
N-Benzylated 5-Hydroxybenzothiophene-2-carboxamides as Multi-Targeted Clk/Dyrk Inhibitors and Potential Anticancer Agents

 , , , , , and
, , , , , and 
Abstract
Simple Summary
Abstract
1. Introduction
2. Results and Discussion
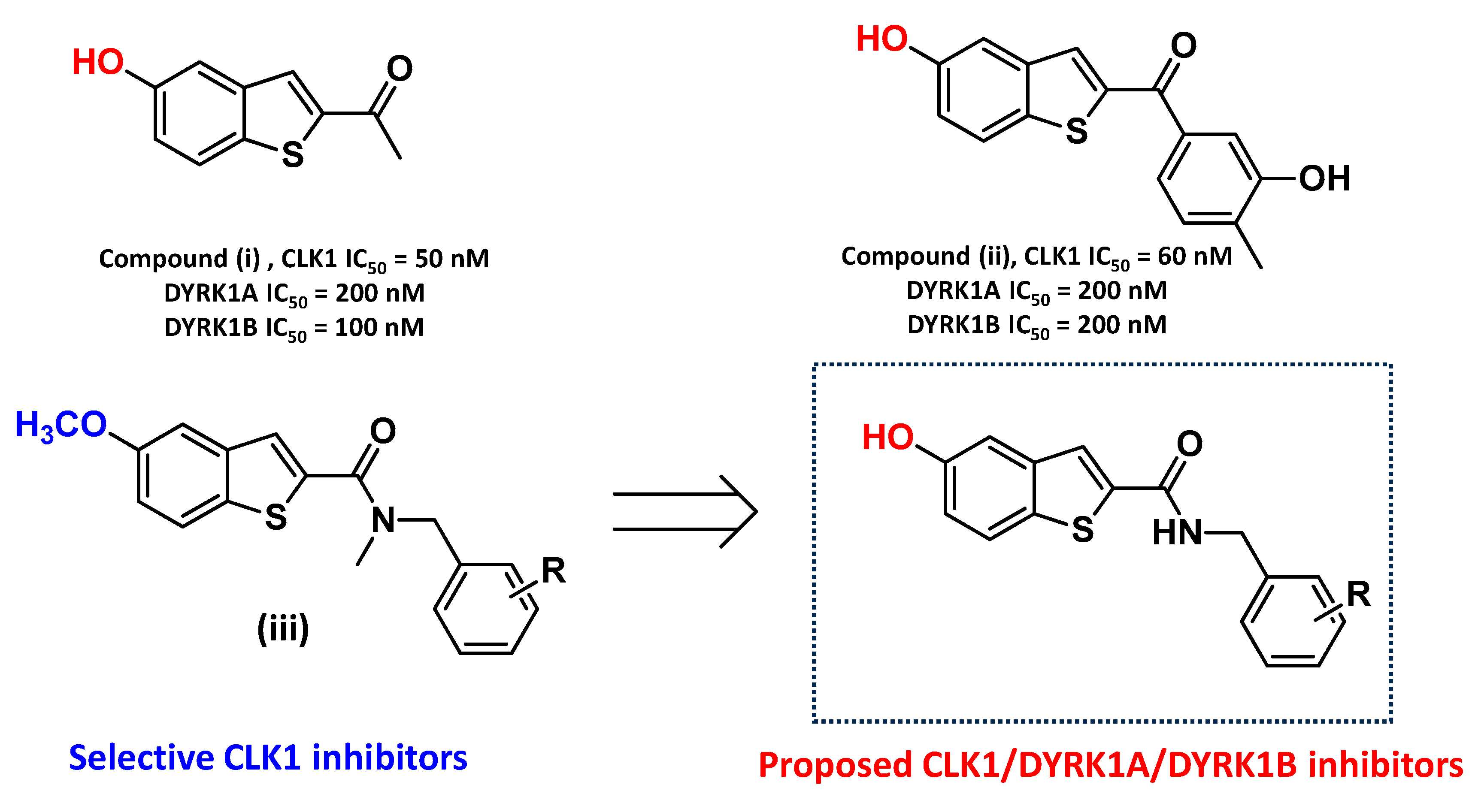
2.1. Compound Design
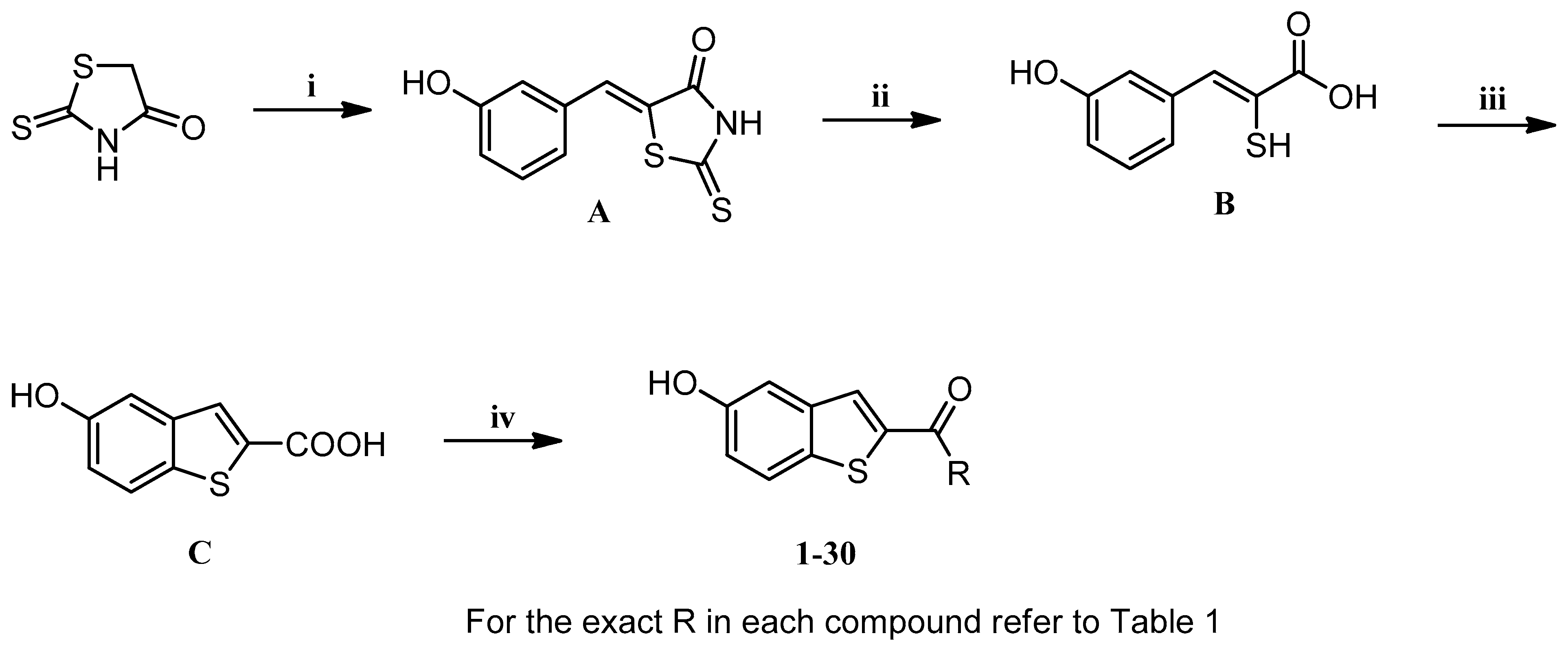
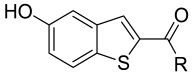
2.2. Chemistry
2.3. Biological Evaluation
2.3.1. Structure–Activity Relationships (SARs) for Dyrk1A, Dyrk1B, and Clk1 Inhibition
2.3.2. Screening against Relevant Kinases That Are Overexpressed in Several Types of Cancer
2.3.3. Inhibition of Tumor Cell Viability in a Panel of Human Cancer Cell Lines
2.3.4. Evaluation of Safety against Normal Cell Lines
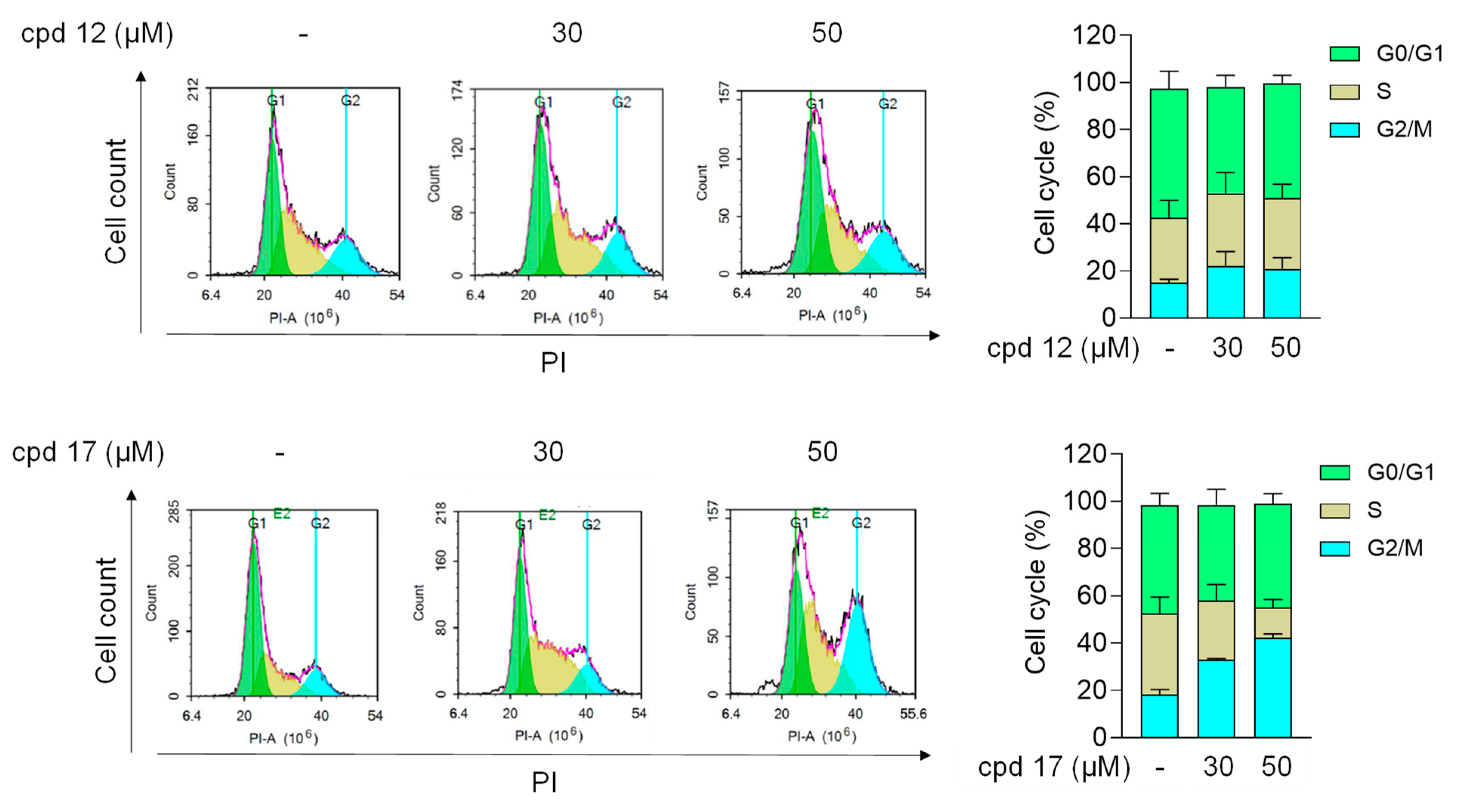
2.3.5. Effect of Compounds 12 and 17 on Cell Cycle in T24 Cells
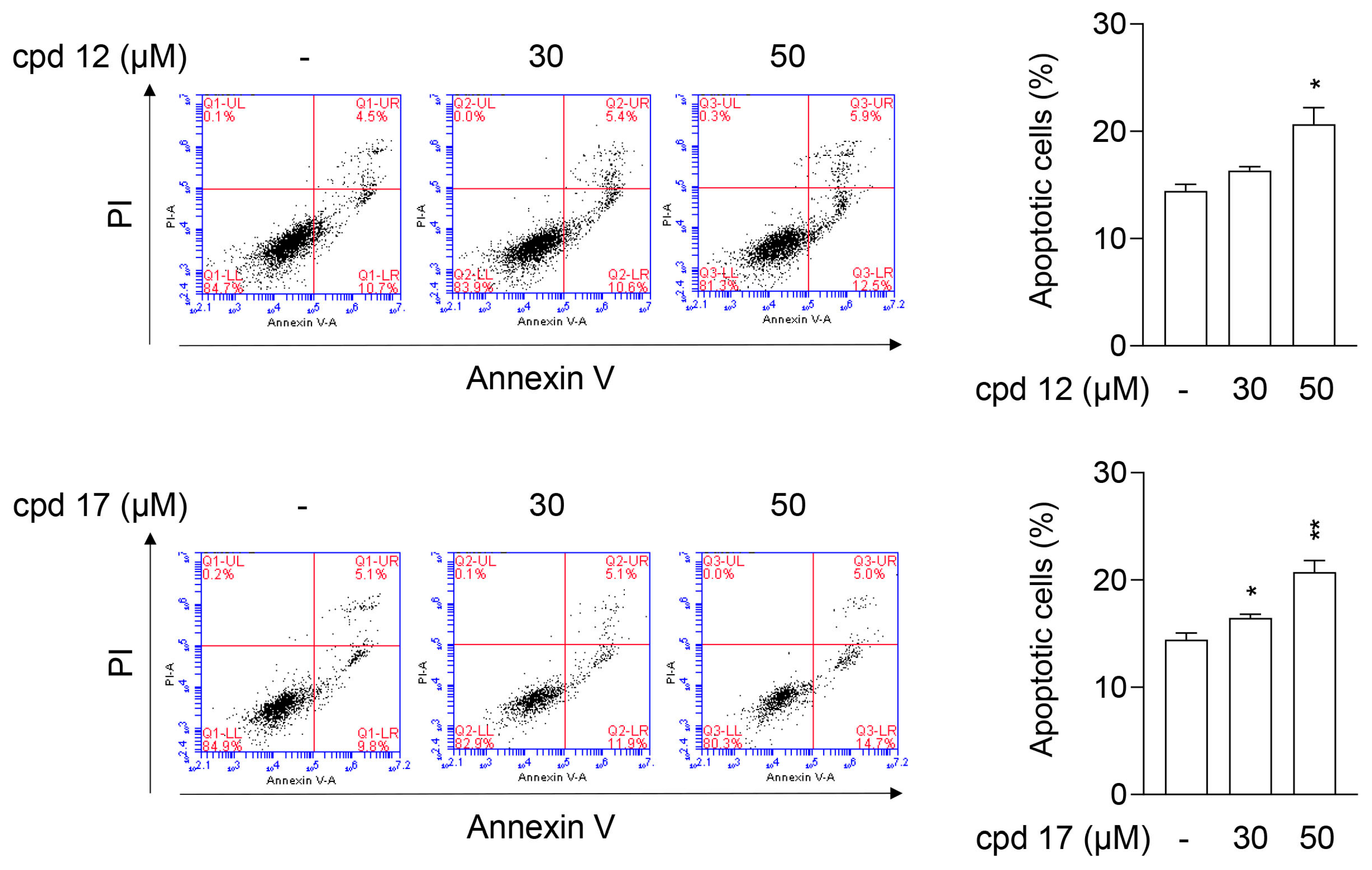
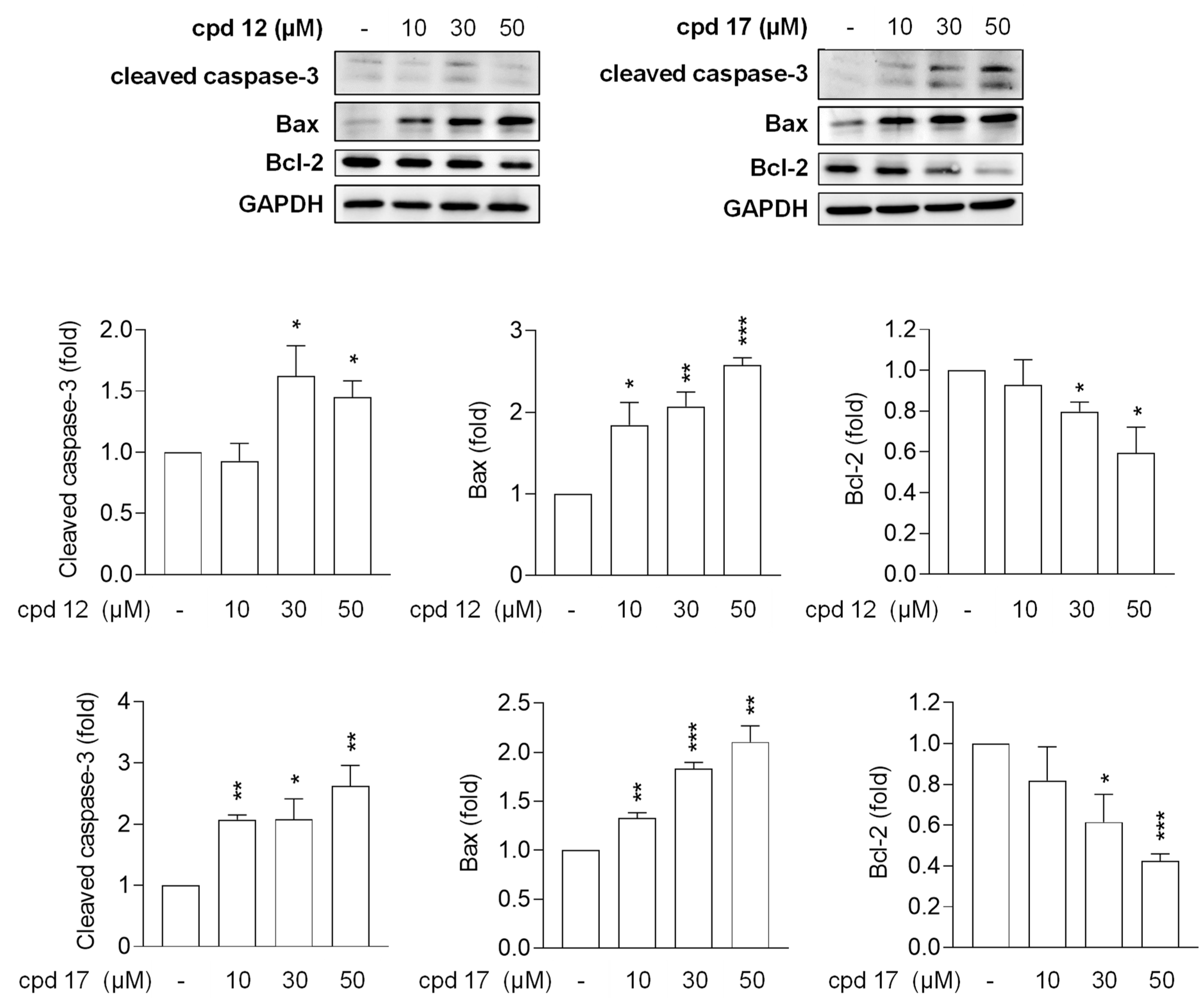
2.3.6. Compounds 12 and 17 Lead to Apoptotic Cell Death in T24 Cells
2.3.7. Compounds 12 and 17 Trigger Proapoptotic Pathways in T24 Cells
3. Conclusions
4. Experimental
4.1. Chemistry
4.1.1. General Synthetic Procedures and Experimental Details
Procedure for Synthesis of Compound 1–30 (Scheme 1)
N-Benzyl-5-hydroxybenzo[b]thiophene-2-carboxamide (1)
N-(2-Chlorobenzyl)-5-hydroxybenzo[b]thiophene-2-carboxamide (2)
N-(3-Chlorobenzyl)-5-hydroxybenzo[b]thiophene-2-carboxamide (3)
N-(4-Chlorobenzyl)-5-hydroxybenzo[b]thiophene-2-carboxamide (4)
N-(2-Fluorobenzyl)-5-hydroxybenzo[b]thiophene-2-carboxamide (5)
N-(3-Fluorobenzyl)-5-hydroxybenzo[b]thiophene-2-carboxamide (6)
N-(4-Fluorobenzyl)-5-hydroxybenzo[b]thiophene-2-carboxamide (7)
5-Hydroxy-N-(2-methylbenzyl)benzo[b]thiophene-2-carboxamide (8)
5-Hydroxy-N-(3-methylbenzyl)benzo[b]thiophene-2-carboxamide (9)
5-Hydroxy-N-(4-methylbenzyl)benzo[b]thiophene-2-carboxamide (10)
5-Hydroxy-N-(2-methoxybenzyl)benzo[b]thiophene-2-carboxamide (11)
5-Hydroxy-N-(3-methoxybenzyl)benzo[b]thiophene-2-carboxamide (12)
5-Hydroxy-N-(4-methoxybenzyl)benzo[b]thiophene-2-carboxamide (13)
N-(3,5-Dichlorobenzyl)-5-hydroxybenzo[b]thiophene-2-carboxamide (14)
N-(2,3-Difluorobenzyl)-5-hydroxybenzo[b]thiophene-2-carboxamide (15)
N-(2,4-Difluorobenzyl)-5-hydroxybenzo[b]thiophene-2-carboxamide (16)
N-(3,5-Difluorobenzyl)-5-hydroxybenzo[b]thiophene-2-carboxamide (17)
N-(3,5-Bis(trifluoromethyl)benzyl)-5-hydroxybenzo[b]thiophene-2-carboxamide (18)
N-(3,5-Dimethylbenzyl)-5-hydroxybenzo[b]thiophene-2-carboxamide (19)
N-(2,3-Dimethoxybenzyl)-5-hydroxybenzo[b]thiophene-2-carboxamide (20)
N-(2,4-Dimethoxybenzyl)-5-hydroxybenzo[b]thiophene-2-carboxamide (21)
N-(3,4-Dimethoxybenzyl)-5-hydroxybenzo[b]thiophene-2-carboxamide (22)
N-(3,5-Dimethoxybenzyl)-5-hydroxybenzo[b]thiophene-2-carboxamide (23)
(3,4-Dihydroisoquinolin-2(1H)-yl)(5-hydroxybenzo[b]thiophen-2-yl)methanone (24)
(5-Chloroisoindolin-2-yl)(5-hydroxybenzo[b]thiophen-2-yl)methanone (25)
5-Hydroxy-N-(pyridin-3-ylmethyl)benzo[b]thiophene-2-carboxamide (26)
5-Hydroxy-N-(thiophen-2-ylmethyl)benzo[b]thiophene-2-carboxamide (27)
N-(Furan-2-Ylmethyl)-5-hydroxybenzo[b]thiophene-2-carboxamide (28)
5-Hydroxy-N-phenethylbenzo[b]thiophene-2-carboxamide (29)
N-(2-(1H-Indol-3-yl)ethyl)-5-hydroxybenzo[b]thiophene-2-carboxamide (30)
4.2. Biological Assays
4.2.1. Protein Kinases and Inhibition Assays
4.2.2. Cell Culture
4.2.3. Cell Viability
4.2.4. Cell Cycle Assay
4.2.5. Apoptosis Assay
4.2.6. Western Blot
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Bray, F.; Laversanne, M.; Weiderpass, E.; Soerjomataram, I. The Ever-increasing Importance of Cancer as a Leading Cause of Premature Death Worldwide. Cancer 2021, 127, 3029–3030. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Fu, R.; Sun, Y.; Sheng, W.; Liao, D. Designing Multi-Targeted Agents: An Emerging Anticancer Drug Discovery Paradigm. Eur. J. Med. Chem. 2017, 136, 195–211. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Ma, S. Recent Advances in the Discovery of Multitargeted Tyrosine Kinase Inhibitors as Anticancer Agents. ChemMedChem 2021, 16, 600–620. [Google Scholar] [CrossRef] [PubMed]
- Mokhtari, R.B.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination Therapy in Combating Cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef] [PubMed]
- Dhokne, P.; Sakla, A.P.; Shankaraiah, N. Structural Insights of Oxindole Based Kinase Inhibitors as Anticancer Agents: Recent Advances. Eur. J. Med. Chem. 2021, 216, 113334. [Google Scholar] [CrossRef] [PubMed]
- Kentrup, H.; Becker, W.; Heukelbach, J.; Wilmes, A.; Schürmann, A.; Huppertz, C.; Kainulainen, H.; Joost, H.-G. Dyrk, a Dual Specificity Protein Kinase with Unique Structural Features Whose Activity Is Dependent on Tyrosine Residues between Subdomains VII and VIII. J. Biol. Chem. 1996, 271, 3488–3495. [Google Scholar] [CrossRef] [PubMed]
- Dekel, N.; Eisenberg-Domovich, Y.; Karlas, A.; Meyer, T.F.; Bracher, F.; Lebendiker, M.; Danieli, T.; Livnah, O. Expression, Purification and Crystallization of CLK1 Kinase—A Potential Target for Antiviral Therapy. Protein Expr. Purif. 2020, 176, 105742. [Google Scholar] [CrossRef]
- Walte, A.; Rüben, K.; Birner-Gruenberger, R.; Preisinger, C.; Bamberg-Lemper, S.; Hilz, N.; Bracher, F.; Becker, W. Mechanism of Dual Specificity Kinase Activity of DYRK1A. FEBS J. 2013, 280, 4495–4511. [Google Scholar] [CrossRef]
- Lindberg, M.F.; Meijer, L. Dual-Specificity, Tyrosine Phosphorylation-Regulated Kinases (DYRKs) and Cdc2-Like Kinases (CLKs) in Human Disease, an Overview. Int. J. Mol. Sci. 2021, 22, 6047. [Google Scholar] [CrossRef]
- Soppa, U.; Becker, W. DYRK Protein Kinases. Curr. Biol. 2015, 25, R488–R489. [Google Scholar] [CrossRef]
- Pozo, N.; Zahonero, C.; Fernández, P.; Liñares, J.M.; Ayuso, A.; Hagiwara, M.; Pérez, A.; Ricoy, J.R.; Hernández-Laín, A.; Sepúlveda, J.M.; et al. Inhibition of DYRK1A Destabilizes EGFR and Reduces EGFR-Dependent Glioblastoma Growth. J. Clin. Investig. 2013, 123, 2475–2487. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Zheng, Z.; Rawal, B.; Schell, M.J.; Bepler, G.; Haura, E.B. Mirk/Dyrk1B, a Novel Therapeutic Target, Mediates Cell Survival in Non-Small Cell Lung Cancer Cells. Cancer Biol. Ther. 2009, 8, 1671–1679. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Yang, X.; Yin, P.; Hu, W.; Liao, H.; Miao, Z.; Pan, C.; Li, N. The Involvement of FoxO in Cell Survival and Chemosensitivity Mediated by Mirk/Dyrk1B in Ovarian Cancer. Int. J. Oncol. 2011, 40, 1203–1209. [Google Scholar] [CrossRef] [PubMed]
- Seifert, A.; Allan, L.A.; Clarke, P.R. DYRK1A Phosphorylates Caspase 9 at an Inhibitory Site and Is Potently Inhibited in Human Cells by Harmine. FEBS J. 2008, 275, 6268–6280. [Google Scholar] [CrossRef] [PubMed]
- Becker, W. A Wake-up Call to Quiescent Cancer Cells—Potential Use of DYRK1B Inhibitors in Cancer Therapy. FEBS J. 2018, 285, 1203–1211. [Google Scholar] [CrossRef] [PubMed]
- Becker, W. Emerging Role of DYRK Family Protein Kinases as Regulators of Protein Stability in Cell Cycle Control. Cell Cycle 2012, 11, 3389–3394. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.; Medda, F.; Gokhale, V.; Dunckley, T.; Hulme, C. Recent Advances in the Design, Synthesis, and Biological Evaluation of Selective DYRK1A Inhibitors: A New Avenue for a Disease Modifying Treatment of Alzheimers? ACS Chem. Neurosci. 2012, 3, 857–872. [Google Scholar] [CrossRef] [PubMed]
- Tarpley, M.; Oladapo, H.O.; Strepay, D.; Caligan, T.B.; Chdid, L.; Shehata, H.; Roques, J.R.; Thomas, R.; Laudeman, C.P.; Onyenwoke, R.U.; et al. Identification of Harmine and β-Carboline Analogs from a High-Throughput Screen of an Approved Drug Collection; Profiling as Differential Inhibitors of DYRK1A and Monoamine Oxidase A and for in Vitro and in Vivo Anti-Cancer Studies. Eur. J. Pharm. Sci. 2021, 162, 105821. [Google Scholar] [CrossRef]
- Loaëc, N.; Attanasio, E.; Villiers, B.; Durieu, E.; Tahtouh, T.; Cam, M.; Davis, R.; Alencar, A.; Roué, M.; Bourguet-Kondracki, M.-L.; et al. Marine-Derived 2-Aminoimidazolone Alkaloids. Leucettamine B-Related Polyandrocarpamines Inhibit Mammalian and Protozoan DYRK & CLK Kinases. Mar. Drugs 2017, 15, 316. [Google Scholar] [CrossRef]
- Bain, J.; McLauchlan, H.; Elliott, M.; Cohen, P. The Specificities of Protein Kinase Inhibitors: An Update. Biochem. J. 2003, 371, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Lamoral-Theys, D.; Pottier, L.; Dufrasne, F.; Neve, J.; Dubois, J.; Kornienko, A.; Kiss, R.; Ingrassia, L. Natural Polyphenols That Display Anticancer Properties through Inhibition of Kinase Activity. Curr. Med. Chem. 2010, 17, 812–825. [Google Scholar] [CrossRef] [PubMed]
- Abe, A.; Kokuba, H. Harmol Induces Autophagy and Subsequent Apoptosis in U251MG Human Glioma Cells through the Downregulation of Survivin. Oncol. Rep. 2013, 29, 1333–1342. [Google Scholar] [CrossRef]
- Abe, A.; Yamada, H. Harmol Induces Apoptosis by Caspase-8 Activation Independently on Fas/Fas Ligand Interaction in Human Lung Carcinoma H596 Cells. Anticancer Drugs 2009, 20, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Lee Walmsley, D.; Murray, J.B.; Dokurno, P.; Massey, A.J.; Benwell, K.; Fiumana, A.; Foloppe, N.; Ray, S.; Smith, J.; Surgenor, A.E.; et al. Fragment-Derived Selective Inhibitors of Dual-Specificity Kinases DYRK1A and DYRK1B. J. Med. Chem. 2021, 64, 8971–8991. [Google Scholar] [CrossRef]
- Powell, C.E.; Hatcher, J.M.; Jiang, J.; Vatsan, P.S.; Che, J.; Gray, N.S. Selective Macrocyclic Inhibitors of DYRK1A/B. ACS Med. Chem. Lett. 2022, 13, 577–585. [Google Scholar] [CrossRef]
- Bourahla, K.; Guihéneuf, S.; Limanton, E.; Paquin, L.; Le Guével, R.; Charlier, T.; Rahmouni, M.; Durieu, E.; Lozach, O.; Carreaux, F.; et al. Design and Microwave Synthesis of New (5Z) 5-Arylidene-2-Thioxo-1,3-Thiazolinidin-4-One and (5Z) 2-Amino-5-Arylidene-1,3-Thiazol-4(5H)-One as New Inhibitors of Protein Kinase DYRK1A. Pharmaceuticals 2021, 14, 1086. [Google Scholar] [CrossRef] [PubMed]
- Ewton, D.Z.; Lee, K.; Deng, X.; Lim, S.; Friedman, E. Rapid Turnover of Cell-Cycle Regulators Found in Mirk/Dyrk1B Transfectants. Int. J. Cancer 2003, 103, 21–28. [Google Scholar] [CrossRef]
- Ashford, A.L.; Oxley, D.; Kettle, J.; Hudson, K.; Guichard, S.; Cook, S.J.; Lochhead, P.A. A Novel DYRK1B Inhibitor AZ191 Demonstrates That DYRK1B Acts Independently of GSK3β to Phosphorylate Cyclin D1 at Thr286, Not Thr288. Biochem. J. 2014, 457, 43–56. [Google Scholar] [CrossRef]
- Deng, X.; Mercer, S.E.; Shah, S.; Ewton, D.Z.; Friedman, E. The Cyclin-Dependent Kinase Inhibitor P27Kip1 Is Stabilized in G0 by Mirk/Dyrk1B Kinase. J. Biol. Chem. 2004, 279, 22498–22504. [Google Scholar] [CrossRef]
- Friedman, E. Mirk/Dyrk1B in Cancer. J. Cell. Biochem. 2007, 102, 274–279. [Google Scholar] [CrossRef] [PubMed]
- Correa-Sáez, A.; Jiménez-Izquierdo, R.; Garrido-Rodríguez, M.; Morrugares, R.; Muñoz, E.; Calzado, M.A. Updating Dual-Specificity Tyrosine-Phosphorylation-Regulated Kinase 2 (DYRK2): Molecular Basis, Functions and Role in Diseases. Cell. Mol. Life Sci. 2020, 77, 4747–4763. [Google Scholar] [CrossRef] [PubMed]
- Jain, P.; Karthikeyan, C.; Moorthy, N.S.; Waiker, D.; Jain, A.; Trivedi, P. Human CDC2-Like Kinase 1 (CLK1): A Novel Target for Alzheimer’s Disease. Curr. Drug Targets 2014, 15, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Muraki, M.; Ohkawara, B.; Hosoya, T.; Onogi, H.; Koizumi, J.; Koizumi, T.; Sumi, K.; Yomoda, J.; Murray, M.V.; Kimura, H.; et al. Manipulation of Alternative Splicing by a Newly Developed Inhibitor of Clks. J. Biol. Chem. 2004, 279, 24246–24254. [Google Scholar] [CrossRef] [PubMed]
- ElHady, A.K.; El-Gamil, D.S.; Abadi, A.H.; Abdel-Halim, M.; Engel, M. An Overview of Cdc2-like Kinase 1 (Clk1) Inhibitors and Their Therapeutic Indications. Med. Res. Rev. 2023, 43, 343–398. [Google Scholar] [CrossRef] [PubMed]
- da Silva, M.R.; Moreira, G.A.; Gonçalves da Silva, R.A.; de Almeida Alves Barbosa, É.; Pais Siqueira, R.; Teixera, R.R.; Almeida, M.R.; Silva Júnior, A.; Fietto, J.L.R.; Bressan, G.C. Splicing Regulators and Their Roles in Cancer Biology and Therapy. BioMed Res. Int. 2015, 2015, 150514. [Google Scholar] [CrossRef] [PubMed]
- ElHady, A.K.; El-Gamil, D.S.; Chen, P.-J.; Hwang, T.-L.; Abadi, A.H.; Abdel-Halim, M.; Engel, M. 5-Methoxybenzothiophene-2-Carboxamides as Inhibitors of Clk1/4: Optimization of Selectivity and Cellular Potency. Molecules 2021, 26, 1001. [Google Scholar] [CrossRef]
- ElHady, A.K.; Abdel-Halim, M.; Abadi, A.H.; Engel, M. Development of Selective Clk1 And-4 Inhibitors for Cellular Depletion of Cancer-Relevant Proteins. J. Med. Chem. 2017, 60, 5377–5391. [Google Scholar] [CrossRef] [PubMed]
- El-Gamil, D.S.; ElHady, A.K.; Chen, P.-J.; Hwang, T.-L.; Abadi, A.H.; Abdel-Halim, M.; Engel, M. Discovery of Novel 5-Methoxybenzothiophene Hydrazides as Metabolically Stable Clk1 Inhibitors with High Potency and Unprecedented Clk1 Isoenzyme Selectivity. Eur. J. Med. Chem. 2023, 247, 115019. [Google Scholar] [CrossRef]
- El-Gamil, D.S.; ElHady, A.K.; Chen, P.-J.; Hwang, T.-L.; Abadi, A.H.; Abdel-Halim, M.; Engel, M. Development of Novel Conformationally Restricted Selective Clk1/4 Inhibitors through Creating an Intramolecular Hydrogen Bond Involving an Imide Linker. Eur. J. Med. Chem. 2022, 238, 114411. [Google Scholar] [CrossRef]
- Zhang, Y.; Xia, A.; Zhang, S.; Lin, G.; Liu, J.; Chen, P.; Mu, B.; Jiao, Y.; Xu, W.; Chen, M.; et al. Discovery of 3,6-Disubstutited-Imidazo [1,2-a]Pyridine Derivatives as a New Class of CLK1 Inhibitors. Bioorg. Med. Chem. Lett. 2021, 41, 127881. [Google Scholar] [CrossRef] [PubMed]
- Massey, A.J.; Benwell, K.; Burbridge, M.; Kotschy, A.; Walmsley, D.L. Targeting DYRK1A/B Kinases to Modulate P21-cyclin D1-p27 Signalling and Induce Anti-tumour Activity in a Model of Human Glioblastoma. J. Cell. Mol. Med. 2021, 25, 10650–10662. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Phoa, A.F.; Abbassi, R.H.; Hoque, M.; Reekie, T.A.; Font, J.S.; Ryan, R.M.; Stringer, B.W.; Day, B.W.; Johns, T.G.; et al. Structural Optimization and Pharmacological Evaluation of Inhibitors Targeting Dual-Specificity Tyrosine Phosphorylation-Regulated Kinases (DYRK) and CDC-like Kinases (CLK) in Glioblastoma. J. Med. Chem. 2017, 60, 2052–2070. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, C.; Kail, D.; Mariano, M.; Empting, M.; Weber, N.; Paul, T.; Hartmann, R.W.; Engel, M. Design and Synthesis of a Library of Lead-like 2,4-Bisheterocyclic Substituted Thiophenes as Selective Dyrk/Clk Inhibitors. PLoS ONE 2014, 9, e87851. [Google Scholar] [CrossRef]
- Schmitt, C.; Miralinaghi, P.; Mariano, M.; Hartmann, R.W.; Engel, M. Hydroxybenzothiophene Ketones Are Efficient Pre-MRNA Splicing Modulators Due to Dual Inhibition of Dyrk1A and Clk1/4. ACS Med. Chem. Lett. 2014, 5, 963–967. [Google Scholar] [CrossRef] [PubMed]
- Corr, B.R.; Moroney, M.R.; Woodruff, E.; Watson, Z.L.; Jordan, K.R.; Danhorn, T.; Bailey, C.; Wolsky, R.J.; Bitler, B.G. Combination CDC-like Kinase Inhibition (CLK)/Dual-Specificity Tyrosine-Regulated Kinase (DYRK) and Taxane Therapy in CTNNB1-Mutated Endometrial Cancer. bioRxiv 2023. [Google Scholar] [CrossRef] [PubMed]
- Walter, A.; Chaikuad, A.; Helmer, R.; Loaëc, N.; Preu, L.; Ott, I.; Knapp, S.; Meijer, L.; Kunick, C. Molecular Structures of Cdc2-like Kinases in Complex with a New Inhibitor Chemotype. PLoS ONE 2018, 13, e0196761. [Google Scholar] [CrossRef] [PubMed]
- Meine, R.; Becker, W.; Falke, H.; Preu, L.; Loaëc, N.; Meijer, L.; Kunick, C. Indole-3-Carbonitriles as DYRK1A Inhibitors by Fragment-Based Drug Design. Molecules 2018, 23, 64. [Google Scholar] [CrossRef]
- Szamborska-Gbur, A.; Rutkowska, E.; Dreas, A.; Frid, M.; Vilenchik, M.; Milik, M.; Brzózka, K.; Król, M. How to Design Potent and Selective DYRK1B Inhibitors? Molecular Modeling Study. J. Mol. Model. 2019, 25, 41. [Google Scholar] [CrossRef]
- Rüben, K.; Wurzlbauer, A.; Walte, A.; Sippl, W.; Bracher, F.; Becker, W. Selectivity Profiling and Biological Activity of Novel β-Carbolines as Potent and Selective DYRK1 Kinase Inhibitors. PLoS ONE 2015, 10, e0132453. [Google Scholar] [CrossRef]
- Yoshida, T.; Kim, J.H.; Carver, K.; Su, Y.; Weremowicz, S.; Mulvey, L.; Yamamoto, S.; Brennan, C.; Mei, S.; Long, H.; et al. CLK2 Is an Oncogenic Kinase and Splicing Regulator in Breast Cancer. Cancer Res. 2015, 75, 1516–1526. [Google Scholar] [CrossRef] [PubMed]
- Duncan, P.I.; Stojdl, D.F.; Marius, R.M.; Scheit, K.H.; Bell, J.C. The Clk2 and Clk3 Dual-Specificity Protein Kinases Regulate the Intranuclear Distribution of SR Proteins and Influence Pre-MRNA Splicing. Exp. Cell Res. 1998, 241, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Lin, G.; Chen, B.; Yuan, J.; Zhuang, Y. CLK2 Expression Is Associated with the Progression of Colorectal Cancer and Is a Prognostic Biomarker. BioMed Res. Int. 2022, 2022, 7250127. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Piao, Y.; Thomas, C.; Fuller, G.N.; de Groot, J.F. Cdc2-like Kinase 2 Is a Key Regulator of the Cell Cycle via FOXO3a/P27 in Glioblastoma. Oncotarget 2016, 7, 26793–26805. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Mi, S.; Zhang, Z.; Wang, H.; Chen, W.; Wei, W.; Lou, G. MFAP2, Upregulated by M1A Methylation, Promotes Colorectal Cancer Invasiveness via CLK3. Cancer Med. 2023, 12, 8403–8414. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Cui, X.; Hu, Q.; Chen, X.; Zhou, P. CLK3 Is A Direct Target Of MiR-144 And Contributes To Aggressive Progression In Hepatocellular Carcinoma. OncoTargets Ther. 2019, 12, 9201–9213. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Hua, X.; Bryner, Y.H.; Liu, S.; Gitter, C.B.; Dai, J. Haspin Inhibition Delays Cell Cycle Progression through Interphase in Cancer Cells. J. Cell. Physiol. 2020, 235, 4508–4519. [Google Scholar] [CrossRef] [PubMed]
- Aboelfotouh, H.G.; Abdallah, M.; Khalifa, H.; Aboushady, Y.; Abadi, A.H.; Engel, M.; Abdel-Halim, M. N1-Benzoylated 5-(4-pyridinyl) Indazole-based Kinase Inhibitors: Attaining Haspin and Clk4 Selectivity via Modulation of the Benzoyl Substituents. Arch. Pharm. 2024, e2400020. [Google Scholar] [CrossRef] [PubMed]
- Shawky, M.M.; Abdallah, M.; Khalifa, H.; Aboushady, Y.; Abadi, A.H.; Engel, M.; Abdel-Halim, M. Synthesis and Evaluation of Novel N1-Acylated 5-(4-Pyridinyl) Indazole Derivatives as Potent and Selective Haspin Inhibitors. Bioorg. Chem. 2024, 145, 107235. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, H.; Fang, Y.; Xing, Y.; Pang, X.; Li, Y.; Zhang, Y.; Liu, Y. Function and Inhibition of Haspin Kinase: Targeting Multiple Cancer Therapies by Antimitosis. J. Pharm. Pharmacol. 2023, 75, 445–465. [Google Scholar] [CrossRef]
- Ye, Z.; Zhang, Z.; Fang, L.; Tian, D.; Liu, X. Bioinformatic Analysis Reveals GSG2 as a Potential Target for Breast Cancer Therapy. Open Life Sci. 2019, 14, 688–698. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Kuang, T.; Ren, Y.; Lu, Z.; Liao, Q.; Chen, W. Haspin Knockdown Can Inhibit Progression and Development of Pancreatic Cancer in Vitro and Vivo. Exp. Cell Res. 2019, 385, 111605. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-E.; Lee, S.-Y.; Jang, M.; Choi, H.-K.; Kim, J.H.; Chen, H.; Lim, T.-G.; Dong, Z.; Lee, K.W. Coumestrol Epigenetically Suppresses Cancer Cell Proliferation: Coumestrol Is a Natural Haspin Kinase Inhibitor. Int. J. Mol. Sci. 2017, 18, 2228. [Google Scholar] [CrossRef]
- Wang, P.; Hua, X.; Sun, Y.; Li, H.; Bryner, Y.H.; Hsung, R.P.; Dai, J. Loss of Haspin Suppresses Cancer Cell Proliferation by Interfering with Cell Cycle Progression at Multiple Stages. FASEB J. 2021, 35, e21923. [Google Scholar] [CrossRef]
- Chen, Y.; Fu, D.; Zhao, H.; Cheng, W.; Xu, F. GSG2 (Haspin) Promotes Development and Progression of Bladder Cancer through Targeting KIF15 (Kinase-12). Aging 2020, 12, 8858–8879. [Google Scholar] [CrossRef]
- Boni, J.; Rubio-Perez, C.; López-Bigas, N.; Fillat, C.; de la Luna, S. The DYRK Family of Kinases in Cancer: Molecular Functions and Therapeutic Opportunities. Cancers 2020, 12, 2106. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, D.; Tsai, Y.-H.; Weatheritt, R.; Wang, Y.; Blencowe, B.J.; Wang, Z. An Extensive Program of Periodic Alternative Splicing Linked to Cell Cycle Progression. eLife 2016, 5, e10288. [Google Scholar] [CrossRef] [PubMed]
- Huertas, D.; Soler, M.; Moreto, J.; Villanueva, A.; Martinez, A.; Vidal, A.; Charlton, M.; Moffat, D.; Patel, S.; McDermott, J.; et al. Antitumor Activity of a Small-Molecule Inhibitor of the Histone Kinase Haspin. Oncogene 2012, 31, 1408–1418. [Google Scholar] [CrossRef]
- Fahmy, S.A.; Sedky, N.K.; Ramzy, A.; Abdelhady, M.M.M.; Alabrahim, O.A.A.; Shamma, S.N.; Azzazy, H.M.E.-S. Green Extraction of Essential Oils from Pistacia Lentiscus Resins: Encapsulation into Niosomes Showed Improved Preferential Cytotoxic and Apoptotic Effects against Breast and Ovarian Cancer Cells. J. Drug Deliv. Sci. Technol. 2023, 87, 104820. [Google Scholar] [CrossRef]
- Hassan, Z.; Hassan, M.; Hellström-Lindberg, E. The Pharmacodynamic Effect of Busulfan in the P39 Myeloid Cell Line in Vitro. Leukemia 2001, 15, 1240–1247. [Google Scholar] [CrossRef]
- Sleiman, R.J.; Stewart, B.W. Early Caspase Activation in Leukemic Cells Subject to Etoposide-Induced G2-M Arrest: Evidence of Commitment to Apoptosis Rather than Mitotic Cell Death. Clin. Cancer Res. 2000, 6, 3756–3765. [Google Scholar] [PubMed]
- Mercer, S.E.; Ewton, D.Z.; Deng, X.; Lim, S.; Mazur, T.R.; Friedman, E. Mirk/Dyrk1B Mediates Survival during the Differentiation of C2C12Myoblasts. J. Biol. Chem. 2005, 280, 25788–25801. [Google Scholar] [CrossRef] [PubMed]
- Uzor, S.; Porazinski, S.R.; Li, L.; Clark, B.; Ajiro, M.; Iida, K.; Hagiwara, M.; Alqasem, A.A.; Perks, C.M.; Wilson, I.D.; et al. CDC2-like (CLK) Protein Kinase Inhibition as a Novel Targeted Therapeutic Strategy in Prostate Cancer. Sci. Rep. 2021, 11, 7963. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-I.; Chen, Z.-C.; Chen, C.-H.; Chang, Y.-H.; Lee, T.-C.; Tang, T.-T.; Yu, T.-W.; Yang, C.-M.; Tsai, M.-C.; Huang, C.-C.; et al. Co-Inhibition of Aurora A and Haspin Kinases Enhances Survivin Blockage and P53 Induction for Mitotic Catastrophe and Apoptosis in Human Colorectal Cancer. Biochem. Pharmacol. 2022, 206, 115289. [Google Scholar] [CrossRef]
- Salton, M.; Misteli, T. Small Molecule Modulators of Pre-MRNA Splicing in Cancer Therapy. Trends Mol. Med. 2016, 22, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Chow, C.K.; Atkins, E.R.; Hillis, G.S.; Nelson, M.R.; Reid, C.M.; Schlaich, M.P.; Hay, P.; Rogers, K.; Billot, L.; Burke, M.; et al. Initial Treatment with a Single Pill Containing Quadruple Combination of Quarter Doses of Blood Pressure Medicines versus Standard Dose Monotherapy in Patients with Hypertension (QUARTET): A Phase 3, Randomised, Double-Blind, Active-Controlled Trial. Lancet 2021, 398, 1043–1052. [Google Scholar] [CrossRef]
- Lehár, J.; Krueger, A.S.; Avery, W.; Heilbut, A.M.; Johansen, L.M.; Price, E.R.; Rickles, R.J.; Short III, G.F.; Staunton, J.E.; Jin, X.; et al. Synergistic Drug Combinations Tend to Improve Therapeutically Relevant Selectivity. Nat. Biotechnol. 2009, 27, 659–666. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||
|---|---|---|---|---|---|---|---|
| Cpd. No. | R | Dyrk1A | Dyrk1B | Clk1 | |||
| % Inh. At 1 µM a | IC50 (nM) a | % Inh. At 1 µM a | IC50 (nM) a | % Inh. At 1 µM a | IC50 (nM) a | ||
| 1 | 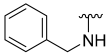 | 46 | ND | 60 | 833 | 60 | 740 |
| 2 | 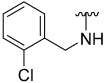 | 61 | 700 | 46 | ND | 61 | 769 |
| 3 | 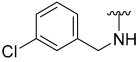 | 60 | 751 | 66 | 530 | 79 | 257 |
| 4 |  | 7 | ND | 0 | ND | 40 | ND |
| 5 | 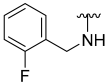 | 48 | ND | 62 | 722 | 63 | 778 |
| 6 |  | 60 | 666 | 71 | 406 | 72 | 410 |
| 7 |  | 26 | ND | 38 | ND | 50 | ND |
| 8 | 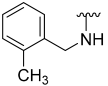 | 61 | 853 | 48 | ND | 63 | 594 |
| 9 | 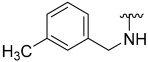 | 33 | ND | 40 | ND | 61 | 787 |
| 10 | 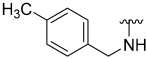 | 8 | ND | 0 | ND | 30 | ND |
| 11 | 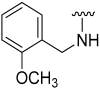 | 64 | 565 | 64 | 578 | 60 | 725 |
| 12 | 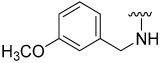 | 65 | 536 | 81 | 238 | 86 | 153 |
| 13 | 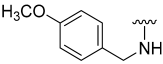 | 9 | ND | 16 | ND | 17 | ND |
| 14 |  | 41 | ND | 12 | ND | 61 | 744 |
| 15 |  | 53 | ND | 61 | 645 | 61 | 779 |
| 16 |  | 40 | ND | 53 | ND | 50 | ND |
| 17 | 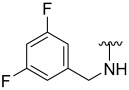 | 67 | 495 | 81 | 242 | 85 | 168 |
| 18 | 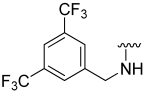 | 8 | ND | 0 | ND | 26 | ND |
| 19 | 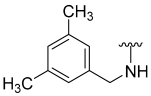 | 22 | ND | 0 | ND | 53 | ND |
| 20 |  | 14 | ND | 19 | ND | 21 | ND |
| 21 | 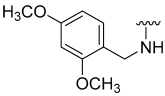 | 9 | ND | 17 | ND | 43 | ND |
| 22 | 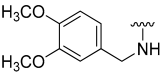 | 8 | ND | 16 | ND | 18 | ND |
| 23 |  | 12 | ND | 22 | ND | 22 | ND |
| 24 | 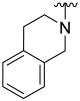 | 12 | ND | 21 | ND | 33 | ND |
| 25 |  | 13 | ND | 24 | ND | 62 | 608 |
| 26 | 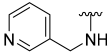 | 34 | ND | 39 | ND | 47 | ND |
| 27 | 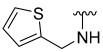 | 53 | ND | 61 | 703 | 60 | 705 |
| 28 | 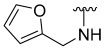 | 66 | 779 | 52 | ND | 61 | 585 |
| 29 |  | 29 | ND | 48 | ND | 61 | 753 |
| 30 | 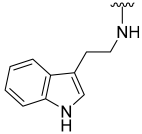 | 20 | ND | 40 | ND | 74 | 350 |
| Cpd No. | % Inhibition at 1 µM a (IC50 in nM) b | ||
|---|---|---|---|
| Clk2 | Clk3 | Haspin | |
| 3 | 46 | 22 | 54 |
| 6 | 38 | 27 | 83 (207) |
| 11 | 50 | 24 | 46 |
| 12 | 62 (617) | 26 | 85 (167) |
| 17 | 62 (708) | 26 | 80 (256) |
| Cpd. | HCT-116 % Inh. at 20 μM a (IC50 in µM) b | MCF-7 % Inh. at μM a (IC50 in µM) b | T24 % Inh. at μM a (IC50 in µM) b | A549 % Inh. at μM a (IC50 in µM) b | U87 % Inh. at μM a (IC50 in µM) b | Hela % Inh. at μM a (IC50 in µM) b |
|---|---|---|---|---|---|---|
| 3 | 25.9 ± 2.54 | 15.6 ± 3.85 | 68.6 ± 3.95 (14) | 54.7 ± 3.77 | 32.17 ± 4.76 | 50.5 ± 3.93 |
| 6 | 60.1 ± 3.11 | 16.2 ± 2.20 | 49.3 ± 6.04 | 36.5 ± 1.57 | 21.04 ± 5.07 | 53.3 ± 2.22 |
| 11 | 0 | 56.4 ± 4.47 | 55.3 ± 3.30 | 34.7 ± 3.91 | 18.09 ± 0.81 | 62.1 ± 1.97 (19.3) |
| 12 | 75.7 ± 0.31 (6.4) | 93.5 ± 0.17 | 100 (6) | 41.0 ± 8.96 | 60.12 ± 6.16 (16.5) | 50.7 ± 2.41 |
| 17 | 67.6 ± 0.58 (8.1) | 20.9 ± 2.02 | 59.9 ± 2.05 (14.9) | 38.8 ± 4.06 | 47.28 ± 5.65 | 60.9 ± 2.82 (18.1) |
| Cpd. | IEC-6 | HaCaT | ||||||
|---|---|---|---|---|---|---|---|---|
| Viability (%) a | IC50 (μM) b | Viability (%) a | IC50 (μM) b | |||||
| 12 | 95.99 | ± | 2.22 | >20 | 67.63 | ± | 6.09 | >20 |
| 17 | 76.83 | ± | 3.00 | >20 | 64.90 | ± | 1.97 | >20 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mostafa, N.; Chen, P.-J.; Darwish, S.S.; Su, Y.-C.; Shiao, M.-H.; Piazza, G.A.; Abadi, A.H.; Engel, M.; Abdel-Halim, M. N-Benzylated 5-Hydroxybenzothiophene-2-carboxamides as Multi-Targeted Clk/Dyrk Inhibitors and Potential Anticancer Agents. Cancers 2024, 16, 2033. https://doi.org/10.3390/cancers16112033
Mostafa N, Chen P-J, Darwish SS, Su Y-C, Shiao M-H, Piazza GA, Abadi AH, Engel M, Abdel-Halim M. N-Benzylated 5-Hydroxybenzothiophene-2-carboxamides as Multi-Targeted Clk/Dyrk Inhibitors and Potential Anticancer Agents. Cancers. 2024; 16(11):2033. https://doi.org/10.3390/cancers16112033
Chicago/Turabian StyleMostafa, Noha, Po-Jen Chen, Sarah S. Darwish, Yu-Chieh Su, Ming-Hua Shiao, Gary A. Piazza, Ashraf H. Abadi, Matthias Engel, and Mohammad Abdel-Halim. 2024. "N-Benzylated 5-Hydroxybenzothiophene-2-carboxamides as Multi-Targeted Clk/Dyrk Inhibitors and Potential Anticancer Agents" Cancers 16, no. 11: 2033. https://doi.org/10.3390/cancers16112033
APA StyleMostafa, N., Chen, P.-J., Darwish, S. S., Su, Y.-C., Shiao, M.-H., Piazza, G. A., Abadi, A. H., Engel, M., & Abdel-Halim, M. (2024). N-Benzylated 5-Hydroxybenzothiophene-2-carboxamides as Multi-Targeted Clk/Dyrk Inhibitors and Potential Anticancer Agents. Cancers, 16(11), 2033. https://doi.org/10.3390/cancers16112033