

When DNA Mutations Interplay with Cellular Proliferation: A Narrative History of Theories of Carcinogenesis




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
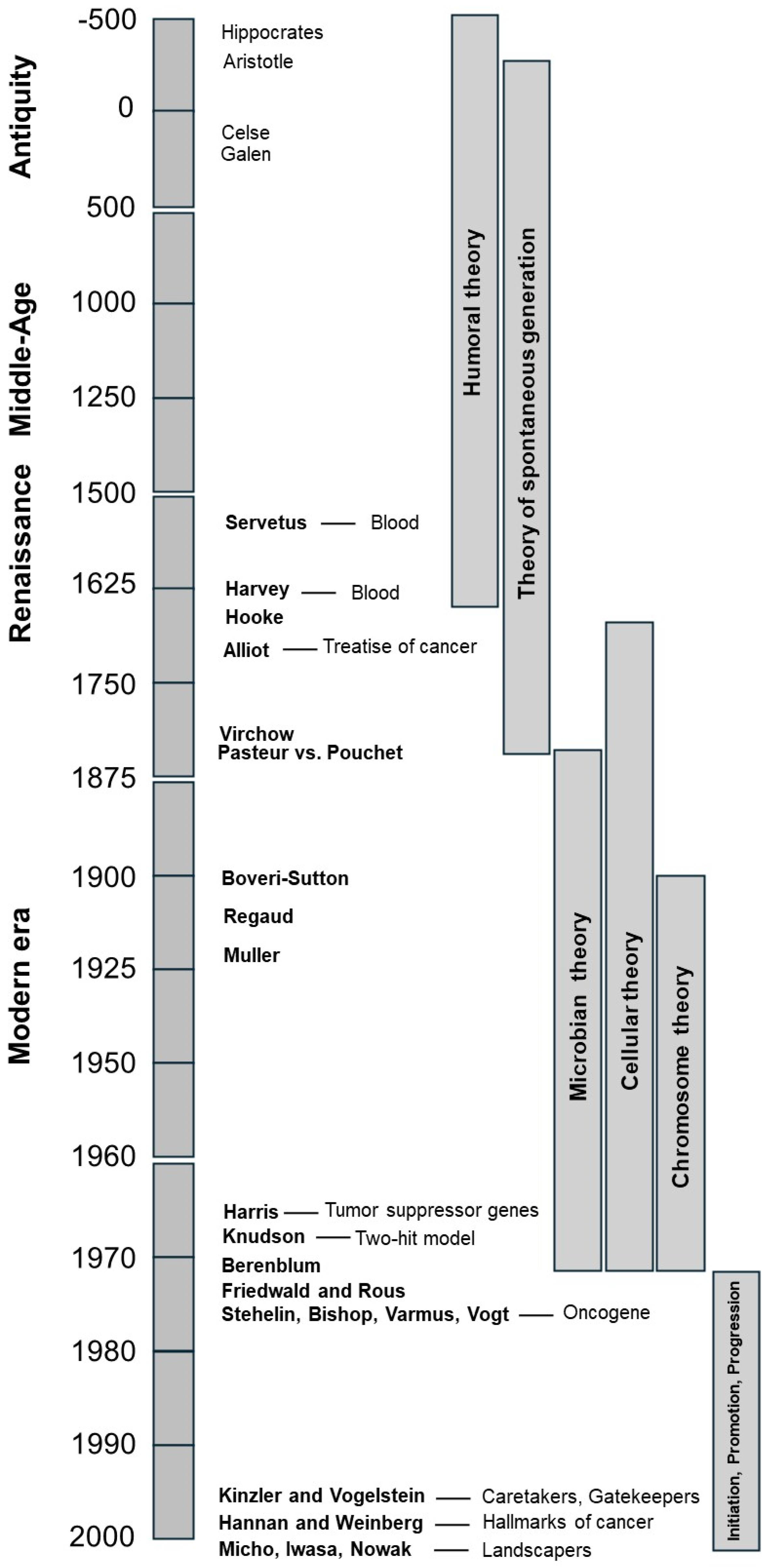
1. Introduction
2. From Antiquity to 1850, Cancer Was Explained by the Humoral Theory
2.1. The Cancer Origin in Antiquity: The Legacy of Hippocrates
2.2. The Triumph of Humoral Theory: The Definitions of Galen
“A tumor is an increase in length, width and depth occurring in a part of the body. There is obviously no question of calling the natural distension of fat and plump crops that way; what characterizes the unnatural tumor is that it thwarts the action of the part of the body that it affects.”
“If the bile spreads into the flesh, if it is acrid, it eats away the surrounding tissue and ulcerates it. If it is without acridity, it produces cancer without ulceration.”
2.3. From the Dark Ages to the Age of Enlightenment: The Breast Cancer of Ann of Austria
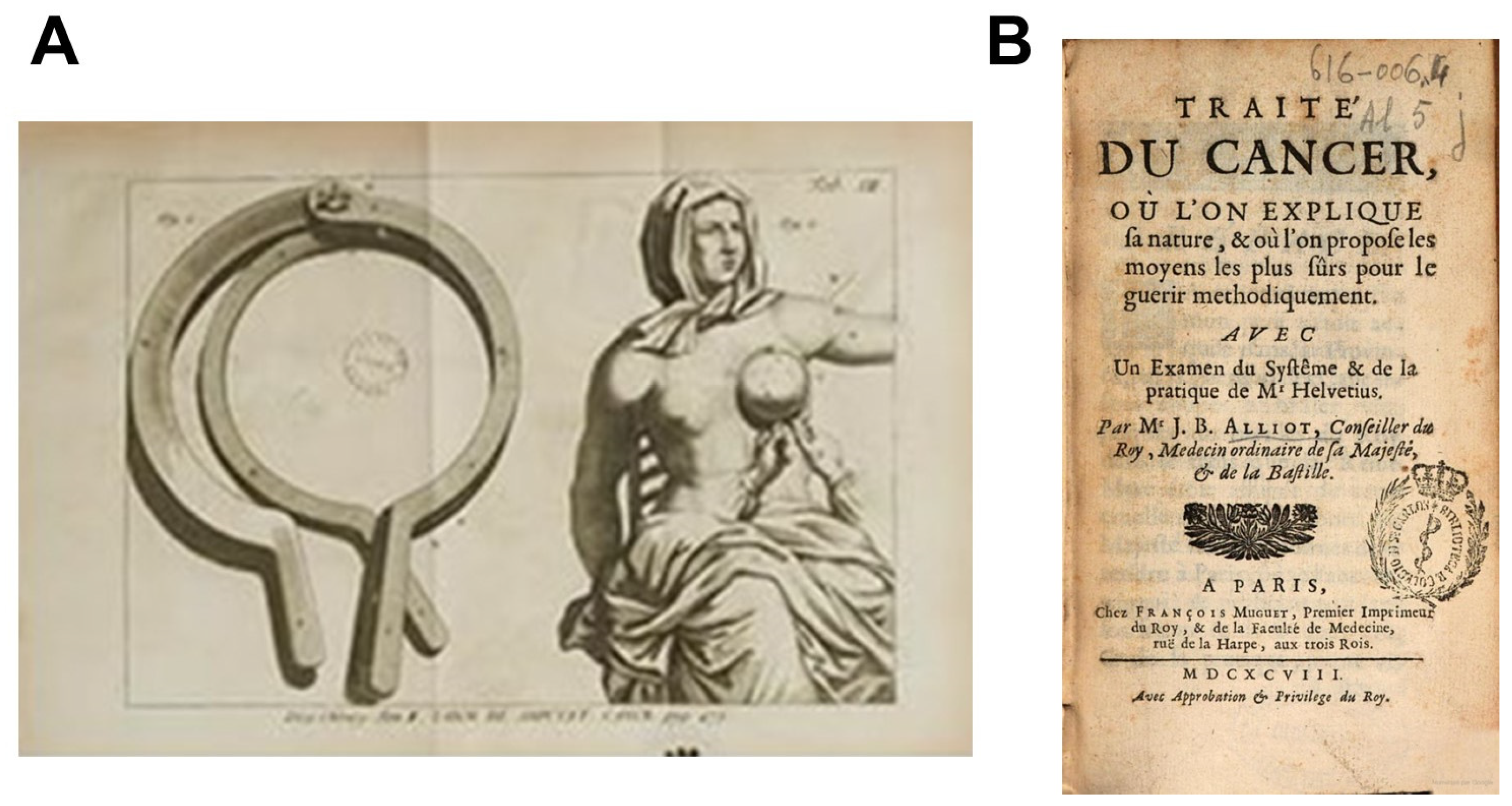
2.4. The First Treatise on Cancer
“cancer is a tumor, very hard, stony sometimes uneven and livid, always accompanied by pain more or less violent, depending on the circumstances that arise encounter, are more or less annoying.”
- -
- External (after a physical or emotional shock). He evoked the environmental influence (through polluted air or a contaminated food) that changes humor composition.
- -
- Internal (misbalance between two humors). He evoked inheritance by describing some cases of similar symptoms between mothers and children [10].
3. From 1850 to 1885, Cancer Was Explained by the Theory of Spontaneous Generation
3.1. When the Circulation of Blood Puts an End to the Bloodletting Practice
3.2. When a Theory Chases the Other: From Humoral Theory to Spontaneous Generation
4. From 1895 to 1940, Cancer Was Explained by the Microbian Theory
4.1. The Death of the Spontaneous Generation Theory
4.2. The First Anti-Cancer Radiotherapy: An Example of the Predominance of the Microbian Theory
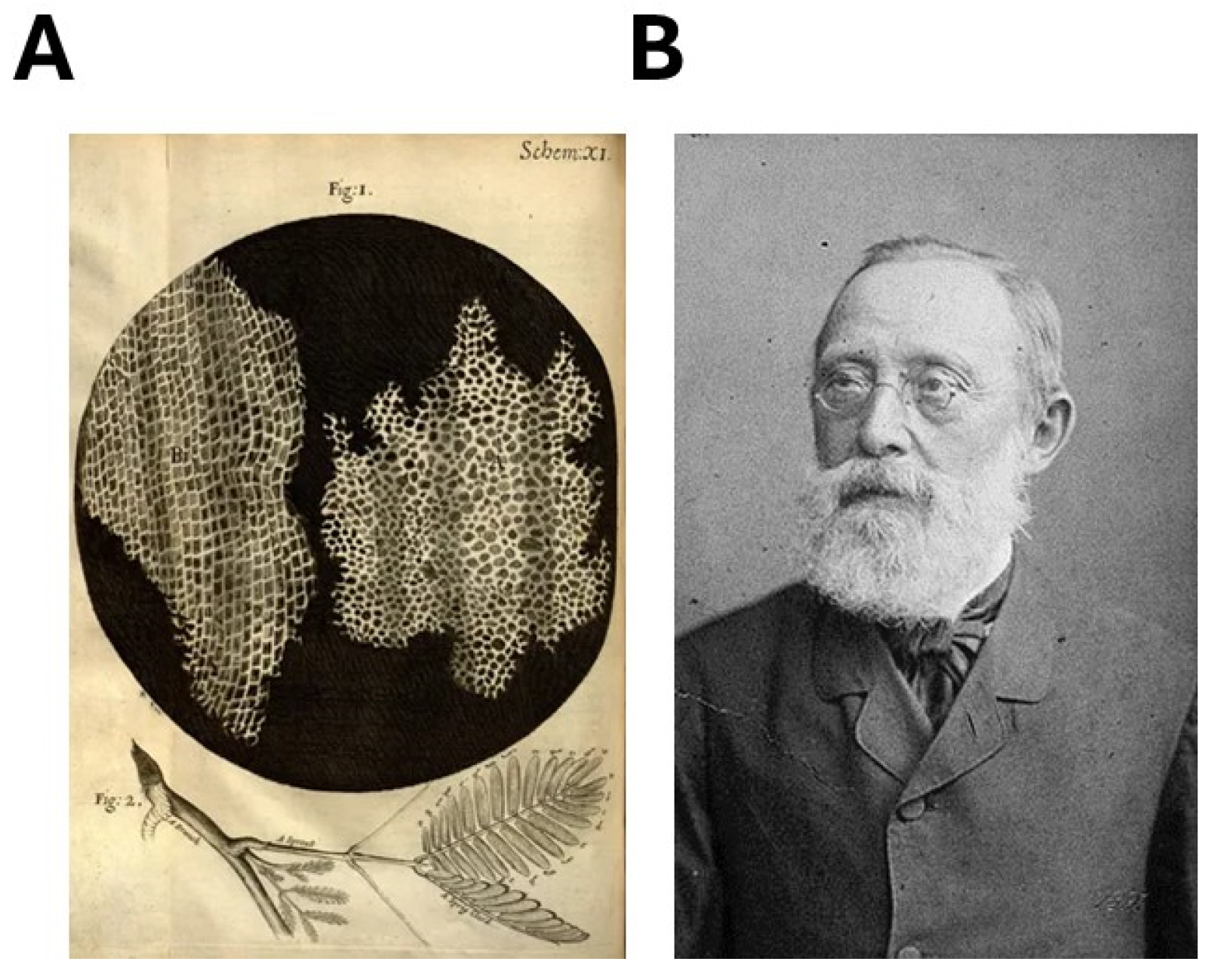
4.3. The Cellular Theory, an Emerging Paradigm to Be Opposed to the Microbian Theory
5. From 1895 to 1940, Cancer Was Also Explained by the Cellular Theory
5.1. First Identifications of Carcinogenic Agents
5.2. When the Chromosome Appeared and When X-rays Changed the Situation Again
- -
- Cancer is known to follow several ordered steps of evolution.
- -
- Some cancer types may be caused by micro-organisms (like stomach cancer) but the majority of them appeared to be caused by one of several deriving cells.
- -
- Some scientists hypothesized the existence of tumor germ cells that may derive preferentially. However, investigations with X-rays called into question the existence of a pool of germ cells since exposure to X-rays induces cancer but impacts cells indifferently.
- -
- The chromosome theory suggested that, whether in the tumor germ cells or in any cell, the primum movens of the formation of tumor is explained by one chromosome aberration per cell.
6. From 1895 to Today, Cancer Is Also Explained by Chromosome Theory
6.1. The Tribondeau and Bergonié Law: A Nice Formula That Propagates a False Idea
“… in our experiments, on the testicles of the rat, we have been able to destroy the germinal cells whereas the interstitial tissue and the Sertoli syncytium remained intact. Thank to these results, it has been possible to formulate the following law: X-rays act on cells inasmuch efficiently as cells have a greater reproductive activity, their karyokinetic fate is longer, their morphology and function are least definitively fixed.”…”Hence, from this law it is easy to understand that roentgenisation destroys tumors without destroying healthy tissues…”.
“It seems pretty clear that the practice of delivering small and repeated doses, in contradiction to the technique with less frequent and heavy doses, is more apt to produce these nondestructive irritations resulting in giant cells and probably malignant transformation.”
6.2. The Works of Claudius Regaud and Hermann Joseph Muller with X-rays
“I was tempted to publish this dissertation in a collection of oncology work: this would have had the advantage of making my ideas available to all researchers. I will not do it, because the fabric of my hypotheses is perhaps only a work of imagination; and, even if the future were to demonstrate that such a fear was unfounded, I will not do so because, having undertaken this work too late, I have not been and will no longer be able to give it the substance and form, which are necessary in a scientific publication. In particular, I was unable to gather the bibliographic documentation which would allow me to link my own ideas to the numerous works of my predecessors on the same subject…. Intermediate between publication and silence, the form of this writing seemed to me to correspond to the meaning of what it contains.”
7. From the 1940s to the 2000s, Initiation, Promotion, and Progression Became the Holy Trinity of Carcinogenesis
7.1. The Definition of the Multi-Step Formation of Cancer
- -
- A preneoplastic phase (or precarcinogenic action);
- -
- A “wart stage” (or epicarcinogenic action);
- -
- A malignant transformation of these warts (or metacarcinogenic action).
- -
- Rule I: Progression occurs independently in different tumors in the same animal.
- -
- Rule II: Progression occurs independently in different characters in the same tumor.
- -
- Rule III: Progression is independent of growth. It occurs in latent tumor cells and in tumors whose growth is arrested.
- -
- Rule IV: Progression is continuous or discontinuous, by gradual change or by abrupt steps.
- -
- Rule V: Progression follows one of alternative paths of development.
- -
- Rule VI: Progression does not always reach an end point within the lifetime of the host.
7.2. New Concepts and the Two-Hit Hypothesis
7.3. The Major Hypotheses of the Initiation Step
- -
- The stochastic submodel based on the hypothesis that each cell is potentially able to form a tumorigenic clone. The hypothesis that the primum movens, whether due to a micro-organism or occurring in a cell, was one of several DNA/chromosome mutation(s) (according to the chromosome theory) and was at the center of this submodel. Some endogenous factors (like genomic instability and individual predisposition) may be involved to lead to a subset of cells that are able to be amplified and form the “wart” stage.
- -
- The tumor stem cells (or hierarchy) submodel was based on the hypothesis that only a specific subset of cells is able to reach the “wart” stage. This submodel summarized the previous tumor germ cells or precancerous cells models cited above. The notion of tumor stem cells was notably refined and born from the studies about the reproductive or hematopoietic systems in which a hierarchy in the cellular differentiation was observed (e.g., Regaud’s works) [65]. The tumor stem cells should be auto reproductive to ensure, at any time, a certain pool of cells (reservoir). However, the notion of tumor stem cells was not consensual. A number of specific biomarkers of tumor stem cells (notably some biomarkers linked to the cellular surface state) were identified and thereafter abandoned [66,67,68]. Furthermore, as mentioned above, ionizing radiation does not impact the tumor stem cells pool preferentially while they induce cancer, which raises questions about the existence of tumor stem cells. In addition to this, a viral or bacterian infection raised similar questions, suggesting that the microbian theory was not necessarily compatible with the tumor stem cells submodel.
- -
- One mutation per cell is not sufficient to initiate cancer;
- -
- Three to seven mutations per cell is the minimum number of mutations required to initiate cancer;
- -
- Cell proliferation is needed to amplify the number of mutations per cell;
- -
- The frequency of carcinoma increases according to the sixth exponent of age.
7.4. Caretakers, Gatekeepers, and Landscapers
- -
- Caretaker genes are involved in the maintenance of genome integrity and stability and include genes implicated in DNA repair: when mutated, caretaker genes explain the accumulation of mutations in cells.
- -
- Gatekeeper genes directly regulate cell growth by either stimulating or inhibiting proliferation, differentiation, or apoptosis.
8. Since the 2000s, Refinement of an Existing Model or New Theory?
8.1. Addition of Cell Hallmarks to Current Models
“Hence, the prospect is raised that phenotypic plasticity and disrupted differentiation is a discrete hallmark capability, and that nonmutational epigenetic reprogramming and polymorphic microbiomes both constitute distinctive enabling characteristics that facilitate the acquisition of hallmark capabilities. Additionally, senescent cells, of varying origins, may be added to the roster of functionally important cell types in the tumor microenvironment.”
8.2. Specific Complements for the Progression Step
- -
- Tumor microenvironment (TME): In a tissue, cells communicate together and with the surrounding stroma, either directly, through junctional complexes, or indirectly through factors secreted in the environment extracellular (hormones, factors of growth, chemokines, etc.). Some of these factors may favor cancer progression. Notably, tissues in proximity of some invasive cancers (e.g., adipose tissue), may contribute to cancer development through their capability to secrete pro-inflammatory cytokines [74,75].
- -
- Tumor blood supply: Cavallaro and Christofori (2000) addressed the important role of angiogenesis in the growth of solid primary tumors as well as their metastases. Induction of angiogenesis precedes malignant tumor formation, and increased vascularization appears to be associated with the invasive properties of tumors, thus defining the malignant tumor phenotype [76]. Although the discovery of tumor-derived angiogenesis modulators has greatly improved our understanding of tumor regulation of angiogenesis, the connection between angiogenesis and tumor progression remains misunderstood. In parallel to angiogenesis, blood supply could be addressed by the de novo vessels channel developed from cancer cells transdifferentiated into endothelial cells [77].
- -
- Epithelial–mesenchymal transition (EMT): EMT is an embryonic capability through which epithelial cells lose many characteristics insuring homeostasis and acquire new characteristics of mesenchymal cells. EMT mechanisms may be reactivated during cancer progression, allowing tumor cells to enhance their motility and invasiveness. In vitro and in vivo observations suggest that EMT could favor both tumor development and metastatic dissemination [78,79].
8.3. The Bad Luck Hypothesis
- -
- The lifetime risk of patients being diagnosed with specific cancers can be very different according to the nature of the tissues and according to factors of exposure to certain physical (e.g., UV, radiation), chemical (e.g., drugs, smoking, alcohol) or biological (e.g., virus) agents. For example, lifetime risk is 6.9% for lung cancer in smokers. It is drastically reduced for non-smokers.
- -
- The genetic factor enhances the risk of cancer occurrence but only 5 to 15% of cancers are considered to have an inheritable component.
8.4. The Epigenetic Investigations
9. Another Vision of the Carcinogenesis Process via the Interplay between Mutations and Proliferation
9.1. Cancer as a Protein Biokinetics Dysfunction
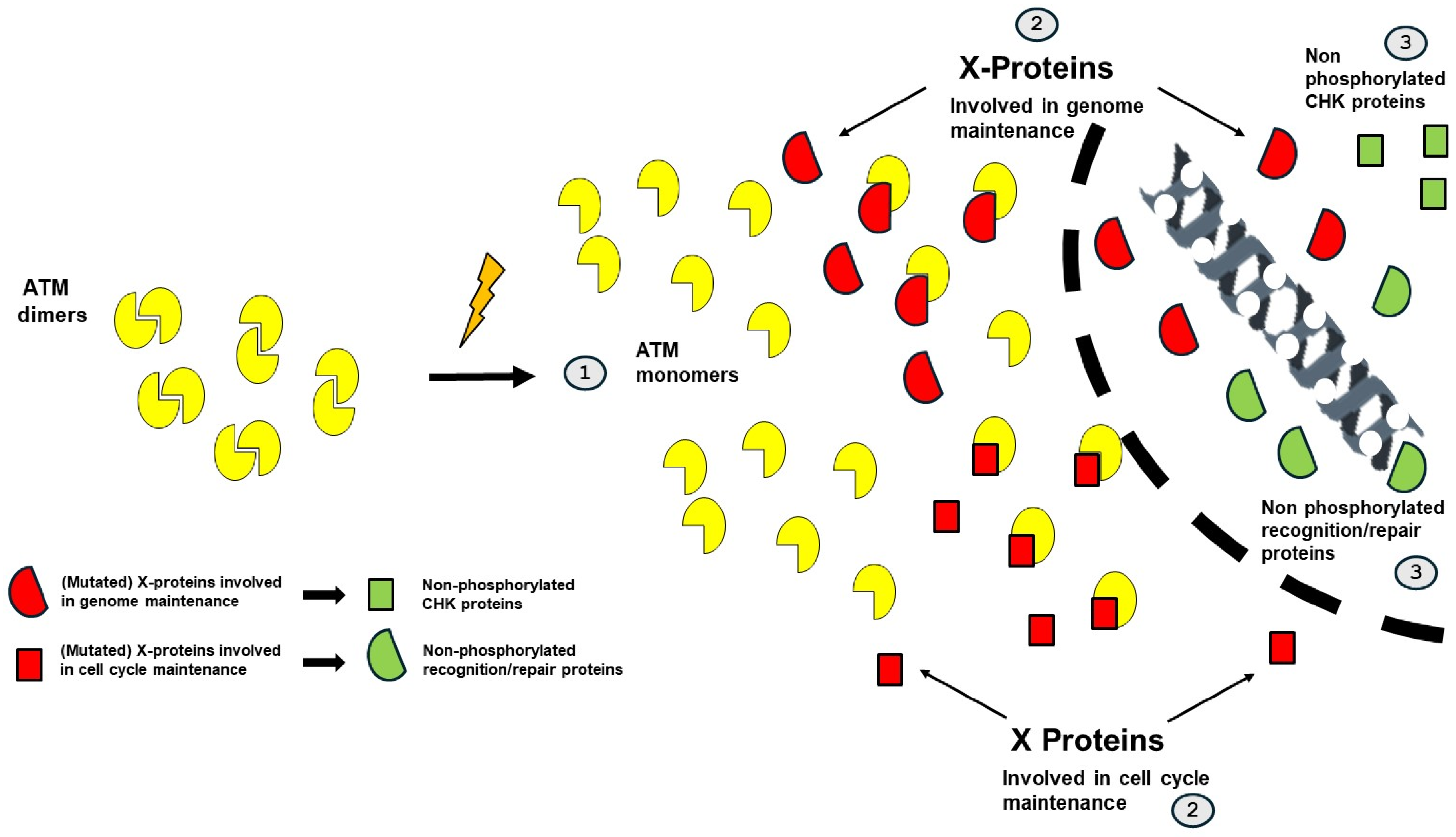
9.2. The RIANS/SIANS Model and Carcinogenesis
- -
- The impairment of the ATM-dependent DNA damage recognition, signaling, and repair may cause misrepaired DNA damage that may be either endogenous (e.g., spontaneous DNA breaks) or exogenous (e.g., from the environment). Their formation can be considered as an initiation step in carcinogenesis.
- -
- The impairment of the ATM-dependent cell cycle checkpoint control: impaired G2/M and/or G1 arrests may contribute to the propagation of the errors described above. This step can be considered as a promotion step in carcinogenesis.
- -
- Category 1: Proteins whose mutations cause category 1 cancer syndromes are crucial for the integrity of the genome. In cells derived from these syndromes, the genomic instability is so high that numerous mutations are produced. In parallel, the cytoplasmic forms of the mutated protein, as X-protein, sequestrate ATM monomers in cytoplasm, which limits the phosphorylation of CHK1 and CHK2 proteins and inhibits the G2 and G1 arrests, respectively. Cellular proliferation is favored. A representative example of category 1 syndrome is Nijmegen’s syndrome, caused by NBS1 mutations.
- -
- Category 2: Proteins whose mutations cause category 2 cancer syndromes are crucial for the control of the cell cycle. In cells derived from these syndromes, cellular proliferation is favored. In parallel, the cytoplasmic forms of the mutated protein, as X-protein, sequestrates ATM monomers in cytoplasm, which limits the DNA damage recognition and repair and favors genomic instability and numerous mutations. A representative example of category 2 syndrome is Li-Fraumeni syndrome, caused by p53 mutations.
- -
- Category 3: Proteins whose mutations cause category 3 cancer syndromes are not necessarily crucial for both integrity of the genome and control of the cell cycle but, when combined with the role of the mutated proteins as X-proteins in cytoplasm, the resulting delay of ATM nucleoshuttling concerns both the integrity of the genome and the control of the cell cycle and favors cellular transformation. A representative example of category 3 syndrome is neurofibromatosis type 1 syndrome, caused by NF1 mutations.
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Weiss, L. Metastasis of cancer: A conceptual history from antiquity to the 1990s. Cancer Metastasis Rev. 2000, 19, 193–383. [Google Scholar] [CrossRef]
- Sugano, H. The cancer problem—Carcinogenesis and prevention from the viewpoint of the natural history of cancer. Anticancer. Res. 1999, 19, 3787–3790. [Google Scholar]
- Di Lonardo, A.; Nasi, S.; Pulciani, S. Cancer: We should not forget the past. J. Cancer 2015, 6, 29–39. [Google Scholar] [CrossRef]
- Hippocrates. Complete Works; Union Latine Editions: Paris, France, 1955. [Google Scholar]
- Celsius, A. De Medicina; Les Belles Lettres: Paris, France, 2002. [Google Scholar]
- Galenus, C. Epitomas; Union Latine Editions: Paris, France, 1962. [Google Scholar]
- Alliot, J.-B. Traité du Cancer; François Muguet: Paris, France, 1698. [Google Scholar]
- Neuss, M.J. Blood money: Harvey’s De motu cordis (1628) as an exercise in accounting. Br. J. Hist. Sci. 2018, 51, 181–203. [Google Scholar] [CrossRef]
- Dorrington, K.L.; Frise, M.C. Lessons of the month 1: Learning from Harvey; improving blood-taking by pointing the needle in the right direction. Clin. Med. 2019, 19, 514–518. [Google Scholar] [CrossRef]
- Harvey, W. De Motu Cordis; Bourgeois, C., Ed.; Editions de “Il giardino di Esculapio”: Paris, France, 1991. [Google Scholar]
- Bainton, R.H. Michael Servetus and the pulmonary transit of the blood. Bull. Hist. Med. 1951, 25, 1–7. [Google Scholar]
- Voltaire. Elemens de la Philosophie de Newton Mis à la Portée de Tout le Monde; Voltaire, Ed.; Etienne Ledet et compagnie: Amsterdam, The Netherlands, 1738. [Google Scholar]
- McNichol, J. Primordial soup, fool’s gold, and spontaneous generation: A brief introduction to the theory, history, and philosophy of the search for the origin of life. Biochem. Mol. Biol. Educ. 2008, 36, 255–261. [Google Scholar] [CrossRef]
- Farley, J. The spontaneous generation controversy (1700–1860): The origin of parasitic worms. J. Hist. Biol. 1972, 5, 95–125. [Google Scholar] [CrossRef]
- Farley, J. The spontaneous generation controversy (1859–1880): British and German reactions to the problem of abiogenesis. J. Hist. Biol. 1972, 5, 285–319. [Google Scholar] [CrossRef]
- Farley, J.; Geison, G.L. Science, politics and spontaneous generation in nineteenth-century France: The Pasteur-Pouchet debate. Bull. Hist. Med. 1974, 48, 161–198. [Google Scholar]
- Martini, M.; Besozzi, G.; Barberis, I. The never-ending story of the fight against tuberculosis: From Koch’s bacillus to global control programs. J. Prev. Med. Hyg. 2018, 59, E241–E247. [Google Scholar] [CrossRef]
- Zetterstrom, R. Robert Koch (1843–1910): Investigations and discoveries in relation to tuberculosis. Acta Paediatr. 2006, 95, 514–516. [Google Scholar] [CrossRef]
- Debre, P. Pasteur at the Academy of Medicine: From hygiene to germ theory. Comptes Rendus Biol. 2022, 345, 83–92. [Google Scholar] [CrossRef]
- Absolon, K.B.; Absolon, M.J.; Zientek, R. From antisepsis to asepsis. Louis Pasteur’s publication on “The germ theory and its application to medicine and surgery”. Rev. Surg. 1970, 27, 245–258. [Google Scholar]
- Foray, N. Victor Despeignes, the forgotten pioneer of radiation oncology. Int. J. Radiat. Oncol. Biol. Phys. 2016, 96, 717–721. [Google Scholar] [CrossRef]
- Despeignes, V. Observation concernant un cas de cancer de l’estomac traité par les rayons Roentgen. Lyon. Med. 1896, 82, 428–430. [Google Scholar]
- Despeignes, V. Observation concernant un cas de cancer de l’estomac traité par les rayons Roentgen Secondes observations. Lyon. Med. 1896, 82, 503–506. [Google Scholar]
- Despeignes, V. Nouvelle observation de cancer traité par les rayons de Roentgen. Lyon. Med. 1896, 83, 550–551. [Google Scholar]
- Rowbury, R. Robert Hooke, 1635–1703. Sci. Prog. 2012, 95, 238–254. [Google Scholar] [CrossRef]
- Gest, H. The remarkable vision of Robert Hooke (1635–1703): First observer of the microbial world. Perspect. Biol. Med. 2005, 48, 266–272. [Google Scholar] [CrossRef]
- Gest, H. The discovery of microorganisms by Robert Hooke and Antoni Van Leeuwenhoek, fellows of the Royal Society. Notes Rec. R. Soc. Lond. 2004, 58, 187–201. [Google Scholar] [CrossRef]
- Danforth, I.N. The Cell Theories of Huxley and Virchow. Chic. Med. J. 1870, 27, 577–583. [Google Scholar]
- Wilson, J.W. Virchow’s contribution to the cell theory. J. Hist. Med. Allied Sci. 1947, 2, 163–178. [Google Scholar] [CrossRef]
- Fernandez-Flores, A.; Fonseca, E. Scrotal cancer, chimney sweepers and Sir Percival Pott. Clin. Dermatol. 2022, 40, 209–220. [Google Scholar] [CrossRef]
- Frumin, E.; Velez, H.; Bingham, E.; Gillen, M.; Brathwaite, M.; LaBarck, R. Occupational bladder cancer in textile dyeing and printing workers: Six cases and their significance for screening programs. J. Occup. Med. 1990, 32, 887–890. [Google Scholar] [CrossRef]
- Frieben, A. Cancroid des rechten Handrückens. Dtsch. Med. Wochenschr. 1902, 28, 335. [Google Scholar]
- Carlsson, F.; Raberg, L. The germ theory revisited: A noncentric view on infection outcome. Proc. Natl. Acad. Sci. USA 2024, 121, e2319605121. [Google Scholar] [CrossRef]
- Baltzer, F. Theodor Boveri. Science 1964, 144, 809–815. [Google Scholar] [CrossRef]
- Stern, C. Boveri and the early days of genetics. Nature 1950, 166, 446. [Google Scholar] [CrossRef]
- Scheer, U. Historical roots of centrosome research: Discovery of Boveri’s microscope slides in Wurzburg. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130469. [Google Scholar] [CrossRef]
- Ried, T. Homage to Theodor Boveri (1862–1915): Boveri’s theory of cancer as a disease of the chromosomes, and the landscape of genomic imbalances in human carcinomas. Environ. Mol. Mutagen. 2009, 50, 593–601. [Google Scholar] [CrossRef]
- Regaud, C. Sur la technique de la coloration des cellules nerveuses par le bleu de méthylène. In Proceedings of the Congrès des Médecins Aliénistes et Neurologistes, Bordeaux, France, 1–7 August 1895; pp. 1–18. [Google Scholar]
- Bergonié, J.; Tribondeau, L. Interprétation de quelques résultats de la radiothérapie et essai de fixation d’une technique rationnelle. Comptes-Rendus de L’académie des Sci. 1906, 143, 983–984. [Google Scholar]
- Vogin, G.; Foray, N. The law of Bergonie and Tribondeau: A nice formula for a first approximation. Int. J. Radiat. Biol. 2013, 89, 2–8. [Google Scholar] [CrossRef]
- Regaud, C. Travaux avec Thomas Nogier. Recherches sur les rayons X et la radiotherapie. Recherches sur les rayons X et la radiotherapie; Fonds. Claudius Regaud (1905–1940); Institut Curie: Paris, France, 1908; CR1B. [Google Scholar]
- Regaud, C. Etudes sur la structure des tubes séminifères et sur la spermatogenèse chez les mammifères. Arch. D’anatomie Microsc. 1901, 4, 101–155. [Google Scholar]
- Regaud, C. Action des rayons de Roentgen sur l’épithélium séminal. Application des résultats à certains problèmes concenrant la structure et les fonctions de cet épithélium. Comptes-Rendus de L’association des Anat.-Pathol. 1907, 9, 30. [Google Scholar]
- Regaud, C. Réflexions et hypothèses au sujet de la pathogénie des cancers. Paris Med. 1941, 31, 125–141. [Google Scholar]
- Calabrese, E.J. Muller’s Nobel Prize Lecture: When ideology prevailed over science. Toxicol. Sci. Off. J. Soc. Toxicol. 2012, 126, 1–4. [Google Scholar] [CrossRef]
- Berenblum, I. The Mechanism of Carcinogenesis. A Study of the Significance of Cocarcinogenic Action and Related Phenomena. Cancer Res. 1941, 1, 807–814. [Google Scholar] [CrossRef]
- Friedewald, W.F.; Rous, P. The Determining Influence of Tar, Benzpyrene, and Methylcholanthrene on the Character of the Benign Tumors Induced Therewith in Rabbit Skin. J. Exp. Med. 1944, 80, 127–144. [Google Scholar] [CrossRef]
- Friedewald, W.F.; Rous, P. The Initiating and Promoting Elements in Tumor Production: An Analysis of the Effects of Tar, Benzpyrene, and Methylcholanthrene on Rabbit Skin. J. Exp. Med. 1944, 80, 101–126. [Google Scholar] [CrossRef]
- Berenblum, I.; Shubik, P. A new, quantitative, approach to the study of the stages of chemical cartinogenesis in the mouse’s skin. Br. J. Cancer 1947, 1, 383–391. [Google Scholar] [CrossRef]
- Rous, P.; Beard, J.W. The Progression to Carcinoma of Virus-Induced Rabbit Papillomas (Shope). J. Exp. Med. 1935, 62, 523–548. [Google Scholar] [CrossRef]
- Foulds, L. The experimental study of tumor progression: A review. Cancer Res. 1954, 14, 327–339. [Google Scholar]
- Foulds, L. Tumor progression. Cancer Res. 1957, 17, 355–356. [Google Scholar]
- Foulds, L. Progression and carcinogenesis. Acta Unio Int. Contra Cancrum 1961, 17, 148–156. [Google Scholar]
- Rubin, H. Experimental control of neoplastic progression in cell populations: Foulds’ rules revisited. Proc. Natl. Acad. Sci. USA 1994, 91, 6619–6623. [Google Scholar] [CrossRef]
- Harris, H.; Miller, O.J.; Klein, G.; Worst, P.; Tachibana, T. Suppression of malignancy by cell fusion. Nature 1969, 223, 363–368. [Google Scholar] [CrossRef]
- Stehelin, D.; Varmus, H.E.; Bishop, J.M.; Vogt, P.K. DNA related to the transforming gene(s) of avian sarcoma viruses is present in normal avian DNA. Nature 1976, 260, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Raju, T.N. The Nobel Chronicles. 1989: John Michael Bishop (b 1936) and Harold Eliot Varmus (b 1939). Lancet 2000, 355, 1106. [Google Scholar] [CrossRef] [PubMed]
- Tu, S.M. Stem Cell Theory of Cancer: Implications of a Viral Etiology in Certain Malignancies. Cancers 2021, 13, 2738. [Google Scholar] [CrossRef] [PubMed]
- Knudson, A.G., Jr. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [PubMed]
- Knudson, A.G. Antioncogenes and human cancer. Proc. Natl. Acad. Sci. USA 1993, 90, 10914–10921. [Google Scholar] [CrossRef]
- Cousineau, I.; Belmaaza, A. BRCA1 haploinsufficiency, but not heterozygosity for a BRCA1-truncating mutation, deregulates homologous recombination. Cell Cycle 2007, 6, 962–971. [Google Scholar] [CrossRef] [PubMed]
- Belmar-Lopez, C.; Mancheno-Corvo, P.; Saornil, M.A.; Baril, P.; Vassaux, G.; Quintanilla, M.; Martin-Duque, P. Uveal vs. cutaneous melanoma. Origins and causes of the differences. Clin. Transl. Oncol. 2008, 10, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Saini, V.; Hose, C.D.; Monks, A.; Nagashima, K.; Han, B.; Newton, D.L.; Millione, A.; Shah, J.; Hollingshead, M.G.; Hite, K.M.; et al. Identification of CBX3 and ABCA5 as putative biomarkers for tumor stem cells in osteosarcoma. PLoS ONE 2012, 7, e41401. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Hu, X.; Huang, X.; He, C. Quantitative analyses of CD133 expression facilitate researches on tumor stem cells. Biol. Pharm. Bull. 2010, 33, 738–742. [Google Scholar] [CrossRef] [PubMed]
- Gholamzad, A.; Khakpour, N.; Khosroshahi, E.M.; Asadi, S.; Koohpar, Z.K.; Matinahmadi, A.; Jebali, A.; Rashidi, M.; Hashemi, M.; Sadi, F.H.; et al. Cancer stem cells: The important role of CD markers, Signaling pathways, and MicroRNAs. Pathol. Res. Pract. 2024, 256, 155227. [Google Scholar] [CrossRef]
- Nordling, C.O. Evidence regarding the multiple mutation theory of the cancer-inducing mechanism. Acta Genet. Stat. Med. 1955, 5, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Kinzler, K.W.; Vogelstein, B. Cancer-susceptibility genes. Gatekeepers and caretakers. Nature 1997, 386, 761–763. [Google Scholar] [CrossRef] [PubMed]
- Michor, F.; Iwasa, Y.; Nowak, M.A. Dynamics of cancer progression. Nat. Rev. Cancer 2004, 4, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Michor, F.; Iwasa, Y.; Vogelstein, B.; Lengauer, C.; Nowak, M.A. Can chromosomal instability initiate tumorigenesis? Semin. Cancer Biol. 2005, 15, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.Z.; Jin, W.L. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct. Target. Ther. 2020, 5, 166. [Google Scholar] [CrossRef] [PubMed]
- Kelber, J.A.; Iwanicki, M.; Kruithof-de Julio, M.; Spike, B.T.; Martinez-Montemayor, M.M. Editorial: Mechanisms of microenvironment governed plasticity and progression in solid tumors. Front. Cell Dev. Biol. 2024, 12, 1373496. [Google Scholar] [CrossRef] [PubMed]
- Cavallaro, U.; Christofori, G. Molecular mechanisms of tumor angiogenesis and tumor progression. J. Neurooncol. 2000, 50, 63–70. [Google Scholar] [CrossRef]
- Maniotis, A.J.; Folberg, R.; Hess, A.; Seftor, E.A.; Gardner, L.M.; Pe’er, J.; Trent, J.M.; Meltzer, P.S.; Hendrix, M.J. Vascular channel formation by human melanoma cells in vivo and in vitro: Vasculogenic mimicry. Am. J. Pathol. 1999, 155, 739–752. [Google Scholar] [CrossRef]
- Goncharov, A.P.; Vashakidze, N.; Kharaishvili, G. Epithelial-Mesenchymal Transition: A Fundamental Cellular and Microenvironmental Process in Benign and Malignant Prostate Pathologies. Biomedicines 2024, 12, 418. [Google Scholar] [CrossRef]
- Varisli, L.; Vlahopoulos, S. Epithelial-Mesenchymal Transition in Acute Leukemias. Int. J. Mol. Sci. 2024, 25, 2173. [Google Scholar] [CrossRef]
- Tomasetti, C.; Vogelstein, B. Cacer etiology. Variation in cancer risk among tissues can be explainedby the number of stem cell divisions. Science 2015, 347, 78–81. [Google Scholar] [CrossRef]
- Tomasetti, C.; Lu, L.; Vogelstein, B. Stem cell divisions, somatic mutations, cancer etiology and cancer prevention. Science 2017, 355, 1330–1334. [Google Scholar] [CrossRef]
- Wu, S.; Powers, S.; Zhu, W.; Hannun, Y.A. Substantial contribution pf extrinsic factors to cancer development. Nature 2016, 529, 43–47. [Google Scholar] [CrossRef]
- Tomita, M.; Maeda, M.; Maezawa, H.; Usami, N.; Kobayashi, K. Bystander cell killing in normal human fibroblasts is induced by synchrotron X-ray microbeams. Radiat. Res. 2010, 173, 380–385. [Google Scholar] [CrossRef]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef]
- Taby, R.; Issa, J.-P. Cancer Epigenetics. CA Cancer J. Clin. 2010, 60, 376–392. [Google Scholar] [CrossRef]
- Berthel, E.; Foray, N.; Ferlazzo, M.L. The Nucleoshuttling of the ATM Protein: A Unified Model to Describe the Individual Response to High- and Low-Dose of Radiation? Cancers 2019, 11, 905. [Google Scholar] [CrossRef]
- Bodgi, L.; Foray, N. The nucleo-shuttling of the ATM protein as a basis for a novel theory of radiation response: Resolution of the linear-quadratic model. Int. J. Radiat. Biol. 2016, 92, 117–131. [Google Scholar] [CrossRef]
- Granzotto, A.; Benadjaoud, M.A.; Vogin, G.; Devic, C.; Ferlazzo, M.L.; Bodgi, L.; Pereira, S.; Sonzogni, L.; Forcheron, F.; Viau, M.; et al. Influence of Nucleoshuttling of the ATM Protein in the Healthy Tissues Response to Radiation Therapy: Toward a Molecular Classification of Human Radiosensitivity. Int. J. Radiat. Oncol. Biol. Phys. 2016, 94, 450–460. [Google Scholar] [CrossRef]
- Berthel, E.; Pujo-Menjouet, L.; Le Reun, E.; Sonzogni, L.; Al-Choboq, J.; Chekroun, A.; Granzotto, A.; Devic, C.; Ferlazzo, M.L.; Pereira, S.; et al. Toward an early diagnosis for Alzheimer’s disease based on the perinuclear localization of the ATM protein. Cells 2023, 12, 1747. [Google Scholar] [CrossRef]
- El Nachef, L.; Berthel, E.; Ferlazzo, M.L.; Le Reun, E.; Al-Choboq, J.; Restier-Verlet, J.; Granzotto, A.; Sonzogni, L.; Bourguignon, M.; Foray, N. Cancer and Radiosensitivity Syndromes: Is Impaired Nuclear ATM Kinase Activity the Primum Movens? Cancers 2022, 14, 6141. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Nachef, L.; Bouchet, A.; Bourguignon, M.; Foray, N. When DNA Mutations Interplay with Cellular Proliferation: A Narrative History of Theories of Carcinogenesis. Cancers 2024, 16, 2104. https://doi.org/10.3390/cancers16112104
El Nachef L, Bouchet A, Bourguignon M, Foray N. When DNA Mutations Interplay with Cellular Proliferation: A Narrative History of Theories of Carcinogenesis. Cancers. 2024; 16(11):2104. https://doi.org/10.3390/cancers16112104
Chicago/Turabian StyleEl Nachef, Laura, Audrey Bouchet, Michel Bourguignon, and Nicolas Foray. 2024. "When DNA Mutations Interplay with Cellular Proliferation: A Narrative History of Theories of Carcinogenesis" Cancers 16, no. 11: 2104. https://doi.org/10.3390/cancers16112104
APA StyleEl Nachef, L., Bouchet, A., Bourguignon, M., & Foray, N. (2024). When DNA Mutations Interplay with Cellular Proliferation: A Narrative History of Theories of Carcinogenesis. Cancers, 16(11), 2104. https://doi.org/10.3390/cancers16112104