
Visual Intratumor Heterogeneity and Breast Tumor Progression

 , , , , , , , , and
, , , , , , , , and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Color Normalization and Image Segmentation
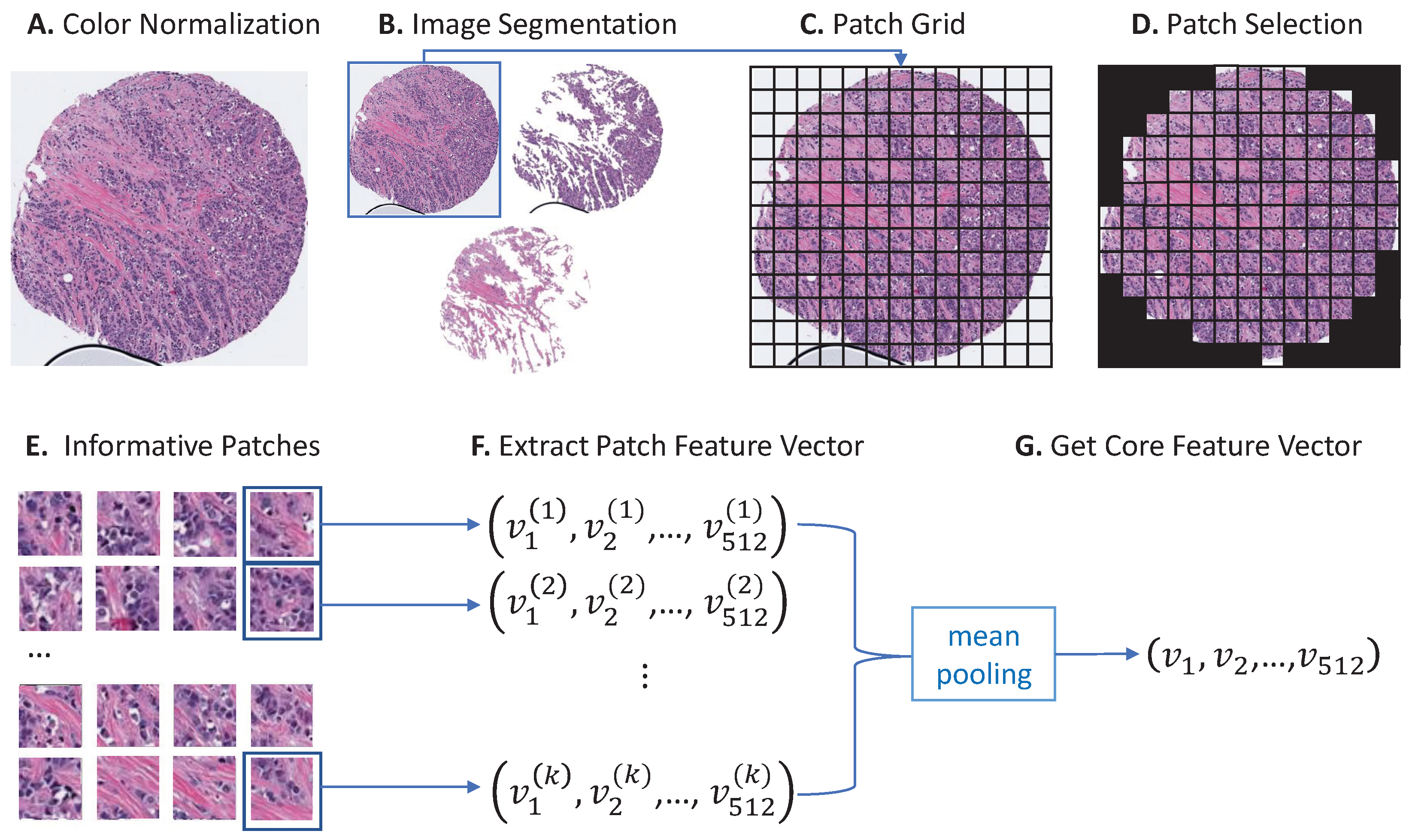
2.3. Core Feature Extraction
2.4. Feature Clustering
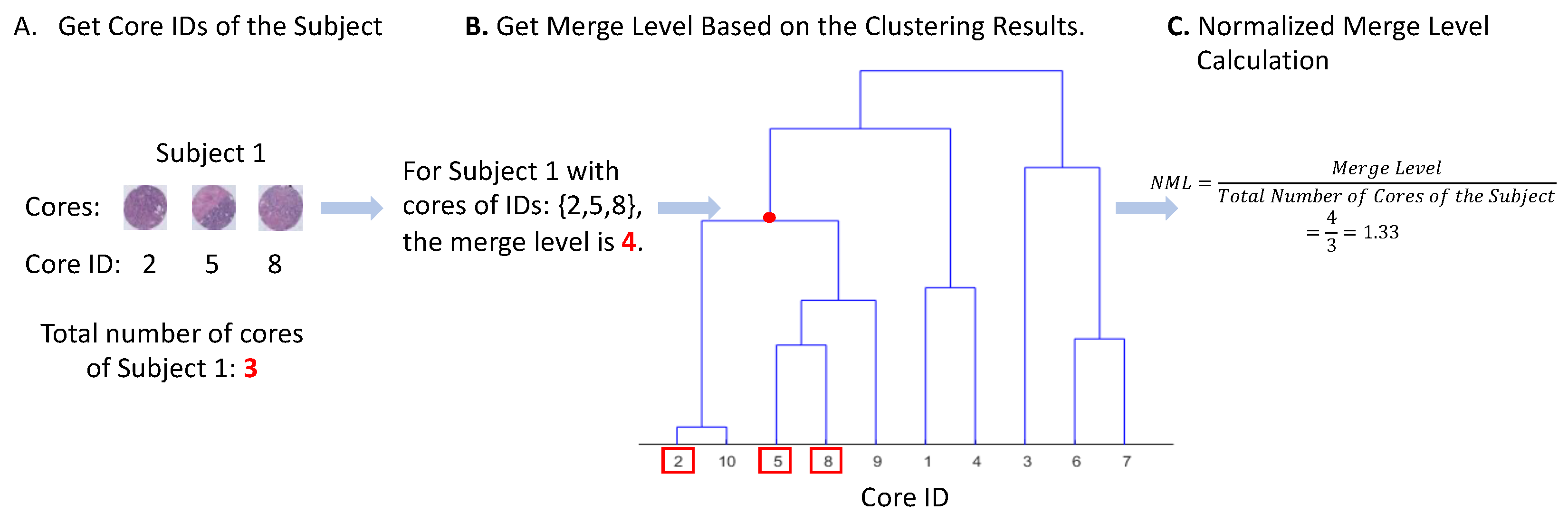
2.5. Measure of Visual Intratumor Heterogeneity
2.6. Measure of Molecular Heterogeneity
2.7. Statistical Analysis
3. Results
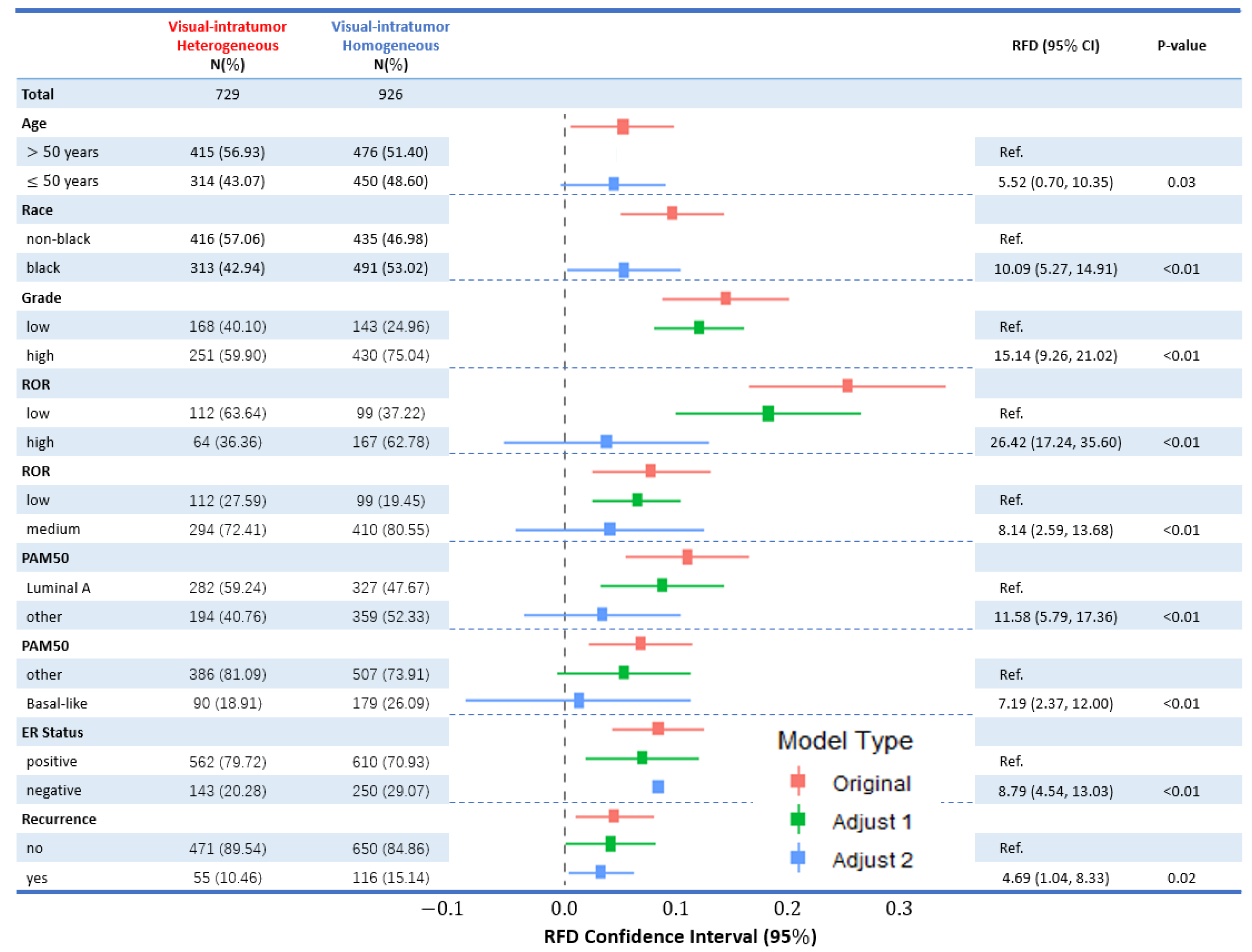
3.1. Visual ITH, Patient, and Tumor Characteristic
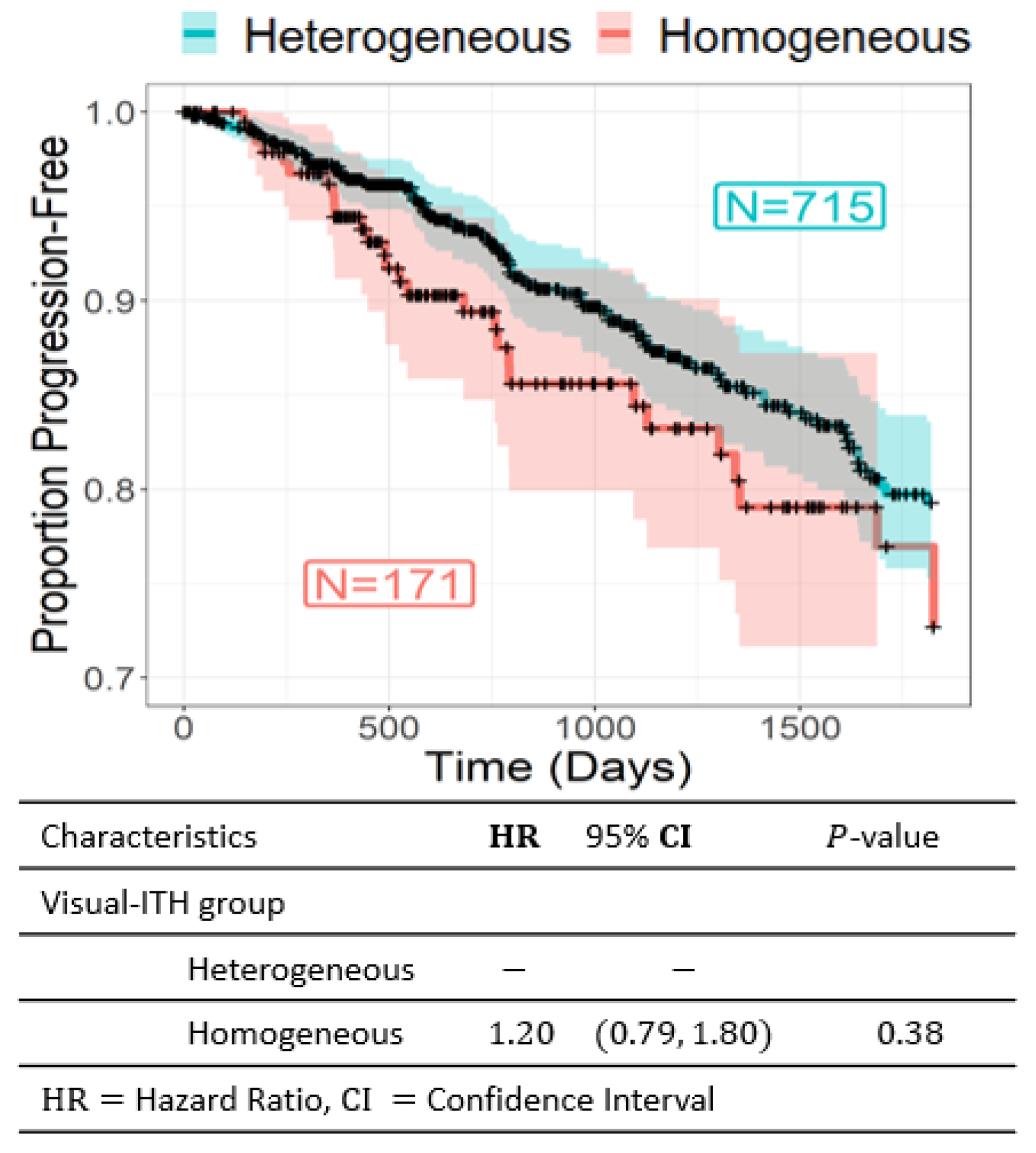
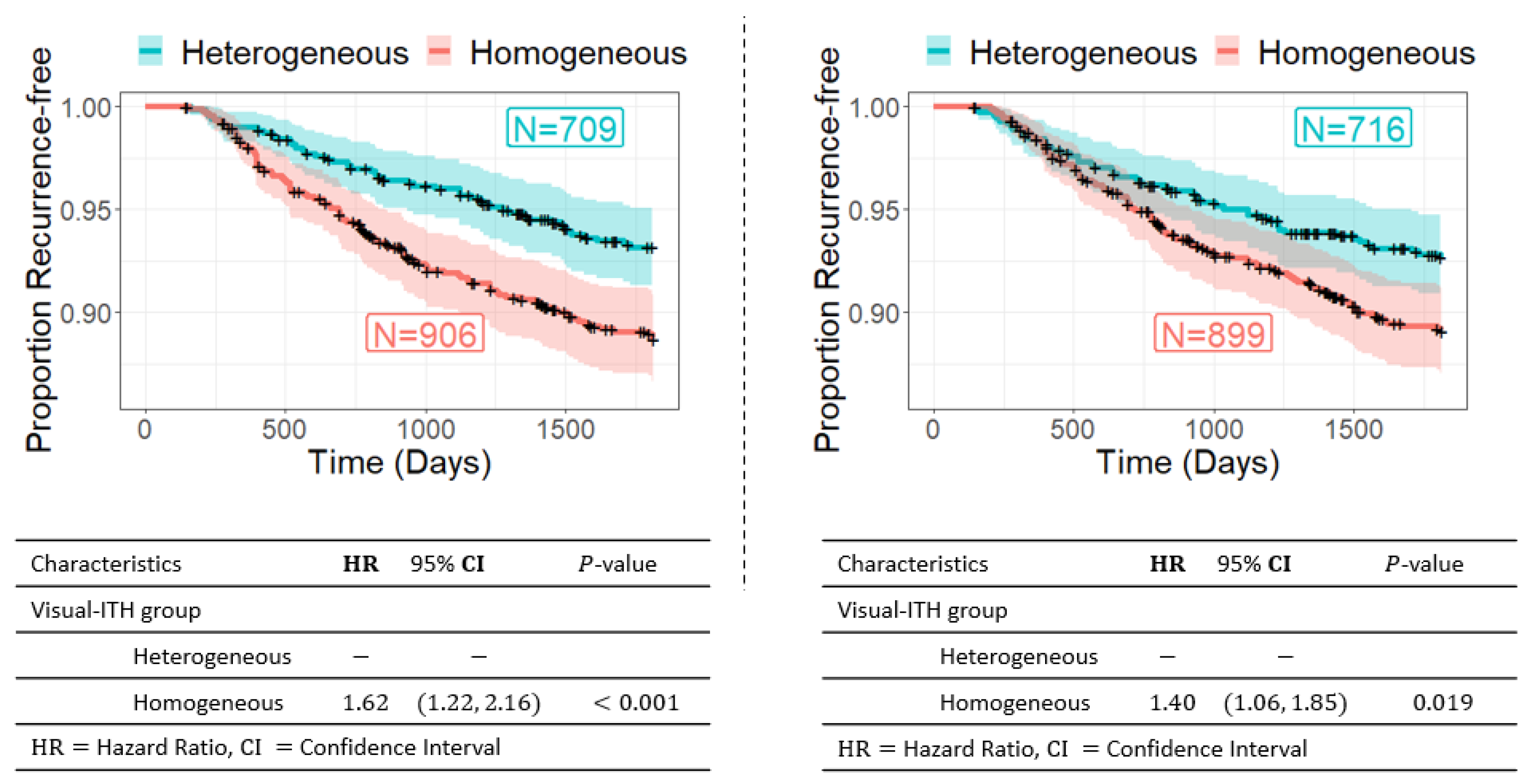
3.2. Visual ITH and Recurrence-Free Survival
3.3. Visual ITH, Genomic Instability, and Clonality by DNA Sequencing
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; Tarpey, P.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef]
- Swanton, C. Intratumor heterogeneity: Evolution through space and time. Cancer Res. 2012, 72, 4875–4882. [Google Scholar] [CrossRef] [PubMed]
- Allott, E.H.; Geradts, J.; Sun, X.; Cohen, S.M.; Zirpoli, G.R.; Khoury, T.; Bshara, W.; Chen, M.; Sherman, M.E.; Palmer, J.R.; et al. Intratumoral heterogeneity as a source of discordance in breast cancer biomarker classification. Breast Cancer Res. 2016, 18, 68. [Google Scholar] [CrossRef]
- Lindström, L.S.; Yau, C.; Czene, K.; Thompson, C.K.; Hoadley, K.A.; Van’t Veer, L.J.; Balassanian, R.; Bishop, J.W.; Carpenter, P.M.; Chen, Y.Y.; et al. Intratumor heterogeneity of the estrogen receptor and the long-term risk of fatal breast cancer. J. Natl. Cancer Inst. 2018, 110, 726–733. [Google Scholar] [CrossRef]
- McGranahan, N.; Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Silva, A.S.; Gillies, R.J.; Frieden, B.R. Adaptive therapy. Cancer Res. 2009, 69, 4894–4903. [Google Scholar] [CrossRef] [PubMed]
- Dentro, S.C.; Leshchiner, I.; Haase, K.; Tarabichi, M.; Wintersinger, J.; Deshwar, A.G.; Yu, K.; Rubanova, Y.; Macintyre, G.; Demeulemeester, J.; et al. Characterizing genetic intra-tumor heterogeneity across 2,658 human cancer genomes. Cell 2021, 184, 2239–2254. [Google Scholar] [CrossRef] [PubMed]
- Van Keymeulen, A.; Lee, M.Y.; Ousset, M.; Brohée, S.; Rorive, S.; Giraddi, R.R.; Wuidart, A.; Bouvencourt, G.; Dubois, C.; Salmon, I.; et al. Reactivation of multipotency by oncogenic PIK3CA induces breast tumour heterogeneity. Nature 2015, 525, 119–123. [Google Scholar] [CrossRef]
- Li, Z.; Seehawer, M.; Polyak, C. Untangling the web of intratumour heterogeneity. Nat. Cell Biol. 2022, 24, 1192–1201. [Google Scholar] [CrossRef] [PubMed]
- Marusyk, A.; Janiszewska, M.; Polyak, C. Intratumor heterogeneity: The Rosetta stone of therapy resistance. Cancer Cell 2020, 37, 471–484. [Google Scholar] [CrossRef]
- Fassler, D.J.; Torre-Healy, L.A.; Gupta, R.; Hamilton, A.M.; Kobayashi, S.; Van Alsten, S.C.; Zhang, Y.; Kurc, T.; Moffitt, R.A.; Troester, M.A.; et al. Spatial characterization of tumor-infiltrating lymphocytes and breast cancer progression. Cancers 2022, 14, 2148. [Google Scholar] [CrossRef] [PubMed]
- Romero-Cordoba, S.; Meneghini, E.; Sant, M.; Iorio, M.V.; Sfondrini, L.; Paolini, B.; Agresti, R.; Tagliabue, E.; Bianchi, F. Decoding immune heterogeneity of triple negative breast cancer and its association with systemic inflammation. Cancers 2019, 11, 911. [Google Scholar] [CrossRef] [PubMed]
- Liegmann, A.S.; Heselmeyer-Haddad, K.; Lischka, A.; Hirsch, D.; Chen, W.D.; Torres, I.; Gemoll, T.; Rody, A.; Thorns, C.; Gertz, E.M.; et al. Single cell genetic profiling of tumors of breast cancer patients aged 50 years and older reveals enormous intratumor heterogeneity independent of individual prognosis. Cancers 2021, 13, 3366. [Google Scholar] [CrossRef] [PubMed]
- Jackson, H.W.; Fischer, J.R.; Zanotelli, V.R.; Ali, H.R.; Mechera, R.; Soysal, S.D.; Moch, H.; Muenst, S.; Varga, Z.; Weber, W.P.; et al. The single-cell pathology landscape of breast cancer. Nature 2020, 578, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Carey, L.A.; Perou, C.M.; Livasy, C.A.; Dressler, L.G.; Cowan, D.; Conway, K.; Karaca, G.; Troester, M.A.; Tse, C.K.; Edmiston, S.; et al. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA 2006, 295, 2492–2502. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, K.M.; Cole, S.R.; Tse, C.K.; Perou, C.M.; Carey, L.A.; Foulkes, W.D.; Dressler, L.G.; Geradts, J.; Millikan, R.C. Intrinsic breast tumor subtypes, race, and long-term survival in the Carolina Breast Cancer Study. Clin. Cancer Res. 2010, 16, 6100–6110. [Google Scholar] [CrossRef] [PubMed]
- Newman, B.; Moorman, P.G.; Millikan, R.; Qaqish, B.F.; Geradts, J.; Aldrich, T.E.; Liu, E.T. The Carolina Breast Cancer Study: Integrating population-based epidemiology and molecular biology. Breast Cancer Res. Treat. 1995, 35, 51–60. [Google Scholar] [CrossRef]
- Razzaghi, H.; Troester, M.A.; Gierach, G.L.; Olshan, A.F.; Yankaskas, B.C.; Millikan, R.C. Mammographic density and breast cancer risk in White and African American Women. Breast Cancer Res. Treat. 2012, 135, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Conway, K.; Parrish, E.; Edmiston, S.N.; Tolbert, D.; Tse, C.K.; Moorman, P.; Newman, B.; Millikan, R.C. Risk factors for breast cancer characterized by the estrogen receptor alpha A908G (K303R) mutation. Breast Cancer Res. 2007, 9, R36. [Google Scholar] [CrossRef]
- Millikan, R.C.; Newman, B.; Tse, C.K.; Moorman, P.G.; Conway, K.; Smith, L.V.; Labbok, M.H.; Geradts, J.; Bensen, J.T.; Jackson, S.; et al. Epidemiology of basal-like breast cancer. Breast Cancer Res. Treat. 2008, 109, 123–139. [Google Scholar] [CrossRef]
- Network, C.G.A. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Ciriello, G.; Gatza, M.L.; Beck, A.H.; Wilkerson, M.D.; Rhie, S.K.; Pastore, A.; Zhang, H.; McLellan, M.; Yau, C.; Kandoth, C.; et al. Comprehensive molecular portraits of invasive lobular breast cancer. Cell 2015, 163, 506–519. [Google Scholar] [CrossRef]
- Macenko, M.; Niethammer, M.; Marron, J.S.; Borland, D.; Woosley, J.T.; Guan, X.; Schmitt, C.; Thomas, N.E. A Method for Normalizing Histology Slides for Quantitative Analysis. In Proceedings of the 2009 IEEE International Symposium on Biomedical Imaging: From Nano to Macro, Boston, MA, USA, 28 June–1 July 2009; pp. 1107–1110. [Google Scholar]
- Klimov, S.; Miligy, I.M.; Gertych, A.; Jiang, Y.; Toss, M.S.; Rida, P.; Ellis, I.O.; Green, A.; Krishnamurti, U.; Rakha, E.A.; et al. A whole slide image-based machine learning approach to predict ductal carcinoma in situ (DCIS) recurrence risk. Breast Cancer Res. 2019, 21, 83. [Google Scholar] [CrossRef] [PubMed]
- Bankhead, P.; Loughrey, M.B.; Fernández, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef] [PubMed]
- Simonyan, K.; Zisserman, A. Very deep convolutional networks for large-scale image recognition. arXiv 2014, arXiv:1409.1556. [Google Scholar]
- Beck, A.H.; Sangoi, A.R.; Leung, S.; Marinelli, R.J.; Nielsen, T.O.; Van De Vijver, M.J.; West, R.B.; Van De Rijn, M.; Koller, D. Systematic analysis of breast cancer morphology uncovers stromal features associated with survival. Sci. Transl. Med. 2011, 3, 108ra113. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Fontenay, G.V.; Han, J.; Cong, G.; Baehner, F.L.; Gray, J.W.; Spellman, P.T.; Parvin, B. Morphometic analysis of TCGA glioblastoma multiforme. BMC Bioinform. 2011, 12, 484. [Google Scholar] [CrossRef] [PubMed]
- Miedema, J.; Marron, J.S.; Niethammer, M.; Borland, D.; Woosley, J.; Coposky, J.; Wei, S.; Reisner, H.; Thomas, N.E. Image and statistical analysis of melanocytic histology. Histopathology 2012, 61, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Cooper, L.A.; Kong, J.; Gutman, D.A.; Wang, F.; Gao, J.; Appin, C.; Cholleti, S.; Pan, T.; Sharma, A.; Scarpace, L.; et al. Integrated morphologic analysis for the identification and characterization of disease subtypes. J. Am. Med. Inform. Assoc. 2012, 19, 317–323. [Google Scholar] [CrossRef]
- Hou, L.; Samaras, D.; Kurc, T.M.; Gao, Y.; Davis, J.E.; Saltz, J.H. Patch-Based Convolutional Neural Network for Whole Slide Tissue Image Classification. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, San Francisco, CA, USA, 18–20 June 1996; pp. 2424–2433. [Google Scholar]
- Xu, J.; Luo, X.; Wang, G.; Gilmore, H.; Madabhushi, A. A deep convolutional neural network for segmenting and classifying epithelial and stromal regions in histopathological images. Neurocomputing 2016, 191, 214–223. [Google Scholar] [CrossRef]
- Deng, J.; Dong, W.; Socher, R.; Li, L.J.; Li, K.; Fei-Fei, L. Imagenet: A Large-Scale Hierarchical Image Database. In Proceedings of the 2009 IEEE Conference on Computer Vision and Pattern Recognition, Miami, FL, USA, 20–25 June 2009; pp. 248–255. [Google Scholar]
- Jain, A.K.; Dubes, R.C. Algorithms for Clustering Data; Prentice-Hall, Inc.: Hoboken, NJ, USA, 1988. [Google Scholar]
- Ward, J.H., Jr. Hierarchical grouping to optimize an objective function. J. Am. Stat. Assoc. 1963, 58, 236–244. [Google Scholar] [CrossRef]
- Mose, L.E.; Perou, C.M.; Parker, J.S. Improved indel detection in DNA and RNA via realignment with ABRA2. Bioinformatics 2019, 35, 2966–2973. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, D.; Sato, T.; Cibulskis, K.; Getz, G.; Stewart, C.; Lichtenstein, L. Calling Somatic SNVs and Indels with Mutect2. bioRxiv 2019, 861054. [Google Scholar] [CrossRef]
- Kim, S.; Scheffler, K.; Halpern, A.L.; Bekritsky, M.A.; Noh, E.; Källberg, M.; Chen, X.; Kim, Y.; Beyter, D.; Krusche, P.; et al. Strelka2: Fast and accurate calling of germline and somatic variants. Nat. Methods 2018, 15, 591–594. [Google Scholar] [CrossRef] [PubMed]
- Talevich, E.; Shain, A.H.; Botton, T.; Bastian, B.C. CNVkit: Genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput. Biol. 2016, 12, e1004873. [Google Scholar] [CrossRef] [PubMed]
- Gillis, S.; Roth, A. PyClone-VI: Scalable inference of clonal population structures using whole genome data. BMC Bioinform. 2020, 21, 571. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.S.; Mullins, M.; Cheang, M.C.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z.; et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 2009, 27, 1160. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, A.M.; Hurson, A.N.; Olsson, L.T.; Walens, A.; Nsonwu-Farley, J.; Kirk, E.L.; Abdou, Y.; Downs-Canner, S.; Serody, J.S.; Perou, C.M.; et al. The landscape of immune microenvironments in racially diverse breast cancer patients. Cancer Epidemiol. Biomarkers Prev. 2022, 31, 1341–1350. [Google Scholar] [CrossRef] [PubMed]
- Olsson, L.T.; Williams, L.A.; Midkiff, B.R.; Kirk, E.L.; Troester, M.A.; Calhoun, B.C. Quantitative analysis of breast cancer tissue composition and associations with tumor subtype. Hum. Pathol. 2022, 123, 84–92. [Google Scholar] [CrossRef]
- Razavi, P.; Chang, M.T.; Xu, G.; Bandlamudi, C.; Ross, D.S.; Vasan, N.; Cai, Y.; Bielski, C.M.; Donoghue, M.T.; Jonsson, P.; et al. The genomic landscape of endocrine-resistant advanced breast cancers. Cancer Cell 2018, 34, 427–438. [Google Scholar] [CrossRef]
- Angus, L.; Smid, M.; Wilting, S.M.; van Riet, J.; Van Hoeck, A.; Nguyen, L.; Nik-Zainal, S.; Steenbruggen, T.G.; Tjan-Heijnen, V.C.; Labots, M.; et al. The genomic landscape of metastatic breast cancer highlights changes in mutation and signature frequencies. Nat. Genet. 2019, 51, 1450–1458. [Google Scholar] [CrossRef] [PubMed]
- Keenan, T.; Moy, B.; Mroz, E.A.; Ross, K.; Niemierko, A.; Rocco, J.W.; Isakoff, S.; Ellisen, L.W.; Bardia, A. Comparison of the genomic landscape between primary breast cancer in African American versus white women and the association of racial differences with tumor recurrence. J. Clin. Oncol. 2015, 33, 3621. [Google Scholar] [CrossRef] [PubMed]
- Pereira, B.; Chin, S.F.; Rueda, O.M.; Vollan, H.K.M.; Provenzano, E.; Bardwell, H.A.; Pugh, M.; Jones, L.; Russell, R.; Sammut, S.J.; et al. The somatic mutation profiles of 2,433 breast cancers refine their genomic and transcriptomic landscapes. Nat. Commun. 2016, 7, 11479. [Google Scholar] [CrossRef] [PubMed]
- Almendro, V.; Marusyk, A.; Polyak, K. Cellular heterogeneity and molecular evolution in cancer. Annu. Rev. Pathol. Mech. Dis. 2013, 8, 277–302. [Google Scholar] [CrossRef] [PubMed]
- Graf, J.F.; Zavodszky, M.I. Characterizing the heterogeneity of tumor tissues from spatially resolved molecular measures. PLoS ONE 2017, 12, e0188878. [Google Scholar] [CrossRef] [PubMed]
- Lineweaver, C.H.; Bussey, K.J.; Blackburn, A.C.; Davies, P.C. Cancer progression as a sequence of atavistic reversions. BioEssays 2021, 43, 2000305. [Google Scholar] [CrossRef] [PubMed]
- Budczies, J.; Bockmayr, M.; Denkert, C.; Klauschen, F.; Lennerz, J.K.; Gyorffy, B.; Dietsel, M.; Loibl, S.; Weichert, W.; Stenzinger, A. Classical pathology and mutational load of breast cancer— Integration of two worlds. J. Pathol. Clin. Res. 2015, 1, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Donehower, L.A.; Soussi, T.; Korkut, A.; Liu, Y.; Schultz, A.; Cardenas, M.; Li, X.; Babur, O.; Hsu, T.K.; Lichtarge, O.; et al. Integrated analysis of TP53 gene and pathway alterations in the cancer genome atlas. Cell Rep. 2019, 28, 1370–1384. [Google Scholar] [CrossRef] [PubMed]
- Fuller, A.M.; Yang, L.; Hamilton, A.M.; Pirone, J.R.; Oldenburg, A.L.; Troester, M.A. Epithelial p53 status modifies stromal-epithelial interactions during basal-like breast carcinogenesis. J. Mammary Gland. Biol. Neoplasia 2021, 26, 89–99. [Google Scholar] [CrossRef]
- Hamilton, A.M.; Van Alsten, S.C.; Gao, X.; Nsonwu-Farley, J.; Calhoun, B.C.; Love, M.I.; Troester, M.A.; Hoadley, K.A. Incorporating RNA-based risk scores for genomic instability to predict breast cancer recurrence and immunogenicity in a diverse population. Cancer Res. Commun. 2023, 3, 12–20. [Google Scholar] [CrossRef]
- Tarabichi, M.; Salcedo, A.; Deshwar, A.G.; Leathlobhair, M.N.; Wintersinger, J.; Wedge, D.C.; Van Loo, P.; Morris, Q.D.; Boutros, P.C. A practical guide to cancer subclonal reconstruction from DNA sequencing. Nat. Methods 2021, 18, 144–155. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Ref. | Original (RFD, 95% CI) | Epithelium (RFD, 95% CI) | Stroma (RFD, 95% CI) | |
|---|---|---|---|---|---|
| Age | >50 | ≤50 | 5.52 (0.70, 10.35) | 1.88 (−2.95, 6.71) | 1.73 (−3.07, 6.53) |
| Race | non-Black | Black | 10.09 (5.27, 14.91) | 6.97 (2.14, 11.80) | 3.51 (−1.30, 8.33) |
| Grade | low | high | 15.14 (9.26, 21.02) | 8.82 (2.96, 14.69) | 9.60 (3.86, 15.35) |
| ROR | low | high | 26.42 (17.24, 35.60) | 25.65 (16.55, 34.75) | 16.74 (7.56, 25.92) |
| ROR | low | medium | 8.14 (2.59, 13.68) | 10.91 (5.38, 16.45) | 6.23 (0.81, 11.66) |
| PAM50 | Luminal A | other | 11.58 (5.79, 17.36) | 12.80 (7.05, 18.56) | 8.29 (2.56, 14.01) |
| PAM50 | other | basal-like | 7.19 (2.37, 12.00) | 7.06 (2.24, 11.87) | 5.10 (0.26, 9.94) |
| ER Status | positive | negative | 8.79 (4.54, 13.03) | 7.04 (2.77, 11.30) | 6.50 (2.20, 10.80) |
| Recurrence | no | yes | 4.69 (1.04, 8.33) | 4.48 (0.84, 8.13) | 3.58 (−0.10, 7.27) |
| Variable | Ref. | Epithelium (RFD, 95% CI) | Stroma (RFD, 95% CI) | |||
|---|---|---|---|---|---|---|
| Reduced | Adjusted | Reduced | Adjusted | |||
| Grade | low | high | 8.82 (2.96, 14.69) | 7.14 (3.19, 11.09) | 9.60 (3.86, 15.35) | 7.95 (4.10, 11.80) |
| ROR | low | high | 25.65 (16.55, 34.75) | 18.72 (10.49, 26.95) | 16.74 (7.56, 25.92) | 11.27 (4.04, 18.50) |
| ROR | low | medium | 10.91 (5.38, 16.45) | 9.65 (5.37, 13.93) | 6.23 (0.81, 11.66) | 5.75 (1.90, 9.60) |
| PAM50 | Luminal A | other | 12.80 (7.05, 18.56) | 10.80 (5.06, 16.53) | 8.29 (2.56, 14.01) | 7.28 (1.78, 12.78) |
| PAM50 | other | basal-like | 7.06 (2.24, 11.87) | 5.75 (−0.44, 11.94) | 5.10 (0.26, 9.94) | 4.37 (−1.80, 10.54) |
| ER Status | positive | negative | 7.04 (2.77, 11.30) | 5.93 (0.62, 11.25) | 6.50 (2.20, 10.80) | 5.88 (0.56, 11.19) |
| Recurrence | no | yes | 4.48 (0.84, 8.13) | 4.15 (0.10, 8.21) | 3.58 (−0.10, 7.27) | 3.39 (−0.91, 7.68) |
| TCGA-BRCA | CBCS | |||||||
|---|---|---|---|---|---|---|---|---|
| Visual-Intratumor | Visual-Intratumor | Visual-Intratumor | Visual-Intratumor | |||||
| Heterogeneous | Homogeneous | RFD (95% CI) | p-Value | Heterogeneous | Homogeneous | RFD (95% CI) | p-Value | |
| N (%) | N (%) | N (%) | N (%) | |||||
| Total | 894 | 208 | 729 | 926 | ||||
| Age | ||||||||
| >50 years | 666 (74.58) | 145 (70.05) | Ref. | 415 (56.93) | 476 (51.40) | Ref. | ||
| ≤50 years | 227 (25.42) | 62 (29.95) | 4.53 (, 11.39) | 0.18 | 314 (43.07) | 450 (48.60) | 5.52 (0.70, 10.35) | 0.03 |
| Race | ||||||||
| non-black | 674 (86.19) | 144 (78.69) | Ref. | 416 (57.06) | 435 (46.98) | Ref. | ||
| black | 108 (13.81) | 39 (21.31) | 7.50 (1.09, 13.91) | 0.01 | 313 (42.94) | 491 (53.02) | 10.09 (5.27, 14.91) | <0.01 |
| ROR | ||||||||
| low | 208 (52.39) | 29 (31.52) | Ref. | 112 (63.64) | 99 (37.22) | Ref. | ||
| high | 189 (47.61) | 63 (68.48) | 20.87 (10.18, 31.56) | <0.01 | 64 (36.36) | 167 (62.78) | 26.42 (17.24, 35.60) | <0.01 |
| ROR | ||||||||
| low | 208 (29.59) | 29 (20.28) | Ref. | 112 (27.59) | 99 (19.45) | Ref. | ||
| medium | 495 (70.41) | 114 (79.72) | 9.31 (1.90, 16.71) | 0.02 | 294 (72.41) | 410 (80.55) | 8.14 (2.59, 13.68) | <0.01 |
| PAM50 | ||||||||
| Luminal A | 470 (52.63) | 93 (44.93) | Ref. | 282 (59.24) | 327 (47.67) | Ref. | ||
| other | 423 (47.37) | 114 (55.07) | 7.70 (0.18, 15.23) | 0.05 | 194 (40.76) | 359 (52.33) | 11.58 (5.79, 17.36) | <0.01 |
| PAM50 | ||||||||
| other | 757 (84.77) | 159 (76.81) | Ref. | 386 (81.09) | 507 (73.91) | Ref. | ||
| Basal-like | 136 (15.23) | 48 (23.19) | 7.96 (1.75, 14.17) | 0.01 | 90 (18.91) | 179 (26.09) | 7.19 (2.37, 12.00) | <0.01 |
| ER Status | ||||||||
| positive | 677 (79.83) | 144 (71.64) | Ref. | 562 (79.72) | 610 (70.93) | Ref. | ||
| negative | 171 (20.17) | 57 (28.36) | 8.19 (1.40, 14.98) | 0.01 | 143 (20.28) | 250 (29.07) | 8.79 (4.54, 13.03) | <0.01 |
| Immune Class | ||||||||
| low | 488 (54.65) | 117 (56.52) | Ref. | 307 (75.25) | 357 (60.92) | Ref. | ||
| high | 405 (45.35) | 90 (43.48) | (, 9.38) | 0.63 | 101 (24.75) | 229 (39.08) | 14.32 (8.57, 20.08) | <0.01 |
| Recurrence | ||||||||
| no | 780 (87.25) | 179 (86.06) | Ref. | 471 (89.54) | 650 (84.86) | Ref. | ||
| yes | 114 (12.75) | 29 (13.94) | 1.19 (, 6.38) | 0.65 | 55 (10.46) | 116 (15.14) | 4.69 (1.04, 8.33) | 0.02 |
| Clonal | Multi-Clonal | Overall OR (95% CI) | |||
|---|---|---|---|---|---|
| Low Visual ITH | High Visual ITH | Low Visual ITH | High Visual ITH | ||
| Original | 113 (66%) | 77 (55%) | 59 (34%) | 62 (45%) | 1.54 (0.98, 2.45) |
| Stromal | 110 (66%) | 80 (55%) | 56 (34%) | 65 (45%) | 1.60 (1.01, 2.53) |
| Epithelial | 107 (62%) | 83 (60%) | 66 (38%) | 55 (40%) | 1.07 (0.68, 1.70) |
| TCGA | 96 (53%) | 426 (58%) | 86 (47%) | 307 (42%) | 0.80 (0.58, 1.12) |
| TP53 Mutation | No TP53 Mutation | Overall OR (95% CI) | |||
| Low Visual ITH | High Visual ITH | Low Visual ITH | High Visual ITH | ||
| Original | 78 (43%) | 44 (27%) | 105 (57%) | 117 (73%) | 1.98 (1.26, 3.13) |
| Stromal | 74 (41%) | 48 (29%) | 105 (59%) | 117 (71%) | 1.72 (1.10, 2.70) |
| Epithelial | 71 (39%) | 51 (32%) | 113 (61%) | 109 (68%) | 1.34 (0.87, 2.10) |
| TCGA | 64 (35%) | 222 (30%) | 118 (65%) | 511 (70%) | 1.25 (0.89, 1.75) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Van Alsten, S.C.; Lee, D.N.; Kim, T.; Calhoun, B.C.; Perou, C.M.; Wobker, S.E.; Marron, J.S.; Hoadley, K.A.; Troester, M.A. Visual Intratumor Heterogeneity and Breast Tumor Progression. Cancers 2024, 16, 2294. https://doi.org/10.3390/cancers16132294
Li Y, Van Alsten SC, Lee DN, Kim T, Calhoun BC, Perou CM, Wobker SE, Marron JS, Hoadley KA, Troester MA. Visual Intratumor Heterogeneity and Breast Tumor Progression. Cancers. 2024; 16(13):2294. https://doi.org/10.3390/cancers16132294
Chicago/Turabian StyleLi, Yao, Sarah C. Van Alsten, Dong Neuck Lee, Taebin Kim, Benjamin C. Calhoun, Charles M. Perou, Sara E. Wobker, J. S. Marron, Katherine A. Hoadley, and Melissa A. Troester. 2024. "Visual Intratumor Heterogeneity and Breast Tumor Progression" Cancers 16, no. 13: 2294. https://doi.org/10.3390/cancers16132294
APA StyleLi, Y., Van Alsten, S. C., Lee, D. N., Kim, T., Calhoun, B. C., Perou, C. M., Wobker, S. E., Marron, J. S., Hoadley, K. A., & Troester, M. A. (2024). Visual Intratumor Heterogeneity and Breast Tumor Progression. Cancers, 16(13), 2294. https://doi.org/10.3390/cancers16132294