

The LDHC-STAT3 Signaling Network Is a Key Regulator of Basal-like Breast Cancer Cell Survival
 , , , ,
, , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. LDHC Silencing
2.3. RNA Extraction and Quality Assessment
2.4. Quantitative Real-Time Reverse Transcription Polymerase Chain Reaction
2.5. RNA Sequencing
2.6. Western Blotting
2.7. Clonogenic Assay
2.8. CellTiter-Glo Cell Viability Assay
2.9. Annexin V/PI Flow Cytometry
2.10. STAT3 Inhibition
2.11. Statistical Analyses
3. Results
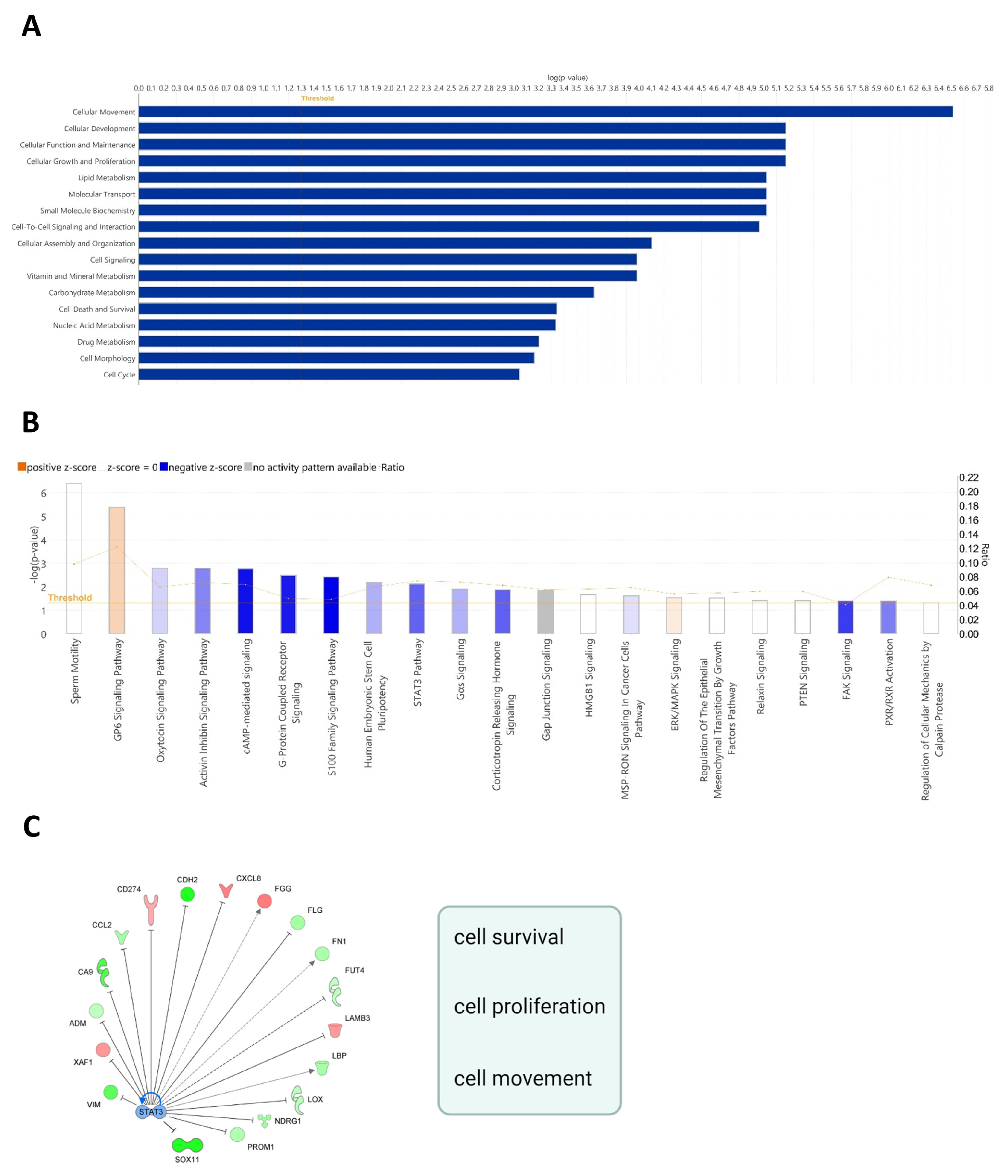
3.1. Depletion of LDHC in MDA-MB-468 Cells Induces Transcriptomic Changes That Affect Cell Cycle Progression and Tumor Cell Survival
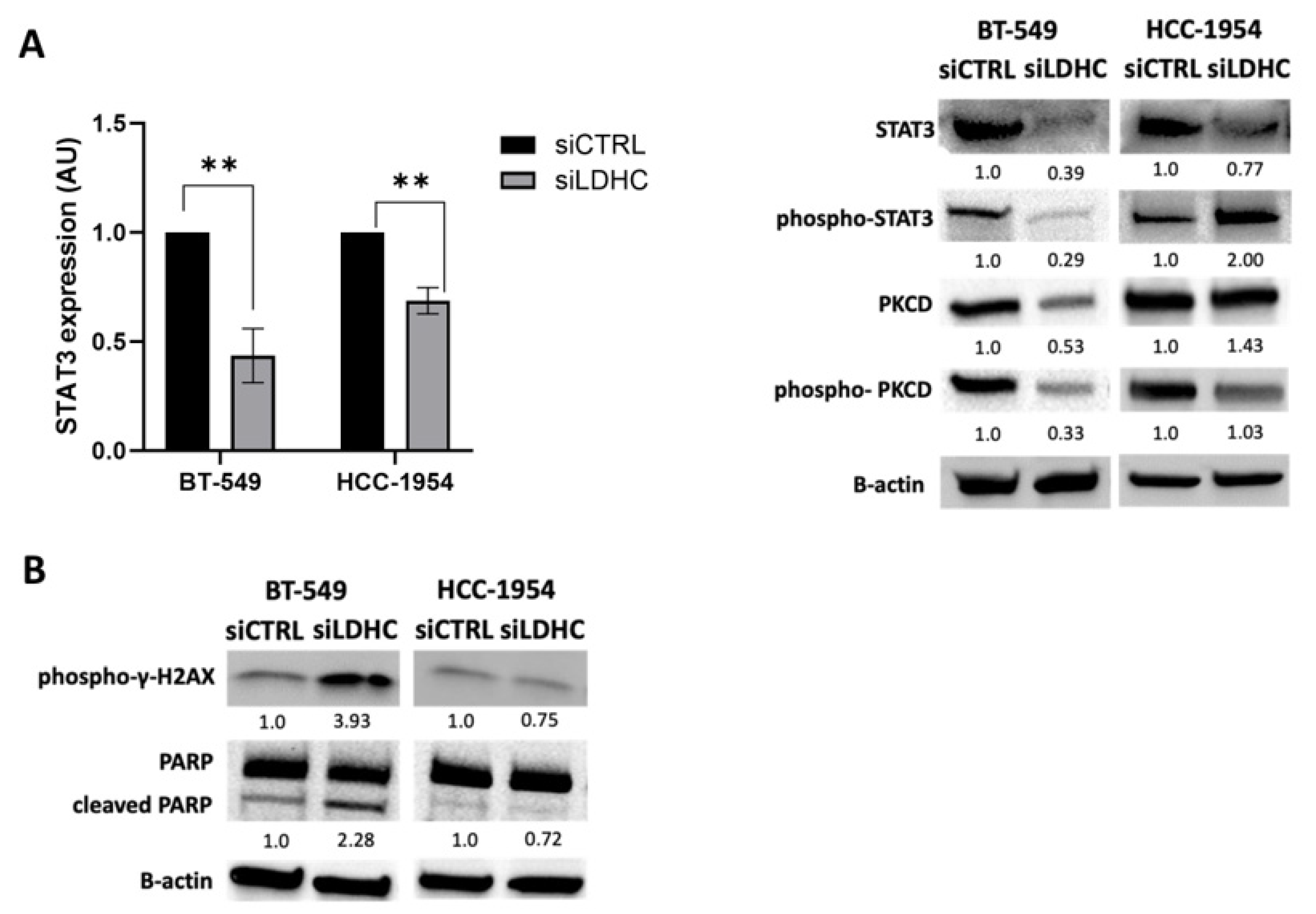
3.2. LDHC Silencing Perturbs STAT3 Pro-Oncogenic Signaling in a Cell Line-Dependent Manner
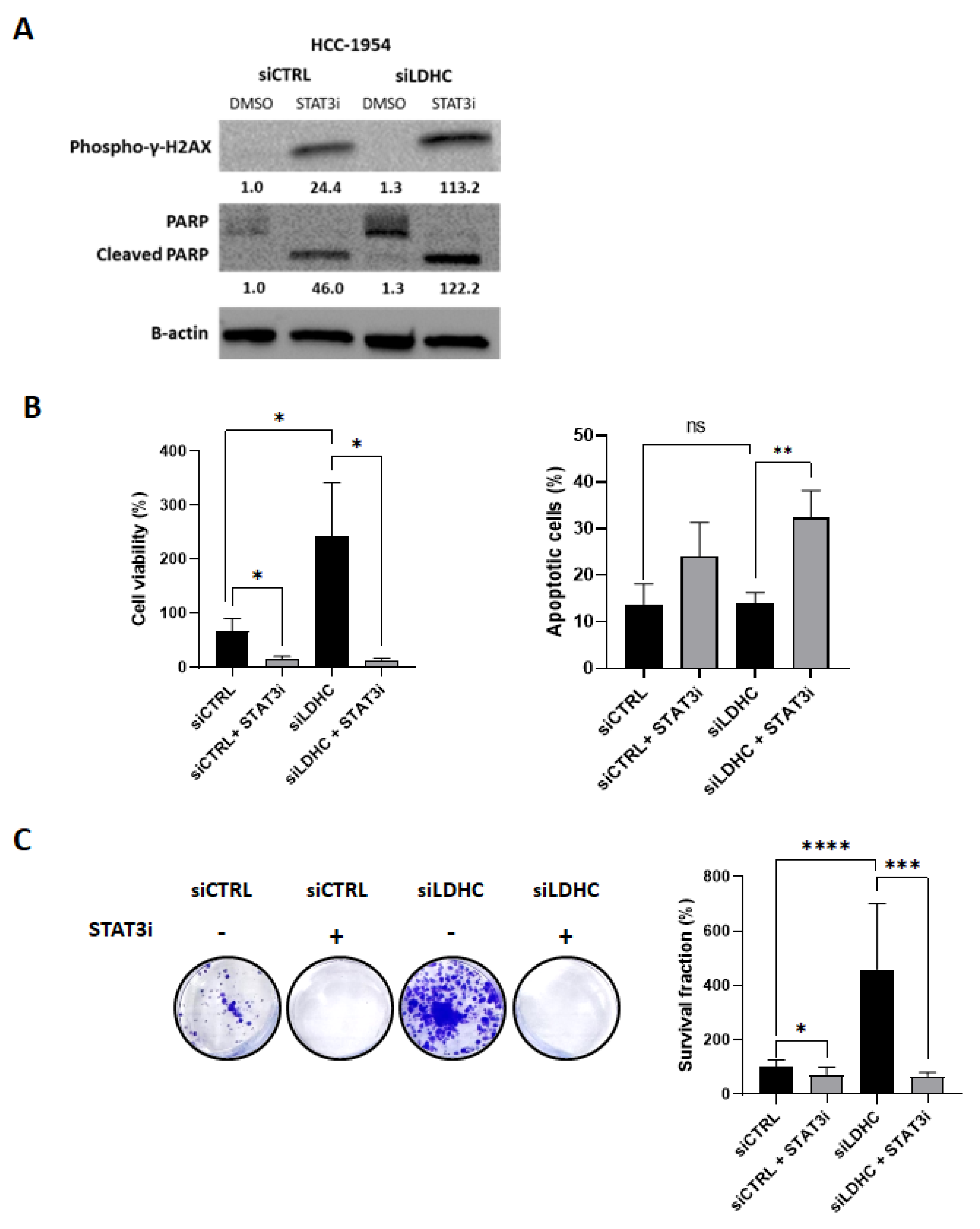
3.3. Role of LDHC-STAT3 Molecular Axis in Tumor Cellular Fitness
3.4. STAT3 Inhibition Reduces Cell Viability in Tumor Cells That Are Resistant to LDHC Silencing
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Ervik, M.; Lam, F.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.; Bray, F. Global Cancer Observatory: Cancer Today. Available online: https://gco.iarc.fr/today (accessed on 7 May 2023).
- Wang, C.; Gu, Y.; Zhang, K.; Xie, K.; Zhu, M.; Dai, N.; Jiang, Y.; Guo, X.; Liu, M.; Dai, J.; et al. Systematic Identification of Genes with a Cancer-Testis Expression Pattern in 19 Cancer Types. Nat. Commun. 2016, 7, 10499. [Google Scholar] [CrossRef]
- da Silva, V.L.; Fonseca, A.F.; Fonseca, M.; da Silva, T.E.; Coelho, A.C.; Kroll, J.E.; de Souza, J.E.S.; Stransky, B.; de Souza, G.A.; de Souza, S.J. Genome-Wide Identification of Cancer/Testis Genes and Their Association with Prognosis in a Pan-Cancer Analysis. Oncotarget 2017, 8, 92966–92977. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Caballero, O.L.; Yung, W.K.; Weinstein, J.N.; Riggins, G.J.; Strausberg, R.L.; Zhao, Q. Tumor Subtype-Specific Cancer-Testis Antigens as Potential Biomarkers and Immunotherapeutic Targets for Cancers. Cancer Immunol. Res. 2014, 2, 371–379. [Google Scholar] [CrossRef] [PubMed]
- van der Bruggen, P.; Traversari, C.; Chomez, P.; Lurquin, C.; Plaen, E.D.; den Eynde, B.V.; Knuth, A.; Boon, T. A Gene Encoding an Antigen Recognized by Cytolytic T Lymphocytes on a Human Melanoma. Science 1991, 254, 1643–1647. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.; Al-Khadairi, G.; Roelands, J.; Hendrickx, W.; Dermime, S.; Bedognetti, D.; Decock, J. NY-ESO-1 Based Immunotherapy of Cancer: Current Perspectives. Front. Immunol. 2018, 9, 947. [Google Scholar] [CrossRef] [PubMed]
- Al-Khadairi, G.; Decock, J. Cancer Testis Antigens and Immunotherapy: Where Do We Stand in the Targeting of PRAME? Cancers 2019, 11, 984. [Google Scholar] [CrossRef] [PubMed]
- Naik, A.; Yeong, J.; Decock, J. Editorial: Cancer Testis Antigens in Cancer: Recent Developments as Cancer Biomarkers and Therapeutic Targets. Front. Oncol. 2022, 12, 1075329. [Google Scholar] [CrossRef]
- Thomas, R.; Shaath, H.; Naik, A.; Toor, S.M.; Elkord, E.; Decock, J. Identification of Two HLA-A*0201 Immunogenic Epitopes of Lactate Dehydrogenase C (LDHC): Potential Novel Targets for Cancer Immunotherapy. Cancer Immunol. Immunother. 2020, 69, 449–463. [Google Scholar] [CrossRef]
- Gjerstorff, M.F.; Andersen, M.H.; Ditzel, H.J. Oncogenic Cancer/Testis Antigens: Prime Candidates for Immunotherapy. Oncotarget 2015, 6, 15772–15787. [Google Scholar] [CrossRef]
- Naik, A.; Decock, J. Targeting of Lactate Dehydrogenase C Dysregulates the Cell Cycle and Sensitizes Breast Cancer Cells to DNA Damage Response Targeted Therapy. Mol. Oncol. 2022, 16, 885–903. [Google Scholar] [CrossRef]
- Chen, L.; Wu, Q.; Xu, X.; Yang, C.; You, J.; Chen, F.; Zeng, Y. Cancer/Testis Antigen LDHC Promotes Proliferation and Metastasis by Activating the PI3K/Akt/GSK-3beta-Signaling Pathway and the in Lung Adenocarcinoma. Exp. Cell Res. 2021, 398, 112414. [Google Scholar] [CrossRef]
- Hua, Y.; Liang, C.; Zhu, J.; Miao, C.; Yu, Y.; Xu, A.; Zhang, J.; Li, P.; Li, S.; Bao, M.; et al. Expression of Lactate Dehydrogenase C Correlates with Poor Prognosis in Renal Cell Carcinoma. Tumour Biol. 2017, 39, 1010428317695968. [Google Scholar] [CrossRef]
- Cui, Z.; Chen, Y.; Hu, M.; Lin, Y.; Zhang, S.; Kong, L.; Chen, Y. Diagnostic and Prognostic Value of the Cancer-Testis Antigen Lactate Dehydrogenase C4 in Breast Cancer. Clin. Chim. Acta 2020, 503, 203–209. [Google Scholar] [CrossRef]
- Cui, Z.; Li, Y.; Gao, Y.; Kong, L.; Lin, Y.; Chen, Y. Cancer-Testis Antigen Lactate Dehydrogenase C4 in Hepatocellular Carcinoma: A Promising Biomarker for Early Diagnosis, Efficacy Evaluation and Prognosis Prediction. Aging (Albany NY) 2020, 12, 19455–19467. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, E.; Eddy, E.M.; Duan, C.; Odet, F. LDHC: The ultimate testis specific gene. J. Androl. 2010, 31, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Du, W.; Cui, Z.; Wang, L.; Yang, Z.; Zhang, H.; Lin, D. Expression of Lactate Dehydrogenase C in MDA-MB-231 Cells and Its Role in Tumor Invasion and Migration. Mol. Med. Rep. 2016, 13, 3533–3538. [Google Scholar] [CrossRef]
- Al-Khadairi, G.; Naik, A.; Thomas, R.; Al-Sulaiti, B.; Rizly, S.; Decock, J. PRAME Promotes Epithelial-to-Mesenchymal Transition in Triple Negative Breast Cancer. J. Transl. Med. 2019, 17, 9. [Google Scholar] [CrossRef] [PubMed]
- Sonnenblick, A.; Brohée, S.; Salgado, R.F.; den Eynden, G.V.; Neven, P.; Loibl, S.; Denkert, C.; Joensuu, H.; Piccart, M.; Sotiriou, C. Constitutively Activated STAT3 Is Predictive for Trastuzumab Resistance in Primary HER2 Positive Breast Cancer. Ann. Oncol. 2015, 26, iii15. [Google Scholar] [CrossRef]
- Chung, S.S.; Giehl, N.; Wu, Y.; Vadgama, J.V. STAT3 Activation in HER2-Overexpressing Breast Cancer Promotes Epithelial-Mesenchymal Transition and Cancer Stem Cell Traits. Int. J. Oncol. 2014, 44, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Verhoeven, Y.; Tilborghs, S.; Jacobs, J.; De Waele, J.; Quatannens, D.; Deben, C.; Prenen, H.; Pauwels, P.; Trinh, X.B.; Wouters, A.; et al. The Potential and Controversy of Targeting STAT Family Members in Cancer. Semin. Cancer Biol. 2020, 60, 41–56. [Google Scholar] [CrossRef]
- Wang, H.-Q.; Man, Q.-W.; Huo, F.-Y.; Gao, X.; Lin, H.; Li, S.-R.; Wang, J.; Su, F.-C.; Cai, L.; Shi, Y.; et al. STAT3 Pathway in Cancers: Past, Present, and Future. MedComm 2022, 3, e124. [Google Scholar] [CrossRef]
- Jerez, A.; Clemente, M.J.; Makishima, H.; Koskela, H.; Leblanc, F.; Peng Ng, K.; Olson, T.; Przychodzen, B.; Afable, M.; Gomez-Segui, I.; et al. STAT3 Mutations Unify the Pathogenesis of Chronic Lymphoproliferative Disorders of NK Cells and T-Cell Large Granular Lymphocyte Leukemia. Blood 2012, 120, 3048–3057. [Google Scholar] [CrossRef]
- Koskela, H.L.M.; Eldfors, S.; Ellonen, P.; van Adrichem, A.J.; Kuusanmäki, H.; Andersson, E.I.; Lagström, S.; Clemente, M.J.; Olson, T.; Jalkanen, S.E.; et al. Somatic STAT3 Mutations in Large Granular Lymphocytic Leukemia. N. Engl. J. Med. 2012, 366, 1905–1913. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Mohammad, I.S.; Liu, Z. Overview of the STAT-3 Signaling Pathway in Cancer and the Development of Specific Inhibitors. Oncol. Lett. 2020, 19, 2585–2594. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Qin, L.; Li, X. Role of STAT3 Signaling Pathway in Breast Cancer. Cell Commun. Signal. 2020, 18, 33. [Google Scholar] [CrossRef] [PubMed]
- Barry, S.P.; Townsend, P.A.; Knight, R.A.; Scarabelli, T.M.; Latchman, D.S.; Stephanou, A. STAT3 Modulates the DNA Damage Response Pathway. Int. J. Exp. Pathol. 2010, 91, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, X.; Qiu, C.; Yang, N. STAT3 Contributes to Radioresistance in Cancer. Front. Oncol. 2020, 10, 1120. [Google Scholar] [CrossRef] [PubMed]
- Barry, S.P.; Townsend, P.A.; McCormick, J.; Knight, R.A.; Scarabelli, T.M.; Latchman, D.S.; Stephanou, A. STAT3 Deletion Sensitizes Cells to Oxidative Stress. Biochem. Biophys. Res. Commun. 2009, 385, 324–329. [Google Scholar] [CrossRef]
- Wen, Z.; Zhong, Z.; Darnell, J.E. Maximal Activation of Transcription by Stat1 and Stat3 Requires Both Tyrosine and Serine Phosphorylation. Cell 1995, 82, 241–250. [Google Scholar] [CrossRef]
- Yokogami, K.; Wakisaka, S.; Avruch, J.; Reeves, S.A. Serine Phosphorylation and Maximal Activation of STAT3 during CNTF Signaling Is Mediated by the Rapamycin Target mTOR. Curr. Biol. 2000, 10, 47–50. [Google Scholar] [CrossRef]
- Zhang, X.; Blenis, J.; Li, H.C.; Schindler, C.; Chen-Kiang, S. Requirement of Serine Phosphorylation for Formation of STAT-Promoter Complexes. Science 1995, 267, 1990–1994. [Google Scholar] [CrossRef] [PubMed]
- Schuringa, J.J.; Dekker, L.V.; Vellenga, E.; Kruijer, W. Sequential Activation of Rac-1, SEK-1/MKK-4, and Protein Kinase Cdelta Is Required for Interleukin-6-Induced STAT3 Ser-727 Phosphorylation and Transactivation. J. Biol. Chem. 2001, 276, 27709–27715. [Google Scholar] [CrossRef] [PubMed]
- Turkson, J.; Bowman, T.; Adnane, J.; Zhang, Y.; Djeu, J.Y.; Sekharam, M.; Frank, D.A.; Holzman, L.B.; Wu, J.; Sebti, S.; et al. Requirement for Ras/Rac1-Mediated P38 and c-Jun N-Terminal Kinase Signaling in Stat3 Transcriptional Activity Induced by the Src Oncoprotein. Mol. Cell. Biol. 1999, 19, 7519–7528. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Zhang, T.; Kee, W.H.; Li, W.; Cao, X. Protein Kinase C Delta Associates with and Phosphorylates Stat3 in an Interleukin-6-Dependent Manner. J. Biol. Chem. 1999, 274, 24392–24400. [Google Scholar] [CrossRef]
- Abe, K.; Hirai, M.; Mizuno, K.; Higashi, N.; Sekimoto, T.; Miki, T.; Hirano, T.; Nakajima, K. The YXXQ Motif in Gp 130 Is Crucial for STAT3 Phosphorylation at Ser727 through an H7-Sensitive Kinase Pathway. Oncogene 2001, 20, 3464–3474. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naik, A.; Thomas, R.; Sikhondze, M.; Babiker, A.; Lattab, B.; Qasem, H.; Jafar, U.; Decock, J. The LDHC-STAT3 Signaling Network Is a Key Regulator of Basal-like Breast Cancer Cell Survival. Cancers 2024, 16, 2451. https://doi.org/10.3390/cancers16132451
Naik A, Thomas R, Sikhondze M, Babiker A, Lattab B, Qasem H, Jafar U, Decock J. The LDHC-STAT3 Signaling Network Is a Key Regulator of Basal-like Breast Cancer Cell Survival. Cancers. 2024; 16(13):2451. https://doi.org/10.3390/cancers16132451
Chicago/Turabian StyleNaik, Adviti, Remy Thomas, Martin Sikhondze, Abeer Babiker, Boucif Lattab, Hanan Qasem, Umar Jafar, and Julie Decock. 2024. "The LDHC-STAT3 Signaling Network Is a Key Regulator of Basal-like Breast Cancer Cell Survival" Cancers 16, no. 13: 2451. https://doi.org/10.3390/cancers16132451
APA StyleNaik, A., Thomas, R., Sikhondze, M., Babiker, A., Lattab, B., Qasem, H., Jafar, U., & Decock, J. (2024). The LDHC-STAT3 Signaling Network Is a Key Regulator of Basal-like Breast Cancer Cell Survival. Cancers, 16(13), 2451. https://doi.org/10.3390/cancers16132451