M.M.; Foltz, J.A.; Becker-Hapak, M.; Neal, C.C.; Foster, M.; Schappe, T.; McClain, E.; Pence, P.P.; et al. Donor memory-like NK cells persist and induce remissions in pediatric patients with relapsed AML after transplant. Blood 2022, 139, 1670–1683. [Google Scholar] [CrossRef] [PubMed]
- Mansour, A.G.; Teng, K.-Y.; Li, Z.; Zhu, Z.; Chen, H.; Tian, L.; Ali, A.; Zhang, J.; Lu, T.; Ma, S.; et al. Off-the-shelf CAR–engineered natural killer cells targeting FLT3 enhance killing of acute myeloid leukemia. Blood Adv. 2023, 7, 6225–6239. [Google Scholar] [CrossRef]
- Bajel, A.; Garciaz, S.; Desai, P.; A Huls, G.; Maiti, A.; Jongen-Lavrencic, M.; Boissel, N.; De Botton, S.; de Leeuw, D.C.; Fleming, S.; et al. First-in-Human Study of the CD123 NK Cell Engager SAR443579 in Relapsed or Refractory Acute Myeloid Leukemia, B-Cell Acute Lymphoblastic Leukemia or High Risk-Myelodysplasia: Updated Safety, Efficacy, Pharmacokinetics and Pharmacodynamics. Blood 2023, 142 (Suppl. S1), 3474. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gavrilova, T.; Schulz, E.; Mina, A. Breaking Boundaries: Immunotherapy for Myeloid Malignancies. Cancers 2024, 16, 2780. https://doi.org/10.3390/cancers16162780
Gavrilova T, Schulz E, Mina A. Breaking Boundaries: Immunotherapy for Myeloid Malignancies. Cancers. 2024; 16(16):2780. https://doi.org/10.3390/cancers16162780
Chicago/Turabian StyleGavrilova, Tatyana, Eduard Schulz, and Alain Mina. 2024. "Breaking Boundaries: Immunotherapy for Myeloid Malignancies" Cancers 16, no. 16: 2780. https://doi.org/10.3390/cancers16162780
APA StyleGavrilova, T., Schulz, E., & Mina, A. (2024). Breaking Boundaries: Immunotherapy for Myeloid Malignancies. Cancers, 16(16), 2780. https://doi.org/10.3390/cancers16162780