

TWIST1 Drives Cytotoxic CD8+ T-Cell Exhaustion through Transcriptional Activation of CD274 (PD-L1) Expression in Breast Cancer Cells
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Mining of Human Breast Cancer Transcriptomes
2.2. Western Blot
2.3. Cell Culture and Generation of Cell Lines
2.4. RT-qPCR
2.5. ChIP-qPCR Assay
2.6. Isolation of OT-1 Mouse CD8+ T Cells
2.7. Breast Cancer Cell Proliferation Assay
2.8. Vitality and Apoptosis Assays of CD8+ T Cells
2.9. Statistical Analyses
3. Results
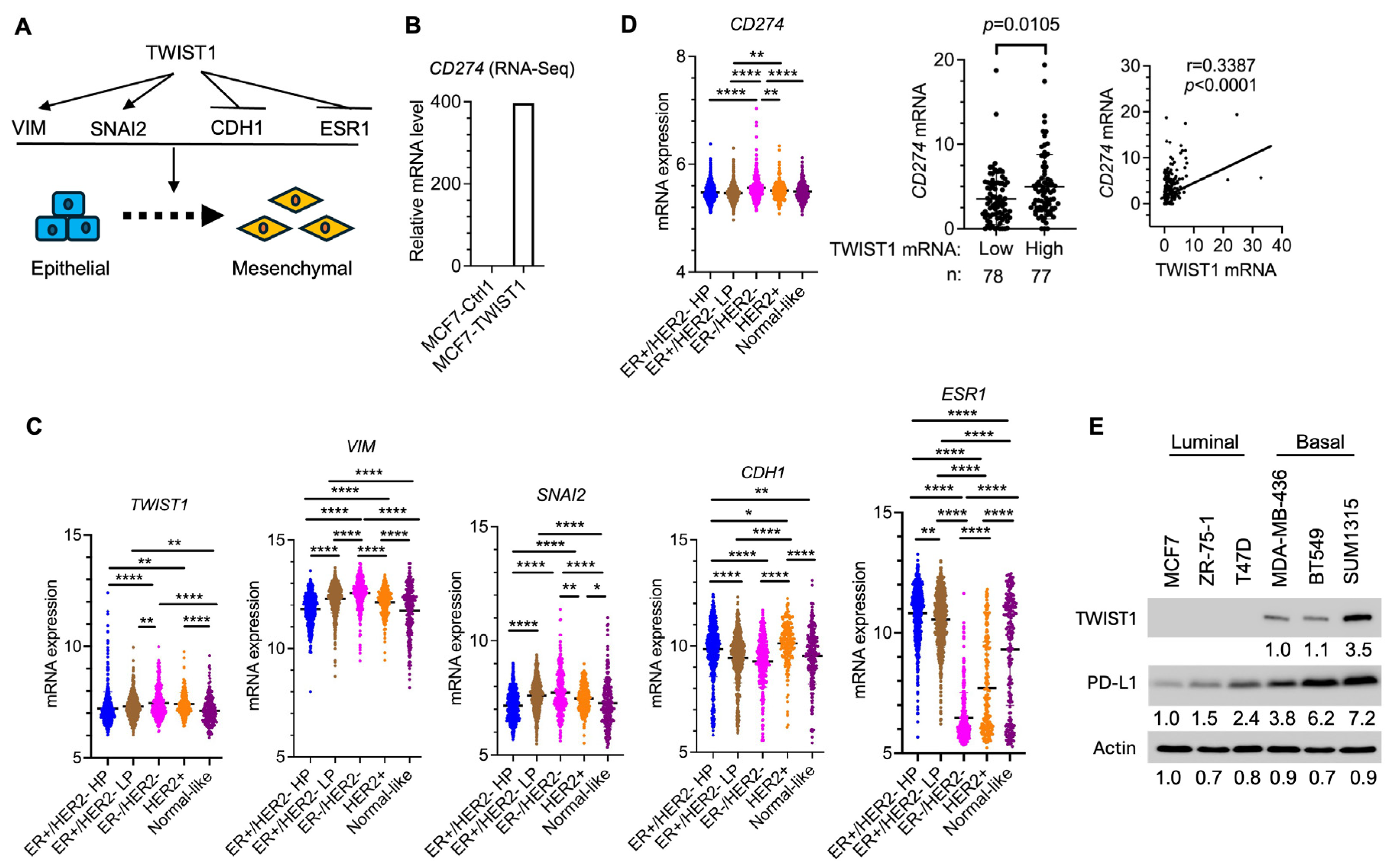
3.1. TWIST1 Robustly Upregulates PD-L1 Expression in Breast Cancer Cells
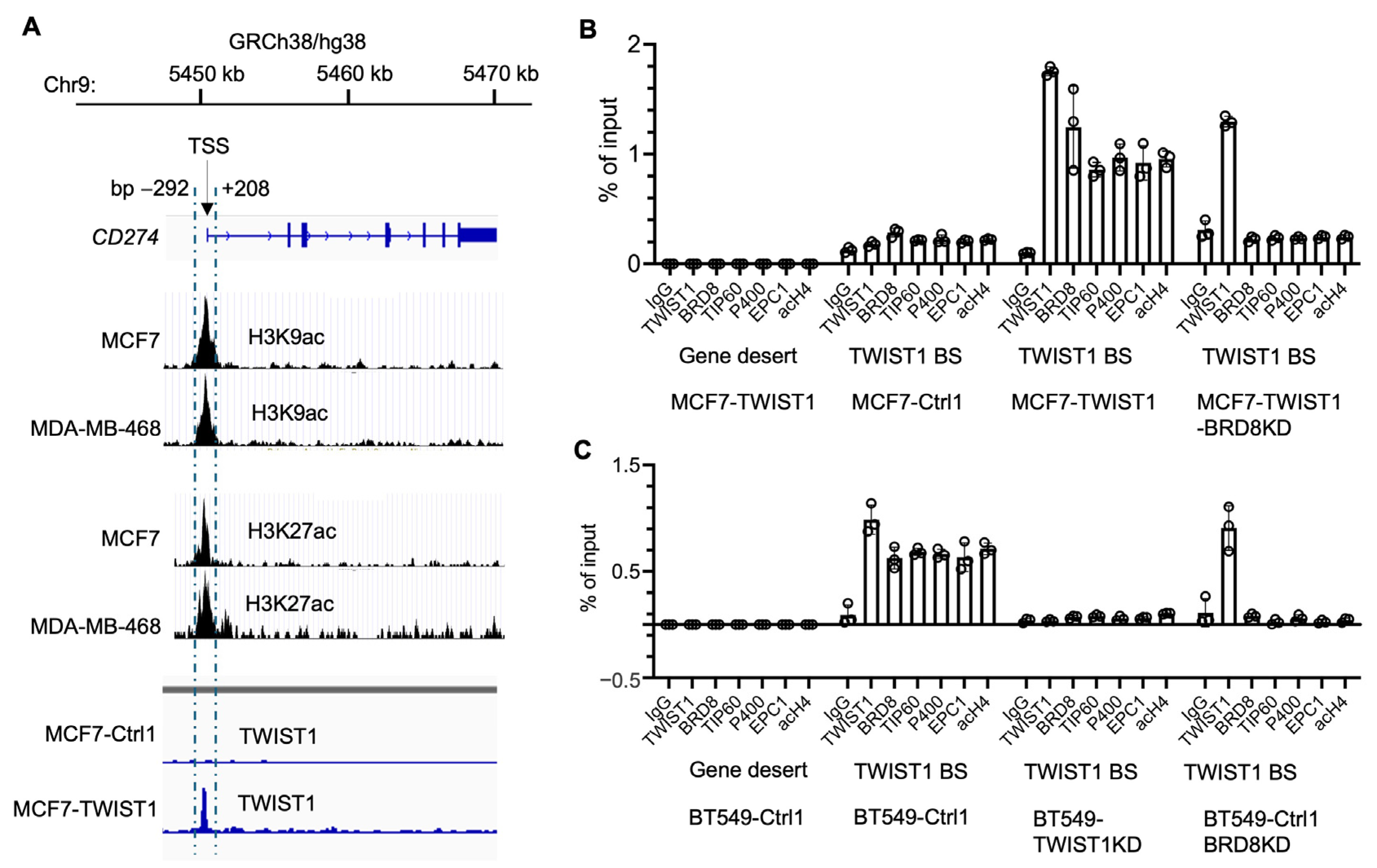
3.2. TWIST1 Binds the CD274 Promoter and Recruits TIP60-Com in a BRD8-Dependent Manner
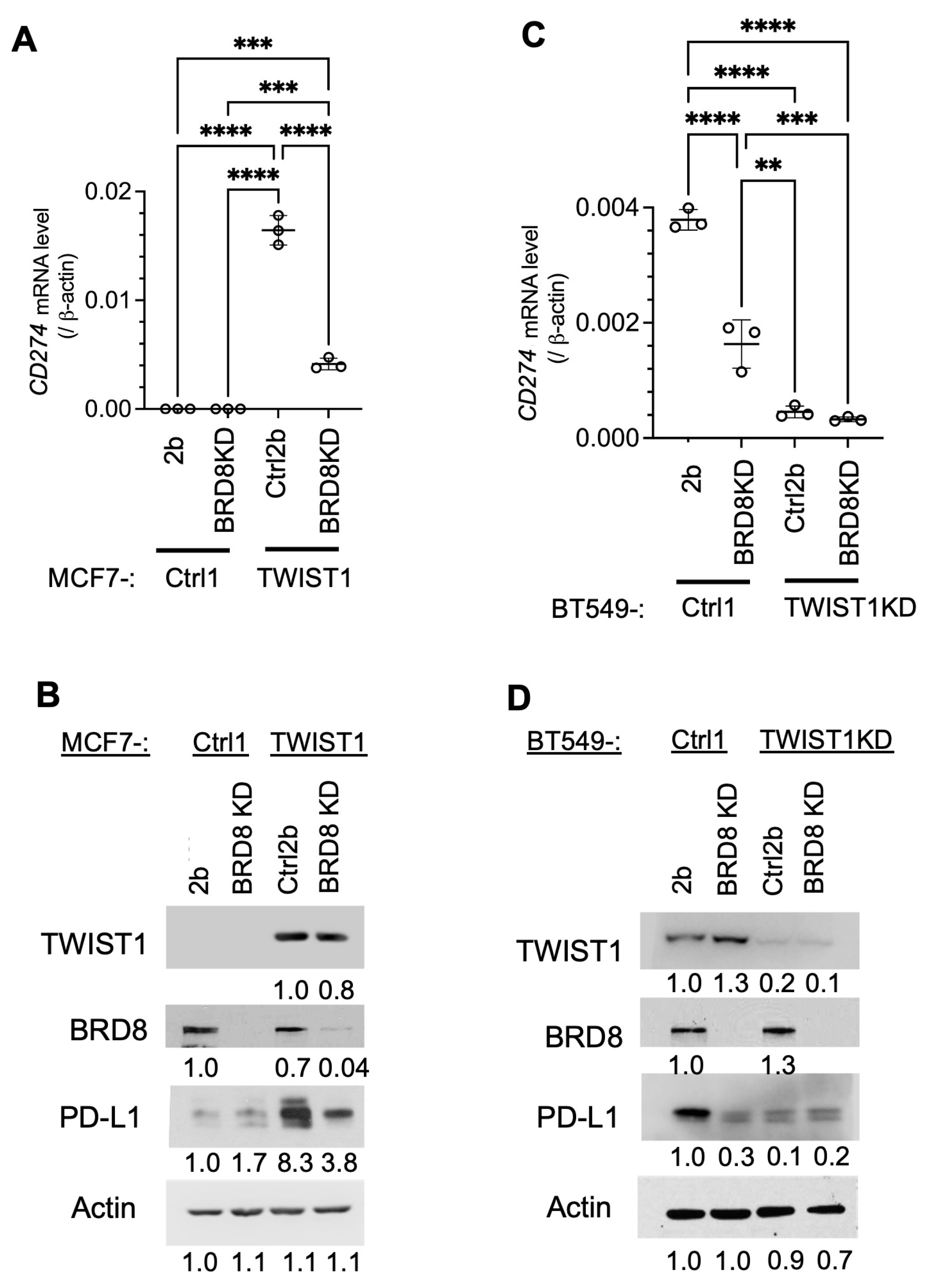
3.3. TWIST1 Requires TIP60-Com to Transcriptionally Activate CD274 Expression
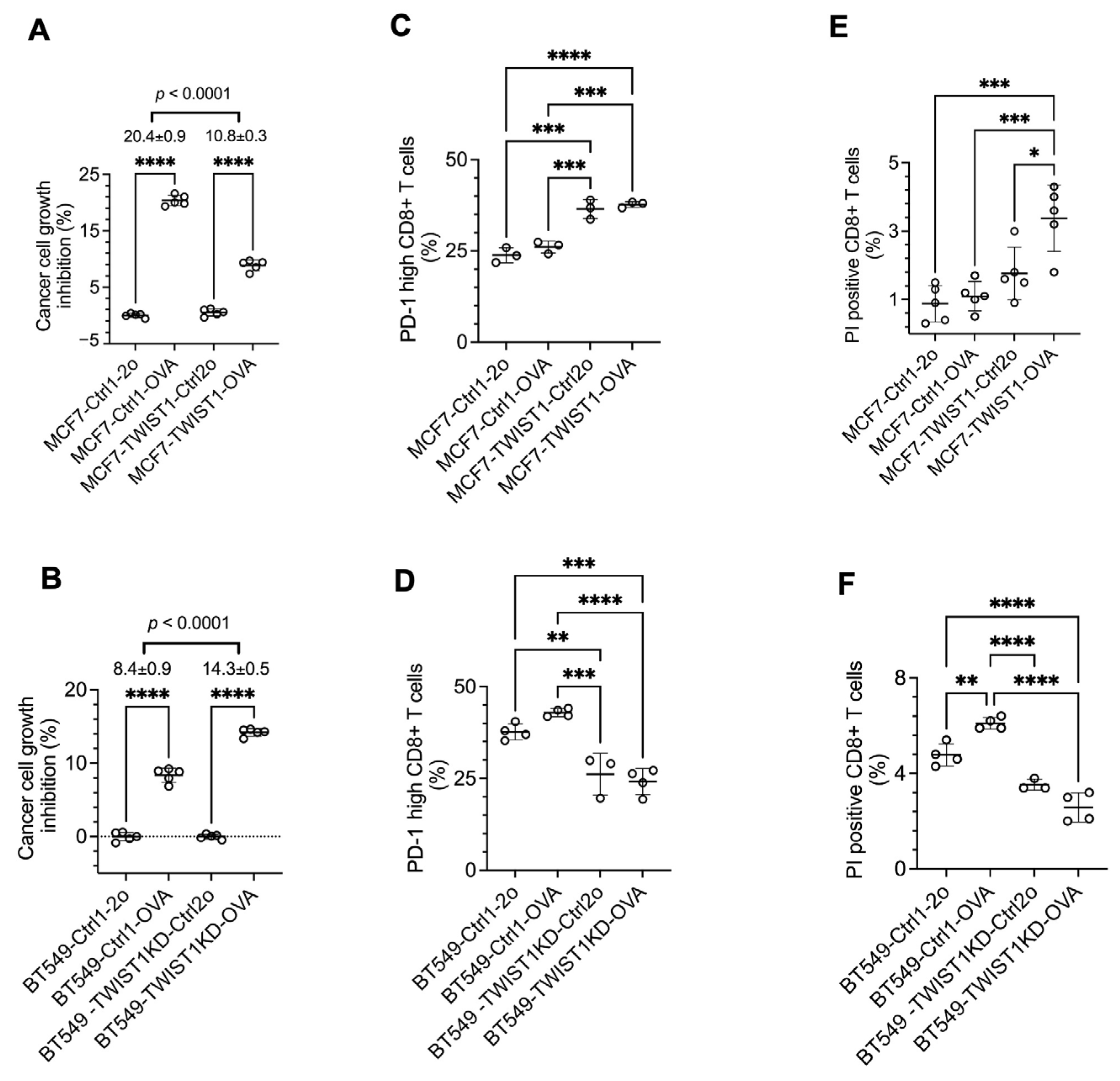
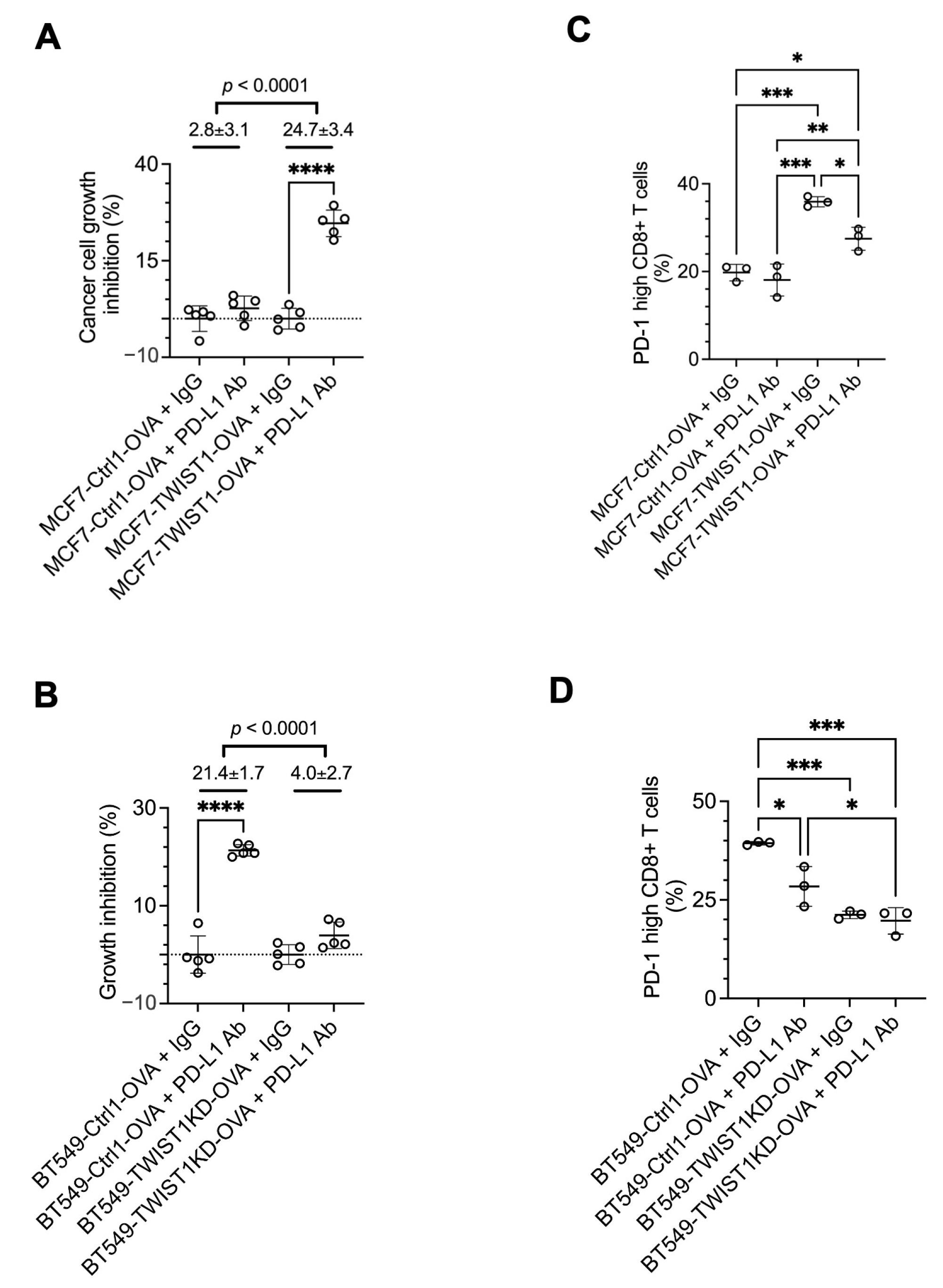
3.4. TWIST1 Expressed in Breast Cancer Cells Promotes Their Immune Evasion in Culture by Accelerating CD8+ T-Cell Exhaustion and Death
3.5. Inhibition of TWIST1-Upregulated PD-L1 (CD274) in Breast Cancer Cells Strongly Sensitizes CD8+ T Cells to Kill Cancer Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Restifo, N.P.; Dudley, M.E.; Rosenberg, S.A. Adoptive immunotherapy for cancer: Harnessing the T cell response. Nat. Rev. Immunol. 2012, 12, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Baessler, A.; Vignali, D.A.A. T Cell Exhaustion. Annu. Rev. Immunol. 2024, 42, 179–206. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Xu, C. Immune checkpoint signaling and cancer immunotherapy. Cell Res. 2020, 30, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Chen, Y.; Li, X.; Long, S.; Shi, Y.; Yu, Y.; Wu, W.; Han, L.; Wang, S. The role of PD-1/PD-L1 and application of immune-checkpoint inhibitors in human cancers. Front. Immunol. 2022, 13, 964442. [Google Scholar] [CrossRef] [PubMed]
- Sinoquet, L.; Jacot, W.; Gauthier, L.; Pouderoux, S.; Viala, M.; Cayrefourcq, L.; Quantin, X.; Alix-Panabieres, C. Programmed Cell Death Ligand 1-Expressing Circulating Tumor Cells: A New Prognostic Biomarker in Non-Small Cell Lung Cancer. Clin. Chem. 2021, 67, 1503–1512. [Google Scholar] [CrossRef] [PubMed]
- Mazel, M.; Jacot, W.; Pantel, K.; Bartkowiak, K.; Topart, D.; Cayrefourcq, L.; Rossille, D.; Maudelonde, T.; Fest, T.; Alix-Panabieres, C. Frequent expression of PD-L1 on circulating breast cancer cells. Mol. Oncol. 2015, 9, 1773–1782. [Google Scholar] [CrossRef]
- Strati, A.; Koutsodontis, G.; Papaxoinis, G.; Angelidis, I.; Zavridou, M.; Economopoulou, P.; Kotsantis, I.; Avgeris, M.; Mazel, M.; Perisanidis, C.; et al. Prognostic significance of PD-L1 expression on circulating tumor cells in patients with head and neck squamous cell carcinoma. Ann. Oncol. 2017, 28, 1923–1933. [Google Scholar] [CrossRef]
- Gu, L.; Chen, M.; Guo, D.; Zhu, H.; Zhang, W.; Pan, J.; Zhong, X.; Li, X.; Qian, H.; Wang, X. PD-L1 and gastric cancer prognosis: A systematic review and meta-analysis. PLoS ONE 2017, 12, e0182692. [Google Scholar] [CrossRef]
- Xu, Y.; Song, G.; Xie, S.; Jiang, W.; Chen, X.; Chu, M.; Hu, X.; Wang, Z.W. The roles of PD-1/PD-L1 in the prognosis and immunotherapy of prostate cancer. Mol. Ther. 2021, 29, 1958–1969. [Google Scholar] [CrossRef] [PubMed]
- Rotte, A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J. Exp. Clin. Cancer Res. 2019, 38, 255. [Google Scholar] [CrossRef] [PubMed]
- Kazandjian, D.; Suzman, D.L.; Blumenthal, G.; Mushti, S.; He, K.; Libeg, M.; Keegan, P.; Pazdur, R. FDA Approval Summary: Nivolumab for the Treatment of Metastatic Non-Small Cell Lung Cancer with Progression on or After Platinum-Based Chemotherapy. Oncologist 2016, 21, 634–642. [Google Scholar] [CrossRef] [PubMed]
- Voena, C.; Chiarle, R. Advances in cancer immunology and cancer immunotherapy. Discov. Med. 2016, 21, 125–133. [Google Scholar] [PubMed]
- Debien, V.; De Caluwe, A.; Wang, X.; Piccart-Gebhart, M.; Tuohy, V.K.; Romano, E.; Buisseret, L. Immunotherapy in breast cancer: An overview of current strategies and perspectives. NPJ Breast Cancer 2023, 9, 7. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.; Rugo, H.S.; Cescon, D.W.; Im, S.A.; Yusof, M.M.; Gallardo, C.; Lipatov, O.; Barrios, C.H.; Perez-Garcia, J.; Iwata, H.; et al. Pembrolizumab plus Chemotherapy in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2022, 387, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.; Cescon, D.W.; Rugo, H.S.; Nowecki, Z.; Im, S.A.; Yusof, M.M.; Gallardo, C.; Lipatov, O.; Barrios, C.H.; Holgado, E.; et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): A randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet 2020, 396, 1817–1828. [Google Scholar] [CrossRef]
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004, 117, 927–939. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Xu, Y.; Lee, D.; Feng, Z.; Xu, Y.; Bu, W.; Li, Y.; Liao, L.; Xu, J. Breast tumor cell-specific knockout of Twist1 inhibits cancer cell plasticity, dissemination and lung metastasis in Mice. Proc. Natl. Acad. Sci. USA 2017, 114, 11494–11499. [Google Scholar] [CrossRef]
- Qin, Q.; Xu, Y.; He, T.; Qin, C.; Xu, J. Normal and disease-related biological functions of Twist1 and underlying molecular mechanisms. Cell Res. 2012, 22, 90–106. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Hou, Y.; Zhou, M.; Wen, S.; Zhou, J.; Xu, L.; Tang, X.; Du, Y.E.; Hu, P.; Liu, M. Twist induces epithelial-mesenchymal transition and cell motility in breast cancer via ITGB1-FAK/ILK signaling axis and its associated downstream network. Int. J. Biochem. Cell Biol. 2016, 71, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Eckert, M.A.; Lwin, T.M.; Chang, A.T.; Kim, J.; Danis, E.; Ohno-Machado, L.; Yang, J. Twist1-induced invadopodia formation promotes tumor metastasis. Cancer Cell 2011, 19, 372–386. [Google Scholar] [CrossRef] [PubMed]
- Battula, V.L.; Evans, K.W.; Hollier, B.G.; Shi, Y.; Marini, F.C.; Ayyanan, A.; Wang, R.Y.; Brisken, C.; Guerra, R.; Andreeff, M.; et al. Epithelial-mesenchymal transition-derived cells exhibit multilineage differentiation potential similar to mesenchymal stem cells. Stem Cells 2010, 28, 1435–1445. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Qin, L.; Sun, T.; Wu, H.; He, T.; Yang, Z.; Mo, Q.; Liao, L.; Xu, J. Twist1 promotes breast cancer invasion and metastasis by silencing Foxa1 expression. Oncogene 2017, 36, 1157–1166. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; He, T.; Tong, Z.; Liao, L.; Huang, S.; Fakhouri, W.D.; Edwards, D.P.; Xu, J. Molecular mechanisms of TWIST1-regulated transcription in EMT and cancer metastasis. EMBO Rep. 2023, 24, e56902. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Qin, L.; He, T.; Qin, J.; Hong, J.; Wong, J.; Liao, L.; Xu, J. The TWIST/Mi2/NuRD protein complex and its essential role in cancer metastasis. Cell Res. 2011, 21, 275–289. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Zhang, L.; He, T.; Xiao, X.; Liu, X.; Wang, L.; Yang, L.; Yang, M.; Zhang, T.; Chen, R.; et al. TWIST represses estrogen receptor-alpha expression by recruiting the NuRD protein complex in breast cancer cells. Int. J. Biol. Sci. 2012, 8, 522–532. [Google Scholar] [CrossRef]
- Cheng, G.Z.; Chan, J.; Wang, Q.; Zhang, W.; Sun, C.D.; Wang, L.H. Twist transcriptionally up-regulates AKT2 in breast cancer cells leading to increased migration, invasion, and resistance to paclitaxel. Cancer Res. 2007, 67, 1979–1987. [Google Scholar] [CrossRef]
- Maestro, R.; Dei Tos, A.P.; Hamamori, Y.; Krasnokutsky, S.; Sartorelli, V.; Kedes, L.; Doglioni, C.; Beach, D.H.; Hannon, G.J. Twist is a potential oncogene that inhibits apoptosis. Genes Dev. 1999, 13, 2207–2217. [Google Scholar] [CrossRef]
- Vesuna, F.; Lisok, A.; van Diest, P.; Raman, V. Twist activates miR-22 to suppress estrogen receptor alpha in breast cancer. Mol. Cell. Biochem. 2021, 476, 2295–2306. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Wang, Y.; Zeng, L.; Wu, Y.; Deng, J.; Zhang, Q.; Lin, Y.; Li, J.; Kang, T.; Tao, M.; et al. Disrupting the interaction of BRD4 with diacetylated Twist suppresses tumorigenesis in basal-like breast cancer. Cancer Cell 2014, 25, 210–225. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, J.; Rho, O.; Youssef, R.M.; DiGiovanni, J. Twist1 regulates keratinocyte proliferation and skin tumor promotion. Mol. Carcinog. 2016, 55, 941–952. [Google Scholar] [CrossRef] [PubMed]
- Allard, S.; Utley, R.T.; Savard, J.; Clarke, A.; Grant, P.; Brandl, C.J.; Pillus, L.; Workman, J.L.; Cote, J. NuA4, an essential transcription adaptor/histone H4 acetyltransferase complex containing Esa1p and the ATM-related cofactor Tra1p. EMBO J. 1999, 18, 5108–5119. [Google Scholar] [CrossRef]
- Doyon, Y.; Selleck, W.; Lane, W.S.; Tan, S.; Cote, J. Structural and functional conservation of the NuA4 histone acetyltransferase complex from yeast to humans. Mol. Cell. Biol. 2004, 24, 1884–1896. [Google Scholar] [CrossRef] [PubMed]
- Jacquet, K.; Fradet-Turcotte, A.; Avvakumov, N.; Lambert, J.P.; Roques, C.; Pandita, R.K.; Paquet, E.; Herst, P.; Gingras, A.C.; Pandita, T.K.; et al. The TIP60 Complex Regulates Bivalent Chromatin Recognition by 53BP1 through Direct H4K20me Binding and H2AK15 Acetylation. Mol. Cell 2016, 62, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Ravens, S.; Yu, C.; Ye, T.; Stierle, M.; Tora, L. Tip60 complex binds to active Pol II promoters and a subset of enhancers and co-regulates the c-Myc network in mouse embryonic stem cells. Epigenetics Chromatin 2015, 8, 45. [Google Scholar] [CrossRef] [PubMed]
- Judes, G.; Rifai, K.; Ngollo, M.; Daures, M.; Bignon, Y.J.; Penault-Llorca, F.; Bernard-Gallon, D. A bivalent role of TIP60 histone acetyl transferase in human cancer. Epigenomics 2015, 7, 1351–1363. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, D.; Cramet, M.; Wang, A.Y.; Steunou, A.L.; Lacoste, N.; Schulze, J.M.; Cote, V.; Monnet-Saksouk, J.; Piquet, S.; Nourani, A.; et al. Eaf5/7/3 form a functionally independent NuA4 submodule linked to RNA polymerase II-coupled nucleosome recycling. EMBO J. 2014, 33, 1397–1415. [Google Scholar] [CrossRef]
- Avvakumov, N.; Cote, J. The MYST family of histone acetyltransferases and their intimate links to cancer. Oncogene 2007, 26, 5395–5407. [Google Scholar] [CrossRef]
- Voss, A.K.; Thomas, T. MYST family histone acetyltransferases take center stage in stem cells and development. Bioessays 2009, 31, 1050–1061. [Google Scholar] [CrossRef] [PubMed]
- Zaware, N.; Zhou, M.M. Bromodomain biology and drug discovery. Nat. Struct. Mol. Biol. 2019, 26, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Terry, S.; Savagner, P.; Ortiz-Cuaran, S.; Mahjoubi, L.; Saintigny, P.; Thiery, J.P.; Chouaib, S. New insights into the role of EMT in tumor immune escape. Mol. Oncol. 2017, 11, 824–846. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Zhan, H. Communication between EMT and PD-L1 signaling: New insights into tumor immune evasion. Cancer Lett. 2020, 468, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Messeha, S.S.; Zarmouh, N.O.; Soliman, K.F.A. Polyphenols Modulating Effects of PD-L1/PD-1 Checkpoint and EMT-Mediated PD-L1 Overexpression in Breast Cancer. Nutrients 2021, 13, 1718. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Gibbons, D.L.; Goswami, S.; Cortez, M.A.; Ahn, Y.H.; Byers, L.A.; Zhang, X.; Yi, X.; Dwyer, D.; Lin, W.; et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat. Commun. 2014, 5, 5241. [Google Scholar] [CrossRef]
- Tsutsumi, S.; Saeki, H.; Nakashima, Y.; Ito, S.; Oki, E.; Morita, M.; Oda, Y.; Okano, S.; Maehara, Y. Programmed death-ligand 1 expression at tumor invasive front is associated with epithelial-mesenchymal transition and poor prognosis in esophageal squamous cell carcinoma. Cancer Sci. 2017, 108, 1119–1127. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Janji, B.; Abdou, A.; Hasmim, M.; Terry, S.; Tan, T.Z.; Mami-Chouaib, F.; Thiery, J.P.; Chouaib, S. The immune checkpoint ligand PD-L1 is upregulated in EMT-activated human breast cancer cells by a mechanism involving ZEB-1 and miR-200. Oncoimmunology 2017, 6, e1263412. [Google Scholar] [CrossRef]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef]
- Pereira, B.; Chin, S.F.; Rueda, O.M.; Vollan, H.K.; Provenzano, E.; Bardwell, H.A.; Pugh, M.; Jones, L.; Russell, R.; Sammut, S.J.; et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat. Commun. 2016, 7, 11479. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Jain, E.; Zañudo, J.G.T.; McGillicuddy, M.; Abravanel, D.L.; Thomas, B.S.; Kim, D.; Balch, S.; Navarro, J.; Weiss, J.H.; Hernandez, T.G.; et al. The Metastatic Breast Cancer Project: Leveraging patient-partnered research to expand the clinical and genomic landscape of metastatic breast cancer and accelerate discoveries. medRxiv 2023. [Google Scholar] [CrossRef]
- Barski, A.; Chepelev, I.; Liko, D.; Cuddapah, S.; Fleming, A.B.; Birch, J.; Cui, K.; White, R.J.; Zhao, K. Pol II and its associated epigenetic marks are present at Pol III-transcribed noncoding RNA genes. Nat. Struct. Mol. Biol. 2010, 17, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Hogquist, K.A.; Jameson, S.C.; Heath, W.R.; Howard, J.L.; Bevan, M.J.; Carbone, F.R. T cell receptor antagonist peptides induce positive selection. Cell 1994, 76, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Casas, E.; Kim, J.; Bendesky, A.; Ohno-Machado, L.; Wolfe, C.J.; Yang, J. Snail2 is an essential mediator of Twist1-induced epithelial mesenchymal transition and metastasis. Cancer Res. 2011, 71, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Gajendran, B.; Sample, K.M.; Wang, C.; Hu, A.; Chen, B.; Li, Y.; Zacksenhaus, E.; Ben-David, Y. A critical ETV4/Twist1/Vimentin axis in Ha-RAS-induced aggressive breast cancer. Cancer Gene Ther. 2022, 29, 1590–1599. [Google Scholar] [CrossRef] [PubMed]
- Consortium, E.P. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.A.; Hitz, B.C.; Sloan, C.A.; Chan, E.T.; Davidson, J.M.; Gabdank, I.; Hilton, J.A.; Jain, K.; Baymuradov, U.K.; Narayanan, A.K.; et al. The Encyclopedia of DNA elements (ENCODE): Data portal update. Nucleic Acids Res. 2018, 46, D794–D801. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, C.; Luong, G.; Sun, Y. A snapshot of the PD-1/PD-L1 pathway. J. Cancer 2021, 12, 2735–2746. [Google Scholar] [CrossRef]
- Kamphorst, A.O.; Wieland, A.; Nasti, T.; Yang, S.; Zhang, R.; Barber, D.L.; Konieczny, B.T.; Daugherty, C.Z.; Koenig, L.; Yu, K.; et al. Rescue of exhausted CD8 T cells by PD-1-targeted therapies is CD28-dependent. Science 2017, 355, 1423–1427. [Google Scholar] [CrossRef]
- Liao, H.; Chang, X.; Gao, L.; Ye, C.; Qiao, Y.; Xie, L.; Lin, J.; Cai, S.; Dong, H. IL-17A promotes tumorigenesis and upregulates PD-L1 expression in non-small cell lung cancer. J. Transl. Med. 2023, 21, 828. [Google Scholar] [CrossRef] [PubMed]
- Bracken, C.P.; Gregory, P.A.; Kolesnikoff, N.; Bert, A.G.; Wang, J.; Shannon, M.F.; Goodall, G.J. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res. 2008, 68, 7846–7854. [Google Scholar] [CrossRef] [PubMed]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Li, Z.; Tao, J.; Hu, H.; Li, Z.; Zhang, Z.; Cheng, F.; Sun, Y.; Zhang, Y.; Yang, J.; et al. Resveratrol induces PD-L1 expression through snail-driven activation of Wnt pathway in lung cancer cells. J. Cancer Res. Clin. Oncol. 2021, 147, 1101–1113. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.H.; Pages, F.; Sautes-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurti, U.; Wetherilt, C.S.; Yang, J.; Peng, L.; Li, X. Tumor-infiltrating lymphocytes are significantly associated with better overall survival and disease-free survival in triple-negative but not estrogen receptor-positive breast cancers. Hum. Pathol. 2017, 64, 7–12. [Google Scholar] [CrossRef]
- Muenst, S.; Schaerli, A.R.; Gao, F.; Daster, S.; Trella, E.; Droeser, R.A.; Muraro, M.G.; Zajac, P.; Zanetti, R.; Gillanders, W.E.; et al. Expression of programmed death ligand 1 (PD-L1) is associated with poor prognosis in human breast cancer. Breast Cancer Res. Treat. 2014, 146, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Liu, Y.; Lee, D.K.; Tong, Z.; Yu, X.; Li, Y.; Xu, Y.; Lanz, R.B.; O’Malley, B.W.; Xu, J. Cell lineage tracing links ERalpha loss in Erbb2-positive breast cancers to the arising of a highly aggressive breast cancer subtype. Proc. Natl. Acad. Sci. USA 2021, 118, e2100673118. [Google Scholar] [CrossRef]
- Xu, Y.; Xu, Y.; Liao, L.; Zhou, N.; Theissen, S.M.; Liao, X.H.; Nguyen, H.; Ludwig, T.; Qin, L.; Martinez, J.D.; et al. Inducible knockout of Twist1 in young and adult mice prolongs hair growth cycle and has mild effects on general health, supporting Twist1 as a preferential cancer target. Am. J. Pathol. 2013, 183, 1281–1292. [Google Scholar] [CrossRef]
- Yochum, Z.A.; Cades, J.; Mazzacurati, L.; Neumann, N.M.; Khetarpal, S.K.; Chatterjee, S.; Wang, H.; Attar, M.A.; Huang, E.H.; Chatley, S.N.; et al. A First-in-Class TWIST1 Inhibitor with Activity in Oncogene-Driven Lung Cancer. Mol. Cancer Res. 2017, 15, 1764–1776. [Google Scholar] [CrossRef]
- Ardiani, A.; Gameiro, S.R.; Palena, C.; Hamilton, D.H.; Kwilas, A.; King, T.H.; Schlom, J.; Hodge, J.W. Vaccine-mediated immunotherapy directed against a transcription factor driving the metastatic process. Cancer Res. 2014, 74, 1945–1957. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| First Generation | Second Generation | Third Generation | |
|---|---|---|---|
| Cell Lines | Selection Marker(s) | ||
| MCF7 | MCF7-Ctrl1 (G418) | MCF7-Ctrl1-2b | G418 and puromycin |
| MCF7-Ctrl1-BRD8KD | |||
| MCF7-Ctrl1-2o | G418 and mCherry | ||
| MCF7-Ctrl1-OVA | |||
| MCF7-TWIST1 (G418) | MCF7-TWIST1-Ctrl2b | G418 and puromycin | |
| MCF7-TWIST1-BRD8KD | |||
| MCF7-TWIST1-Ctrl2o | G418 and mCherry | ||
| MCF7-TWIST1-OVA | |||
| BT549 | BT549-Ctrl1 (puromycin) | BT549-Ctrl1-2b | puromycin |
| BT549-Ctrl1-BRD8KD | |||
| BT549-Ctrl1-2o | puromycin and mCherry | ||
| BT549-Ctrl1-OVA | |||
| BT549 TWIST1KD (puromycin) | BT549-TWIST1KD-Ctrl2b | puromycin | |
| BT549-TWIST1KD-BRD8KD | |||
| BT549-TWIST1KD-Ctrl2o | puromycin and mCherry | ||
| BT549-TWIST1KD-OVA | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, X.; Xu, J. TWIST1 Drives Cytotoxic CD8+ T-Cell Exhaustion through Transcriptional Activation of CD274 (PD-L1) Expression in Breast Cancer Cells. Cancers 2024, 16, 1973. https://doi.org/10.3390/cancers16111973
Yu X, Xu J. TWIST1 Drives Cytotoxic CD8+ T-Cell Exhaustion through Transcriptional Activation of CD274 (PD-L1) Expression in Breast Cancer Cells. Cancers. 2024; 16(11):1973. https://doi.org/10.3390/cancers16111973
Chicago/Turabian StyleYu, Xiaobin, and Jianming Xu. 2024. "TWIST1 Drives Cytotoxic CD8+ T-Cell Exhaustion through Transcriptional Activation of CD274 (PD-L1) Expression in Breast Cancer Cells" Cancers 16, no. 11: 1973. https://doi.org/10.3390/cancers16111973
APA StyleYu, X., & Xu, J. (2024). TWIST1 Drives Cytotoxic CD8+ T-Cell Exhaustion through Transcriptional Activation of CD274 (PD-L1) Expression in Breast Cancer Cells. Cancers, 16(11), 1973. https://doi.org/10.3390/cancers16111973