2. Materials and Methods
2.1. Antibody Generation and Selection
Anti-PSGL-1 antibodies were generated by mouse immunizations via standard hybridoma procedures by methods described below (LakePharma; Belmont, CA, USA). All immunization work involving animals has been performed under the institutional oversight of LakePharma according to AAALAC animal welfare guidelines. A cohort of eight 6- to 8-week-old mice (CD-1, B6;129, and NZB/w strains) were immunized with recombinant human PSGL-1 Fc fusion protein (hPSGL-1-Fc; R&D Systems, Minneapolis, MN, USA, Cat. 3345-PS; GenBank Accession AAC50061.1), consisting of amino acids Q42-V295 of the human PSGL-1 extracellular domain fused to human IgG1 Fc, and boosted with a final boost of HL-60 cells (ATCC, CCL-240), which endogenously express human PSGL-1 on their surface [
26]. Similar cohorts were immunized with a DNA expression vector encoding full-length human PSGL-1, as well as a peptide consisting of the N-terminal 19 amino acids of human PSGL-1 (amino acids 42–60) conjugated to KLH.
The popliteal, iliac, and inguinal lymph nodes and spleens were collected from immunized mice and combined, B-cell-enriched lymphocytes were electrofused with murine myeloma B cells, and the resulting hybridomas were plated at an average of 0.6 cells per well in five 384-well plates. In a primary screen, supernatants from the 384-well plates were evaluated for binding to HEK293T cells stably expressing human PSGL-1 (hPSGL-1-293T) and HL-60 cells (that endogenously express PSGL-1) by flow cytometry, and hybridomas from target positive wells were transferred to 24- or 48-well plates for further expansion. In a confirmatory screen, supernatants from the 24- or 48-well plates were re-assayed for binding to hPSGL-1-293T and HL-60 cells, and target positive wells were further expanded and cryopreserved after the collection of the supernatants. In a final tertiary screen, supernatants were assayed for binding to recombinant His-tagged human PSGL-1 extracellular domain (hPSGL-1-His) and recombinant cynomolgus monkey His-tagged PSGL-1 extracellular domain (cPSGL-1-His) by ELISA, as well as for binding to hPSGL-1-293T cells, parental 293T cells, and HL-60 cells by flow cytometry.
Hybridomas that bound human PSGL-1 expressed on cells and recombinant protein were subcloned by standard limiting dilution methods, and hybridoma supernatants were assessed for binding to hPSGL-1-His by ELISA. Two subclones, A and B, were generated for each hybridoma. Supernatants from 19L04 subclones A and B were confirmed to bind human PSGL-1 as described above. All subsequent studies were performed with the 19L04 hybridoma subclone A. Following subcloning, 19L04 was purified from 100 mL hybridoma supernatant by Protein G affinity chromatography and eluted at pH 3.5. Purified antibody was formulated in 200 mM HEPES, 100 mM sodium chloride, 50 mM sodium acetate, pH 7.0, filtered through a 0.2 µm filter, aliquoted, and frozen. 19L04 was sequenced by standard rapid amplification of cDNA ends (RACE) methods. Recombinant anti-PSGL-1 antibodies were produced in HEK293 cells at ATUM (Newark, CA, USA).
2.2. Antibody Humanization
Humanization designs were generated by CDR grafting of the murine 19L04 CDRs into high-identity human germline acceptor frameworks, followed by back-mutation of framework positions to murine 19L04 residues predicted to be structurally important for maintaining CDR loop conformation and binding to PSGL-1. The human variable heavy chain framework IGHV1-2x02 (IgG4, S228P) and the human variable light chain framework IGKV4-1x01 (Igκ) were chosen as acceptor frameworks based upon identity to murine 19L04. The 19L04 CDRs were grafted into these frameworks, and framework positions were reverted to the murine 19L04 sequence based on their potential importance in maintaining 19L04 CDR loop conformation. This humanized antibody is referred to as VTX-0811.
2.3. PSGL-1 ELISA
Experiments were performed with plate-immobilized hPSGL-1-His or PSGL-1-Fc protein. PSGL-1 was immobilized on half area plates (Corning, Corning, NY, USA, Cat #3690) at a concentration of 2 µg/mL (25 µL per well) in phosphate-buffered saline (PBS), pH 7.4 (Fisher Scientific, Cat # BP665-1) and incubated for 2 h at 37 °C. Plates were washed 3 times (150 µL per well) with wash buffer (PBS containing 0.05% Tween 20, pH 7.4) and then blocked (150 µL per well) with blocking buffer (3% bovine serum albumin (BSA) in PBS, pH 7.4) for 1 h at 37 °C. Plates were washed 3 times with wash buffer, and antibodies diluted in assay buffer (3% BSA in PBS containing 0.05% Tween 20, pH 7.4) were added (50 µL per well) into appropriate wells. The plates were incubated for 45 min at 37 °C, washed 3 times in wash buffer, and HRP-conjugated anti-mouse IgG (Jackson ImmunoResearch, Cat #115-035-071) or anti-human IgG4 (Invitrogen, Carlsbad, CA, USA, Cat #A10654) antibody added (50 µL per well) for 45 min incubation at 37 °C. Plates were washed 3 times in a wash buffer. Plates were developed with 3,3′,5,5′-tetramethylbenzidine (TMB) HRP substrate (50 µL per well) (Seracare Life Sciences Inc., Milford, MA, USA, cat # 5120-0077) at room temperature for approximately 5 min until sufficient color change was noted. The HRP-TMB reaction was stopped with 1 M sulfuric acid (50 µL per well). Plates were read on the BioTek Cytation 5 instrument at an absorbance of 450 nm.
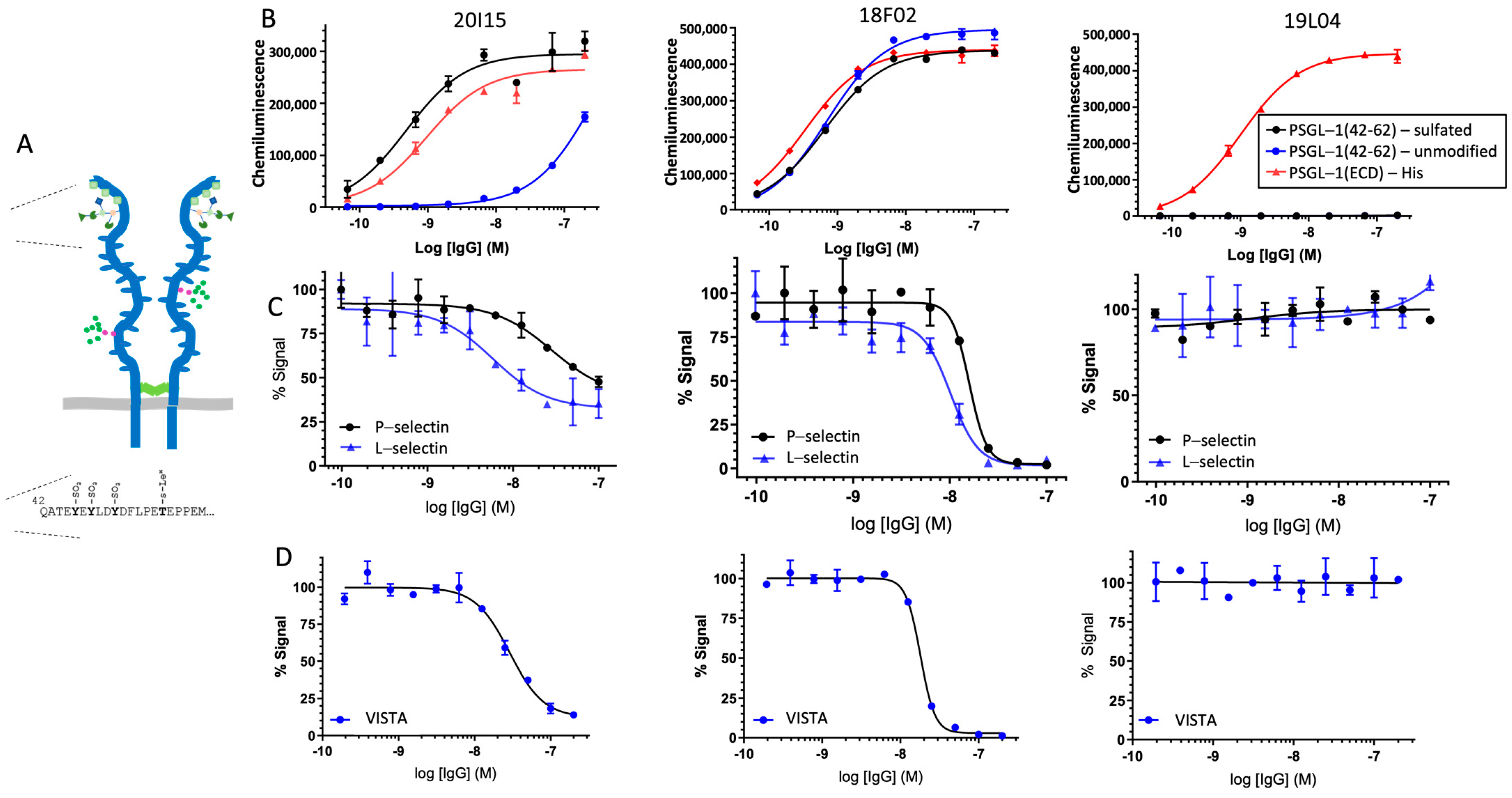
PSGL-1 peptides were synthesized with a C-terminal biotin (Biomer Technologies, Pleasanton, CA, USA), and ELISAs performed via capture on streptavidin-immobilized half-area plates, according to the conditions described above. The peptides used in the studies are as follows, where Y(SO3) denotes sulfated tyrosine:
PSGL-1(42-62)-unmodified: QATEYEYLDYDFLPETEPPEMGGGK-Biotin
PSGL-1(42-62)-sulfated: QATEY(SO3)EY(SO3)LDY(SO3)DFLPETEPPEMGGGK-Biotin.
2.4. Ligand Competition ELISAs
Human PSGL-1, Fc chimera (R&D Systems) was immobilized on half-area 96-well plates (CostarTM, Thermo Fisher, Waltham, MA, USA) at a concentration of 2 µg/mL (25 µL per well) in Dulbecco’s phosphate-buffered saline (DPBS) overnight at 4 °C. Plates were washed 3 times with 150 µL/well of wash buffer (50 mM HEPES, 125 mM NaCl, 1 mM CaCl2, containing 0.05% Tween 20, pH 7.4), then blocked with 150 µL/well of 3% bovine serum albumin (BSA; Thermo Fisher, Waltham, MA USA) in 50 mM HEPES, 125 mM NaCl, 1 mM CaCl2, pH 7.4 for 1 h at 37 °C, and then washed 3 times with wash buffer as described above. Anti-PSGL-1 antibodies and human IgG4 isotype control antibodies were titrated 2-fold from 100 nM to 0.1 nM to generate 11-point dose–response curves for each antibody in ELISA buffer (3% BSA in 50 mM HEPES, 125 mM NaCl, 1 mM CaCl2, and 0.05% Tween 20, pH 7.4). Antibodies (25 µL/well) were added to PSGL-1-coated plates in duplicate and incubated for 45 min at 37 °C. Biotinylated P- and L-selectin (R&D Systems) (25 µL per well) were then directly added to the plates at 2x their EC50 values (2 nM and 80 nM, respectively, to a final concentration of 1 nM and 40 nM, respectively) and plates incubated for another 45 min at 37 °C. Plates were washed as described previously with 150 µL per well wash buffer. The secondary antibody, Streptavidin HRP conjugate (Jackson ImmunoResearch, West Grove, PA, USA), was diluted 1:1000 in ELISA buffer for a final concentration of 1 µg/mL. A secondary antibody (25 µL/well) was added, and the plates were incubated for 45 min at 37 °C, followed by a final 3 washes with 150 µL/well wash buffer. Plates were developed with 25 µL/well of 3,3′,5,5′-Tetramethylbenzidine (TMB) substrate (SeraCare, Seracare Life Sciences Inc., Milford, MA, USA) at room temperature (RT) for approximately 5 min until sufficient color change was noted. The HRP-TMB reaction was stopped with 25 µL/well of 1 N hydrochloric acid (Thermo Fisher, Waltham, MA USA). Plates were read on the BioTek Cytation 5 instrument at an absorbance of 450 nm. A similar assay protocol was used for VISTA competition, using biotinylated VISTA (R&D Systems, Minneapolis, MN, USA), but substituting the following buffers to perform the assay pH 6.0: wash buffer (25 mM ACES, 150 mM NaCl, 1 mM CaCl2, containing 0.05% Tween 20, pH 6.0); blocking buffer (3% BSA in 25 mM ACES, 150 mM NaCl, 1 mM CaCl2, pH 6.0); ELISA buffer (3% BSA in 25 mM ACES, 150 mM NaCl, 1 mM CaCl2, and 0.05% Tween 20, pH 6.0).
2.5. Binding of VTX-0811 to PSGL-1 Expressing and PSGL-1 Knock-Out Cells
The binding of VTX-0811 and isotype control antibodies to HL-60 (ATCC, cat#CCL-240), a PSGL-1-expressing cell line, and to a HL-60 PSGL-1 CRISPR knock-out (Synthego, Redwood City, CA, USA) cell pool was evaluated using flow cytometry. HL-60 or PSGL-1 CRISPR knock-out HL-60 cells were washed twice in flow cytometry (FC) buffer (Dulbecco’s phosphate-buffered saline (DPBS; Gibco/Thermo Fisher, Waltham, MA USA), 5% fetal bovine serum (FBS; Bio Fluid Technologies, Bryn Mawr, PA, USA), 0.05% sodium azide (Ricca Chemical Company, Arlington, TX, USA)), and 50,000 cells/well (at a density of 1 × 106 cells/mL) were plated in a 96 well plate. Antibodies were diluted from a starting concentration of 120 nM to 0.16 nM in 3-fold dilutions in FC buffer. Plated cells were spun at 500× g (RCF) for 3 min and the supernatant was decanted. Antibody dilutions were added to the plated cells (50 mL/well), in duplicate. Plates were covered and incubated for 1 h at 4 °C. After incubation, plates were washed with 100 µL FC buffer. The secondary antibody, mouse huIgG-PE conjugate (Abcam, Waltham, MA, USA), was diluted 1:500 in FC buffer and 50 mL was added to each well. Plates were incubated, protected from light, for 30 min at 4 °C. Plates were washed twice with 100 µL of FC buffer. Fixation buffer (100 µL; 1% Paraformaldehyde (Alfa Aesar, Haverhill, MA, USA) in FC buffer) was added to each well. The plate was covered and protected from light until analysis. Plates were read on the Attune NxT flow cytometer attached to the Attune autosampler to generate geometric mean fluorescent intensity (gMFI) values.
2.6. Macrophage Differentiation and Polarization
Human PBMC samples or whole-blood Leukopaks were purchased from Research Blood Components, LLC. (Watertown, MA, USA). Research Blood Components follows the American Association of Blood Banks guidelines for drawing donors. The donor population consisted of healthy males and females between the ages of 18 and 65. All donors completed a uniform blood donor history questionnaire. An IRB-approved consent form was obtained from each donor giving permission to collect their blood and use or sell it at the Research Blood Components discretion for research purposes. Confidentiality and donor identification were assured. For more information, please see
https://www.researchbloodcomponents.com/about-us (accessed on 30 May 2022).
PBMCs were isolated from leukopaks as per standard methods such as (Protocol for Leukopak Processing and Washing for Downstream Cell Isolation|STEMCELL Technologies). Monocytes were enriched from the PBMCs using a Stemcell monocyte negative selection kit (Stemcell Technologies, Vancouver, BC, Canada, Cat # 19058) following the manufacturer’s recommendation. Enriched monocytes were centrifuged at 300× g for 5 min at RT, and the cell pellet was resuspended in assay media (Iscove’s Modified Dulbecco’s Medium (IMDM) containing 10% FBS) to a final concentration of 4 × 105 cells/mL.
To differentiate and polarize monocytes into M2c macrophage, enriched monocytes from each donor (5 donors total; 1 mL of 4 × 105 cells/mL per well) were added to six 24-well plates and incubated at 37 °C overnight (Day 0). Twenty-four hours after plating monocytes (Day 1), the media was aspirated and 1 mL of fresh assay media containing 50 ng/mL human macrophage colony-stimulating factor (M-CSF; Biolegend, San Diego, CA, USA, Cat #574804) was added per well. The cells were incubated at 37 °C for 72 h. On Day 4, 500 µL of assay media was removed and replaced with 500 µL fresh assay media containing M-CSF, and the cells were incubated at 37 °C for an additional 48 h. On Day 6 of the assay, differentiated M2 cells were polarized to M2c macrophages. Media was removed from cells and replaced with 1 mL of assay media containing 50 ng/mL M-CSF and 10 ng/mL of interleukin-10 (IL-10, Biolegend, San Diego, CA, USA, Cat #574004), and the cells were then incubated at 37 °C for 24 h.
2.7. Macrophage Functional Assay
To evaluate the effect of 19L04c on M2c function, on Day 7, M2c macrophages from 6 donors were treated with 19L04c or isotype control at 10 µg/mL in the 24-well format for 30 min at 37 °C. The cells were then activated with 100 ng/mL LPS (InvivoGen, San Diego, CA, USA, Cat #tlrl-eblps) in assay media containing 50 ng/mL M-CSF and 10 ng/mL IL-10 and further incubated at 37 °C for 24 h. After the incubation period, the plates were centrifuged at 500× g for 5 min at RT, and the supernatants were collected and stored at −80 °C until assessment for mediator production using a cytokine 25-plex human Luminex panel (Invitrogen, Carlsbad, CA, USA, Cat #LHC0009M).
2.8. Functionality in Multi-Cellular Assays
To demonstrate that VTX-0811 activity on macrophages can be translated into a coordinated immune response within a more complex system, two assays were established. In the first assay, human PBMCs were stimulated with Staphylococcus Enterotoxin Type B (SEB), which cross-links MHC-II expressed on antigen-presenting cells with the T-cell receptor on T cells. For the second assay, we established a mixed lymphocyte response assay using M2 macrophages and allogeneic T cells.
For the SEB-PBMC assay, 2 × 105 PBMCs (100 µL) in complete media (RPMI containing 10% fetal bovine serum (FBS), 1X non-essential amino acids, 1 mM sodium pyruvate, 10 mM HEPES and 0.1% 2-mercaptoethanol) were aliquoted per well into a 96-well plate. 19L04c and VTX-0811 (50 µL/well) were added to PBMCs at final concentrations ranging from 60 µg/mL to 9.1 ng/mL. Complete RPMI media (50 µL/well) was also added to each well, and plates were then incubated at 37 °C for 15 min. SEB (Millipore Sigma, Burlington, MA, USA, 324798) was then added at 0.25 µg/mL final concentration per well, a pre-determined sub-optimal concentration that stimulated PBMCs without inducing a maximal stimulatory response. The plate was then incubated at 37 °C for 4 days. Following incubation, the plate was centrifuged at 350× g for 5 min at RT, and the culture supernatants were collected and stored at −80 °C until being analyzed for secreted mediators by Luminex and ELISA (chemokine (C-C motif) ligands 4 (CCL4)).
For the MLR assay, on Day 1, monocytes were isolated as described above, adjusted to 500,000 cells/mL in IMDM + 10% FBS, and 100 µL (50,000 cells) was added per well of a 96-well flat bottom plate. Twenty-four hours later (Day 2), there was a full media change with 100 µL of M0 media (IMDM + 10% FBS + 50 ng/mL M-CSF) followed by the addition of 100 µL of M0 media with 20 µg/mL 19L04c or the isotype control (10 µg/mL final concentration). On Day 5 and Day 7, there was a half media change with 100 µL of M0 media containing 10 µg/mL mAb (10 µg/mL final concentration). On Day 9, T cells were isolated from allogeneic donor PBMCs using a total CD3-negative isolation kit (Stemcell Technologies, EasySep™ Human T Cell Isolation Kit). The T cells were washed 1X with 200 µL of warm T-cell media (RMPI 1640 containing 10% FBS, 1X NEAA, 1 mM sodium pyruvate, 10 mM HEPES, and freshly added 55 µM 2βME (beta-mercaptoethanol)), resuspended at 1 × 106 cells/mL, and 100 µL of T cells were added per well to the M0 macrophages. The MLR reaction was incubated for 4 days and on Day 13, supernatant was collected for Luminex analysis of mediators, and the cells were processed for flow cytometry.
2.9. NSG-SGM3-BLT Humanized Tumor-Bearing Mouse Model
All experiments were performed in accordance with the guidelines of the Institutional Animal Care and Use Committee of the University of Massachusetts Medical School and the recommendations in the Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, National Research Council, National Academy of Sciences, 1996).
To assess the effect of 19L04 on tumor growth, anti-human PSGL-1 19L04c antibody was evaluated in a humanized NSG-SGM3-BLT mouse model of melanoma. The model was set up as previously described [
21]. In brief, 10-week-old NSG-SGM3 mice (Jackson Laboratories) were engrafted with fragments of human fetal liver and thymus under the mouse kidney capsule. Two weeks later, the mice were irradiated (100 cGy gamma) to destroy the mouse bone marrow and humanized with 2 × 10
5 human CD34
+ hematopoietic progenitor cells (HSC) to reconstitute a human immune system. The degree of humanization was assessed 8 weeks post-humanization with CD34+ HSCs by performing flow cytometry on whole blood (200 µL) with anti-mouse CD45, anti-human CD45, anti-human CD3, and anti-human CD20 antibodies. Mice were considered well-humanized and ready for studies when the degree of humanization (human CD45+ cells) was approximately 20–30% of all immune cells. All mice had quantifiable T-cell (12–95% of CD45 population) and B-cell (0.6–46% of CD45 population) populations in serum indicating maturation of human lymphocytes in the model.
At 6 weeks post HSC injection, mice were inoculated with patient-derived xenograft (PDX) melanoma cells (AV17.26) and randomized into groups after tumors grew to a palpable size of approximately 50–100 mm3. Mice were administered 10 mg/kg of 19L04c or Isotype (negative control antibody), while anti-PD-1 (Pembrolizumab) was dosed at an initial dose of 10 mg/kg and all subsequent doses at 5 mg/kg—a regimen optimized by M. Brehm and colleagues in previous experiments (personal communication). Mice were dosed twice weekly for 3.5 weeks, tumor volumes were measured using calipers, and following the last dose mice were euthanized, and tumors were harvested to evaluate infiltrating immune cell composition.
2.10. Flow Cytometry Analysis of Tumors
Explanted tumors were mechanically dissociated by chopping and grinding tumor fragments through mesh screens followed by passing through cell strainers. About 1 mL of 500 × 106 live single-cell suspension cells/mL was stained for viability using Fixable Viability Dye eFluor780 (eBioscience/Thermo Fisher, Waltham, MA USA), followed by blocking unspecific antibody binding by incubating in 1 mL Fc-blocking buffer (human FcX; Biolegend, San Diego, CA, USA) for 10 min. Following the blocking step, 100 µL of cells were combined with 100 µL of the lymphoid or myeloid staining cocktail and incubated for 1 h on ice. The lymphoid staining cocktail contained CD3, CD4, CD8, CD16, CD20, CD25, CD45, CD45RA, CD56, MHCI, PD-1, and PSGL-1. The myeloid staining cocktail contained CD3, CD11b, CD14, CD16, CD33, CD45, CD86, CD163, CD206, CSF1R (CD115), HLA-DR (MHCII), PSGL-1, and VSIG4. Stained cells were resuspended in a fixation buffer containing 0.32% paraformaldehyde (ThermoFisher Scientific). Fixed samples were acquired using an AttuneTM NxT Flow Cytometer using AttuneTM NxT Software (Version 4.2.0) for acquisition. Data were analyzed using FlowJo software version 10.7.1 (Becton Dickinson & Company, Franklin Lakes, NJ, USA). Distinct macrophage populations were defined as CD45+ CD33+ or CD14+CD11b+ gated from CD45+ viable cells. Macrophage populations were then analyzed for the presence of M1 phenotypic markers (MHCII and CD86) or M2 phenotypic markers (CD163 or CD206).
For multivariate analysis of flow cytometric data (UMAP), acquisition files in Flow Cytometry Standard (FCS) format 3.1 were exported from the flow cytometer and analyzed in the R statistical computing environment (v4.0.3). The flowCore (v2.3.1; 9) R package was used to import, compensate, and transform FCS files. For data transformation, a special class of biexponential functions known as the logicle transformation was used. Marginal events, debris, and doublets were removed using the “openCyto” (v2.2.0; 10) and “flowDensity” (v1.24.0; 11) R packages. Marginal events were defined using forward scatter (FSC) FSC-A and side scatter (SSC) SSC-A parameters. Debris was defined using FSC-A. Doublets were defined using FSC-A and FSC-W. CD45+ immune cells were gated based on the “VL3-A CD45-A” (BV605) channel for each sample, then pooled across all samples for downstream multivariate analysis. Analysis of CD45+ Tumor-Infiltrating Immune Cells Algorithms based on clustering, dimensionality reduction, and trajectory inference fully switch from the univariate/bivariate analysis to a multivariate approach. These tools consider the distribution of all markers simultaneously in the whole dataset, overcoming many manual gating limitations. Prior to dimensionality reduction, forward and side scatter parameters (FSC-A, FSC-H, FSC-W, SSC-A, SSC-H, SSC-W) were normalized using the z-score method. Dimensionality reduction using the UMAP (Uniform Manifold Approximation and Projection) algorithm was performed on a 20-dimensional data matrix (3 FSC parameters, 3 SSC parameters, 14 fluorescent markers). The “umap” (v 0.2.7.0) R package was used with the default settings. Manual gating analysis was performed on dimensional-reduced 2D UMAP maps, and phenotypically complex gated cell populations were further subjected to UMAP projection and manual gating. For each unique cell population per sample, the mean fluorescence intensity of each marker was calculated as the arithmetic mean of the logicle-transformed fluorescence intensities. Statistical analyses were performed on tumor volumes at the termination of the study using GraphPad Prism (v9.1.0). Tumor volumes from different groups were compared using two-way ANOVA tests with multiple comparisons using Bonferroni correction.
2.11. Ex Vivo Patient-Derived Human Tumor Cultures
Fresh human tumor tissue originating from the discarded sample was sent on wet ice in DMEM within no more than 24 h after surgery for culture. Tissue samples were provided by the NCI Cooperative Human Tissue Network (CHTN,
https://chtn.cancer.gov/, accessed on 13 June 2022), the National Disease Research Interchange (NDRI,
https://ndriresource.org/, accessed on 13 June 2022), or BioIVT (
https://bioivt.com/, accessed on 13 June 2022). Tissue samples were obtained using all the applicable operating policies, and procedures that protect the subjects from whom specimens are obtained. These policies and procedures are consistent with current regulations and guidance for repositories from the Office of Human Research Protections (OHRP, DHHS).
Eleven fresh primary human tumors (5 kidney, 3 uterine, 1 endometrial, 1 ovarian, and 1 omentum) were dissociated using enzymatic digestion and cultured in the presence of 19L04c, Pembrolizumab, or isotype control antibodies for 48 h. Fresh tumor tissue was placed in DMEM within 60 min of surgical resection and moved to a tissue culture-treated petri dish containing 20 mL of cold Hanks balanced salt solution (HBSS; Gibco). After removing fat, fibrous, and necrotic areas, tumors were cut into small pieces of 2–4 mm and subsequently transferred into the MACS enzyme mix and tumors were further minced. The dissociation enzyme mix was prepared from a MACS tumor dissociation kit (Miltenyi Biotec, Gaithersburg, MD, USA) by adding 200 µL of Enzyme H, 100 µL of Enzyme R, and 25 µL Enzyme A to 4.7 mL of DMEM. Samples were vortexed and incubated at 37 °C for 45 min to 1 h. Digested tumors were filtered with 40 µm cell strainers and subsequently incubated in ice-cold DMEM supplemented with 8% FBS, 2% human serum, 100 IU/mL penicillin/streptomycin, 1 mM Glutamax, 55 µM 2-ME, 1X non-essential amino acids, 1 mM sodium pyruvate, 48 ng/mL human recombinant IL-2 (100 IU/mL, assuming a specific activity of 2.1 × 10
6 IU/mg), 1X insulin/transferrin/selenium, and 4 ng/mL human M-CSF to stop the enzymatic reaction. After centrifugation at 300×
g, for 5 min at RT, cell pellets were resuspended in a culture medium and the cells were counted. About 5 × 10
5 cells/mL were plated/well into 6-well plates and incubated with 10 µg/mL of each antibody (anti-PSGL-1, isotype control, and pembrolizumab) for 48 h. Supernatants from the dissociated tumor cultures were analyzed using a cytokine 25-plex human Luminex panel (Invitrogen) according to the manufacturer’s instructions. Cytokine levels for the duplicate incubations with 19L04c, Isotype, and Pembrolizumab were averaged. The treatment effect was then assessed as percent induction of the treatment arms (19L04c or Pembrolizumab) over the isotype control arm normalized to the isotype control using the following formula:
2.12. NHP Pharmacokinetic and Toxicology Studies
PK was evaluated in monkeys treated with a single dose of VTX-0811. Eighteen naïve cynomolgus monkeys were randomly assigned to 3 groups of 3 females and 3 males per group. VTX-0811 was dosed at 3, 10, and 30 mg/kg once via 10 min intravenous (iv) infusion at a dose volume of 3 mL/kg. Blood samples for PK analysis were collected at pre-dose, 0.17 h (10 min), 6 h, 24 h, 48 h, 72 h, 96 h, 168 h, 264 h, 360 h, 456 h, 552 h, and 648 h post-dose.
Next, a 4-week toxicity and toxicokinetic study of VTX-0811 was performed. VTX-0811 was administered once weekly for 4 consecutive weeks (on Days 1, 8, 15, 22, and 29) via i.v. infusion (30 min at a dose volume of 10 mL/kg) to investigate the reversibility, progression, and/or potential delayed effects during a 4-week recovery period. Naïve monkeys (n = 40) were randomly assigned to 4 groups at 0, 25, 75, and 200 mg/kg/dose. Each group had 5 F and 5 M monkeys with 3 F and 3 M monkeys in each group being sacrificed on Day 30, while 2 F and 2 M monkeys from each group were maintained through the recovery phase, being sacrificed on Day 58 following a 29-day untreated period.
To assess the RO levels of VTX-0811 on peripheral cells after VTX-0811 treatment, a qualified RO method (MQ 99205-201251) was used to determine the RO of VTX-0811 binding to PSGL-1 on the surface of T cells in whole blood in terms of antibody-binding capacity.
Parameters for toxicity evaluations included mortality, clinical observations, body weights and body weight changes, food consumption (qualitative), Draize evaluation at the injection site, body temperature, blood pressure, electrocardiogram (ECG), ophthalmology, clinical pathology (clinical chemistry, hematology, coagulation, and urinalysis), absolute and relative organ weights, gross pathology, and histopathology evaluation.
Blood samples for TK were collected during Week 1, on Day 15, and during Week 4; for cytokine analysis once prior to the first dosing, 2 h post first dosing, 24 h post first dosing, 2 h post last dosing, and on Day 58; for complement (C3 and C4) and globulin (IgA, IgG, and IgM) analysis once prior to the first dosing, 24 h post first dosing and prior to necropsy; for immunophenotyping once prior to the first dosing, 24 h post first dosing and prior to necropsy.
Immunophenotyping was assessed by flow cytometry (FCM). The percentages and absolute numbers of lymphocyte subpopulations of total T cells (CD45+CD16–D3+), cytotoxic T (Tc) cells (CD45+CD16–CD3+CD4–CD8+), double-positive (DP) T cells (CD45+CD16–CD3+CD4+CD8+), double-negative (DN) T cells (CD45+CD16–CD3+CD4–CD8–), helper T cells (CD45+CD16–CD3+CD4+CD8–), B cells (CD45+CD16–CD20+), and NK cells (CD45+CD3–CD16+) in the peripheral blood from cynomolgus monkeys.
This study was conducted in compliance with U.S. FDA Good Laboratory Practice (GLP) Regulations for Nonclinical Laboratory Studies (21 CFR Part 58) and National Medical Products Administration (NMPA) Good Laboratory Practice (GLP), No. 34 (1 September 2017). The characterization of the test article was analyzed under non-GMP conditions. The manufacture of the Test Article and Control Article, VTX-0811 injection, and VTX-0811 injection placebo, respectively, were GMP-compliant.
2.13. Naïve Unstimulated PBMCs Assay
The assay was based on features identified to improve assay sensitivity [
27,
28,
29] for the human CD28 superagonist, TGN1412. Based on these modifications, the agonist potential of VTX-0811 was evaluated as an immobilized or soluble antibody using short-term pre-cultured PBMCs. TGN1412 was used as a positive control in both assay formats, and an isotype control IgG4 antibody (ATUM-produced control anti-respiratory syncytial virus (RSV) antibody based on the palivizumab sequence on an IgG4 backbone) was included as a negative control for VTX-0811.
To prepare the pre-cultured PBMCs, 200 µL (2 × 105 cells) of the PBMCs were aliquoted per well into a 96-well round bottom tissue culture plate, and the cells were cultured for 48 h at 37 °C. After incubation, on Day 3, the PBMCs were harvested, washed with Dulbecco’s phosphate-buffered saline (DPBS), centrifuged at 350× g for 5 min at RT, and resuspended at 1 × 107 cells/mL in complete RPMI media.
For the soluble assay format, on Day 3, 100 µL (1 × 106 cells) of the pre-cultured PBMCs was aliquoted per well in triplicate into a 96-well round bottom plate. VTX-0811 and the IgG4 isotype control antibody were diluted to 2× the final concentrations of 1000, 10, 1, 0.1, and 0.01 mg/mL, and the positive control anti-CD28 antibody was diluted to 2× the final concentrations of 50, 10, and 1 µg/mL. All antibodies were diluted in complete RPMI media, and 100 µL per well of the 2× concentration of each antibody was added to the PBMCs with a final volume in the plate of 200 µL/well. The cells were incubated at 37 °C/5%CO2 for 48 h.
For the plate-bound assay format, on Day 2, tissue culture treated, flat bottom, 96-well plates were coated with 100 µL/well of F(ab′)2 goat anti-human IgG, IgM (H + L) (Invitrogen, Carlsbad, CA, USA, Cat #16-5099-85) in DPBS at a final concentration of 10 µg/mL, and the plates were incubated at 4 °C overnight. After incubation, on Day 3, the F(ab′)2 goat anti-human IgG, IgM (H + L) coated plates were washed twice with 200 µL/well DPBS; 100 µL per well of VTX-0811 and the IgG4 isotype control antibody were added at final concentrations of 1000, 10, 1, 0.1, and 0.01 mg/mL, and the TGN1412 antibody was added at final concentrations of 50, 10, and 1 µg/mL (100 µL/well). All antibodies were diluted in complete RPMI media. The plates were incubated at 37 °C/5% CO2 for 2 h, and then 100 µL (1 × 106 cells) per well of the pre-cultured PBMCs was then added into the antibody-coated, and further incubated at 37 °C for 48 h.
For both assay formats, on Day 5, the cells were centrifuged at 500× g for 5 min at RT, and the supernatants were collected into new 96-well plates and stored at −80 °C until analysis of cytokine levels using a cytokine 25-plex human Luminex panel (Invitrogen, Carlsbad, CA, USA, Cat #LHC0009M). Raw data were generated in FlexMap3D software (Updated in May, 2021) and then exported to Microsoft Excel (Version 2205). Compiled data graphs were then generated in GraphPad Prism for final analysis.
2.14. CD3-Stimulated T-Cell Assay
Tissue culture plates were pre-coated with 5 µg/mL anti-CD3 (Thermo Fisher Scientific, Cat #16-0031-82) in PBS at 4 °C overnight, then removed. T cells were added at a density of 5 × 105/mL in 10% FCS RPMI medium and incubated at 37 degrees for 3 days with anti-CD28 antibody at 5 µg/mL (Thermo Fisher Scientific, Waltham, MA, USA Cat # 16-0289-81). 19L04c or the isotype control was used at 10 µg/mL final concentration and added to the culture with an anti-CD28 antibody. At the end of 3 days, cells were stained for analysis by flow cytometry, and supernatants were collected to be analyzed by ELISA. For flow cytometry, the cells were labeled with a viability dye (Fixable Viability Dye eFluor780 (eBioscience/Thermo Fisher Scientific, Waltham, MA, USA) as per the manufacturer’s recommendation), washed in FACS buffer, and 5 µL of FcX human blocking buffer (5 µL per well, BioLegend, San Diego, CA, USA Cat # 422301) was added per well for 10 min at RT. This step was followed by the addition of 50 µL of the staining panel (CD45, CD3, CD8, and CD69, BioLegend, San Diego, CA, USA). The cells were then washed twice with FACS buffer, resuspended in FACS buffer, and the data were acquired on the Attune flow cytometer. Data are presented as the % CD69+CD8+ T cells from viable total CD3 cells.
2.15. Phagocytosis Assay
To evaluate the effect of anti-PSGL-1 blockade on phagocytosis, M2c macrophages were treated with 19L04c prior to co-incubation with labeled tumor cells. Human M2c macrophages were generated as described above.
SK-MEL-5 was plated in the RMPI/10% FBS on Day 1 and split on Day 3 into multiple T75 flasks and grown to 80–90% confluency in the flasks. Non-adherent cells were removed by washing the cells with 5 mL of PBS, and adherent cells were detached from the plastic with Trypsin/EDTA, transferred to a 50 mL conical, centrifuged for 5 min at 350× g at RT, washed with RMPI/10% FBS, and resuspended in 5 mL of RMPI/10% FBS. Cells were then counted and labeled with pHrodo Red stain by adding 90 µL of pHrodo stock (prepared as per the manufacturer’s recommendation, ThermoFisher Scientific, Waltham, MA, USA, Cat # P35372) to SK-MEL-5 at 1 × 106 cells/mL for a final concentration of 120 ng/mL and incubating the cells for 30 min on plate rotator at RT. Following the 30 min incubation, 10 mL of PBS was added, the cells were then centrifuged, washed with PBS, and resuspended in 5 mL of RMPI/10% FBS. During the 30 min incubation period of pHrodo Red labeling of the SK-MEL-5 cells, test articles and control antibodies were added to the M2c macrophages at 10 µg/mL in the 24-well differentiation/polarization plates.
For the phagocytosis assay, the treated M2c macrophages were co-cultured with the pHrodo Red labeled SK-MEL-5 cells. About 500 µL of pHrodo-labeled SK-MEL-5 cells at 8 × 105 cells/mL (400,000 cells) was added to the macrophages. The macrophages were not detached and counted after the differentiation/polarization step; therefore, the ratio of tumor cells to macrophages was based on the original 400,000 monocytes per well and in a 500 µL volume to obtain a 1:1 ratio. The M2c/SK-MEL-5 co-culture plate was incubated at 37 °C for 2 h to allow the phagocytosis of the labeled cancer cells.
To evaluate the degree of phagocytosis, the media was removed from wells containing the M2c/SK-MEL-5 cells, and the cells were washed 1× with RMPI/10% FBS per well. The plates were placed on ice to slow/stop phagocytosis, the cells were gently scrapped to release them from the plate, and the cell suspension was pipetted into a 1 mL 96-well deep well plate, centrifuged and resuspended in 200 µL per well of FACS buffer. The cells were then incubated with an FcX blocking buffer (5 µL per well, BioLegend, San Diego, CA, USA, Cat # 422301), labeled with a viability dye (Fixable Viability Dye eFluor780, eBioscience, Cat # 65-0865-14 as per the manufacturer’s recommendation), washed with FACS buffer, and 5 µL of FcX human blocking buffer was added per well for 10 min at RT. This step was followed by the addition of 50 µL of either the myeloid staining panel (CD45, CD86, live/dead-viability dye from BioLegend, San Diego, CA, USA and CD163 from R&D Systems, Minneapolis, MN, USA) or the isotype control or FMO stain and incubated for 1 h on ice. After the incubation, 300 µL of FACS buffer was added per well and the cells were centrifuged, washed with FACS buffer, resuspended in 200 µL of fixation buffer per well, and immediately run on the Attune flow cytometer. Phagocytosis of the cancer cells was measured based on CD163+SK-MEL-5+ double positivity. CD163 is a marker of M2 macrophages and SK-MEL-5 positivity is measured by the presence of pHrodo dye in CD163+ macrophages due to the phagocytosis of the labeled SK-MEL-5 cells.
2.16. Neutrophil Activation Assay
Blood from healthy human donors was collected in acid-citrate dextrose (ACD) tubes and processed within 2–3 h of the blood draw. About 490 µL of whole blood was pipetted per well into a 96-deep well plate (ThermoFisher, Waltham, MA, USA, Cat #260252); 5 µL of stimulant or diluted antibody was added per well to the appropriate wells for a total volume of 500 µL. The stimulant fMLP (200 mM stock from Sigma Aldrich, Burlington, MA, USA Cat #F3506) was diluted in PBS and used at a concentration of 0.24 µM, a concentration determined from a previous neutrophil activation assay as the EC80. Antibodies were added at a final concentration of 10 μg/mL, 1 μg/mL, or 0.1 μg/mL. To understand the effect of the antibodies on naïve neutrophils, the cells were treated with the antibodies and incubated for 1 h without stimulation (No stimulation condition). To evaluate the effect of the antibodies on the ability of neutrophils to be activated, the cells were treated with the antibodies for 1 h and then stimulated for 15 min at 37 °C (Pre-stimulation). To evaluate the effect of the antibodies on activated neutrophils, cells were stimulated for 15 min at 37 °C and then treated with the antibodies and incubated at 37 °C for an additional 1 h (Post-stimulation).
At the completion of the assay period, the assay plate was placed on ice to stop the reaction, and the blood suspension was mixed with a 300 µL multi-channel pipette set to 160 µL. About 40 μL of the stimulated whole blood was added to a V-bottom 96-well plate (Corning, Glendale, AZ, USA, Cat. #3797) containing 160 μL of ice-cold neutrophil wash buffer (NWB: 2.5 g BSA (0.25%), 10 mL of Hepes, 1.5% dextrose (~8 mM), brought to liter with 1× PBS without Ca2+ and Mg2+) for staining. The blood and NWB suspension was mixed by setting a pipette to 50 μL and pipetting up and down 3 times, centrifuged at 1200 rpm for 5 min, discarded supernatant, added 50 μL viability dye (Invitrogen, Waltham, MA, USA, Cat. #65-0865-14, 1:1000), and incubated for 10 min on ice followed by the addition of 200 μL of NWB per well. The plate was centrifuged at 1200 rpm for 5 min, the supernatant was discarded by pipetting, and the pellet was resuspended in 10 μL blocking buffer (Human TruStain FcX Biolegend, San Diego, CA, USA, Cat #422302 diluted 1:20 in FACS staining buffer) and incubated for 10 min on ice. Next, a 60 µL staining cocktail (CD66b, CD45, CD15 CXCR2, CD11b CD63 CD62L) or FMOs or isotype cocktail was added per well, and the plate was incubated for 20 min on ice followed by the addition of 120 μL NWB to each well and gentle mixing. The plate was centrifuged, the supernatant discarded by pipetting, and the cells were washed with another 200 μL NWB. After centrifugation, 100 μL of a secondary antibody (Invitrogen, Waltham, MA, USA, Cat. #A55749; diluted 1:10) was added per well, and the cells were incubated for 10 min on ice followed by the addition of 120 μL NWB to each well. The cells were centrifuged and washed with 200 μL NWB as above and then resuspended in 200 μL of 1× Lysis buffer (Becton Dickinson, Franklin Lakes, NJ, USA, Cat #349202 1:10 in ddH20) and incubated for 10 min at room temperature. The cells were again centrifuged and washed 2× with 200 μL each time of NWB, resuspended in 200 μL of NWB, and run on attune. Data are presented as the MFI of the indicated marker (CD11b, CD62L, or CXCR2) on viable CD66b+CD15+ neutrophils.
Note that there is no fixation step and that a positive control for cell death was prepared by adding 100 µL of viability dye to a well containing whole blood, and the plate was placed at −20 °C for 10 min and then maintained at RT and subjected to the lysis step as described above.
4. Discussion
A clinically diagnosed tumor has overcome multiple resistance mechanisms with the biggest one of avoiding immune response to itself under conditions of smoldering tissue damage, lack of nutrients, lack of blood flow, modified pH, and continuous attempts of the immune system to eliminate the transformed tissue. The role of macrophages in the tumor microenvironment should not be underestimated: These cells need to be controlled by the tumor to avoid the initiation of an active pro-inflammatory anti-tumor response, to prevent the initiation of active pro-inflammatory chemotaxis, the tumors need to engage macrophages in tumor niche formation, organize neo-angiogenesis. Macrophages are located deep in the tumor bed or at the perimeter of the tumor, are modulated by the tumor, and are actively involved in phagocytosing tumor cell debris, thereby presenting tumor-associated antigens but are influenced by the tumor to become suppressive macrophages. Several different approaches have been suggested to influence TAMs: depletion, activation of the do-not-eat-me signal, and repolarization [
9]. As evident from CSF-1R inhibition, depletion seems to activate CSF-1 production and quick repopulation [
45]. In addition, the depletion approach robs the tumor of many antigen-presenting cells instrumental in initiating a productive anti-tumor immune response. Activation of the do-not-eat-me signal has led to clinical success when used in combination with antibodies targeting a tumor antigen such as HER2, CD33, or CD20, demonstrating that an additional push towards a pro-inflammatory response delivered by the tumor-targeting antibody is needed in addition to stimulating phagocytic properties [
46,
47]. Specific repolarization approaches are only now entering mid-stage clinical development, with very exciting clinical data from the MK-3840 clinical trial [
25], demonstrating responses in patients who have failed all available therapies, including PD-1/PD-L1 blockade. Clinical responses in solid cancer patients have also been shown for the Clevel-1 targeting antibody Bexmarilimab that induces macrophage repolarization [
48].
We have previously demonstrated that PSGL-1 is an inhibitory checkpoint expressed on suppressive human TAMs and M2 macrophages, which promote a pro-tumorigenic state [
21]. The critical role of PSGL-1 in maintaining the macrophages in a suppressed state was shown by the siRNA knockdown of PSGL-1 on human macrophages. Furthermore, attenuating PSGL-1 activity led to repolarization of the macrophages towards an M1 phenotype, an important quality of an anti-tumor microenvironment [
21].
In this study, we identified an antibody that mimics the functional effect of siRNA knockdown of PSGL-1. Various immunization methods were utilized to make mouse hybridomas including a recombinant full-length PSGL protein, N-terminal peptide, or DNA encoding the full-length protein to yield a broad set of antibodies recognizing multiple epitopes on PSGL-1 molecule. These various methods yielded over 7500 hybridomas, from which 11 representing multiple epitope bins determined by antibody cross-blocking experiments by ForteBio Octet and ELISA, where antibodies that cross-compete for PSGL-1 binding are binned together. Representative antibodies from these 11 hybridomas were sub-cloned and subjected to secondary screening. Based on binding properties to the recombinant PSGL-1 and primary cells and the ability to induce GM-CSF from antibody-treated M2 cells, clone 19L04 was identified as a lead for further development.
19L04c is a chimeric antibody that consists of the murine 19L04 variable regions and human IgG4 (S228P) heavy and kappa light chain constant region sequences. 19L04c bound to recombinant human PSGL1-His EC50 of 0.449 mM. Functionally, 19L04c was shown to repolarize M2-suppressive macrophages towards a potent pro-inflammatory state characterized by secretion of TNFa, IL-1b, IL6, and GM-CSF. This effect on macrophages translated into complex immune response systems including the SEB-PBMC and MLR assays, where 19L04c induced the secretion of the same pro-inflammatory mediators secreted from 19L04c treated human M2c macrophages. Furthermore, in the complex immune cell assays, we also detected effector T-cell mediators such as IL-2 in the SEB-PBMC assay and IFNg in the MLR assay suggesting the repolarized macrophages led to the activation of effector T cells. However, although PSGL-1 is also expressed and has been shown to have direct functional effects on T-cell activation [
14,
15,
17] and cell trafficking [
12,
13], neither of these attributes was impacted by PSGL-1 inhibition by our antibody (
Figure 3 and
Figure 7).
To assess if the observed effects of 19L04c in vitro on macrophages and complex immune responses translated in vivo, and if this modulation of an immune response affected tumor growth, 19L04c was evaluated in a humanized mouse model. Having shown in vitro that VTX-0811 and 19L04c have similar activity, 19L04c was used to evaluate the effect of blocking anti-PSGL-1 in vivo. 19L04c led to a significant decrease in tumor volume and a much deeper inhibition compared to Pembrolizumab. This decreased tumor volume correlated with an increase in total leukocytes and a re-direction of the tumor microenvironment to a pro-inflammatory anti-tumor state. 19L04c led to a decrease in M2-suppressive CD163 expressing macrophages and regulatory T cells and an increase in activated CD8 T effector cells in the tumors. Consistent with the in vitro data, the observed in vivo effects of 19L04c support the hypothesis that anti-PSGL-1 re-polarizes macrophages from an M2 towards an M1 functional profile and switches the tumor microenvironment to a pro-inflammatory phenotype leading to tumor growth inhibition. Similar increases in the effector to regulatory T-cell ratio were recently reported in tumors from syngeneic mouse models, where mice were treated with anti-PGSL-1 [
22]. These effects were often even more pronounced in tumors from mice co-treated with anti-PD-1. Notably, effector T-cell mediators were elevated in the anti-PSGL-1 monotherapy and combination groups. It is important to mention that a different antibody to PSGL-1, as used in the published studies by other groups, might have a different mechanism of action and can lead to different activation mechanisms. Additionally, we have shown that our antibody does not activate T cells directly under the conditions tested and is not affecting naïve or pre-activated neutrophils that express PSGL-1.
From here we further assessed translational effects using human primary tumors. 19L04c was shown to induce a pro-inflammatory profile based on multiple mediator signatures encompassing pro-inflammatory cytokine and chemokines and T-cell activation. Combining all signatures into a final total tumor inflammatory signature score indicated that 19L04c led to an even higher degree of an anti-tumor response than that observed for Pembrolizumab. It should be noted that anti-PSGL-1-treated patient-derived tumor cultures demonstrated anti-tumor immune responses and T-cell activation even in tumors not responding to Pembrolizumab—a high unmet need patient population. As PSGL-1 is present in most patients across multiple indications, this antibody has a very broad potential applicability both as monotherapy and in combination with T-cell checkpoint inhibitors.
To progress 19L04 as the lead candidate, 19L04c was subsequently humanized (VTX-0811) via CDR-grafting into high-identity human germline frameworks and back-mutating to maintain structurally important mouse framework residues and to additionally remove select potential sequence liabilities. VTX-0811 binds to recombinant human bivalent PSGL-1 with a similar binding affinity as 19L04c and shows similar dose–response curves, and the EC50s demonstrate no reduction in binding following the humanization process.
VTX-0811 did not have any appreciable agonist activity on cytokine release from human primary unstimulated PBMCs, which addresses the FDA Guidance for “Immunogenicity Assessment for Therapeutic Protein Products” targeting cell surface receptors on cytokine release. This is a necessary requirement for developing antibodies aimed to enhance anti-tumor immune response.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}