
GAS-Luc2 Reporter Cell Lines for Immune Checkpoint Drug Screening in Solid Tumors


Abstract
Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Surface Protein Profiling via Flow Cytometry
2.3. Plasmid Transduction and Single Cell Cloning
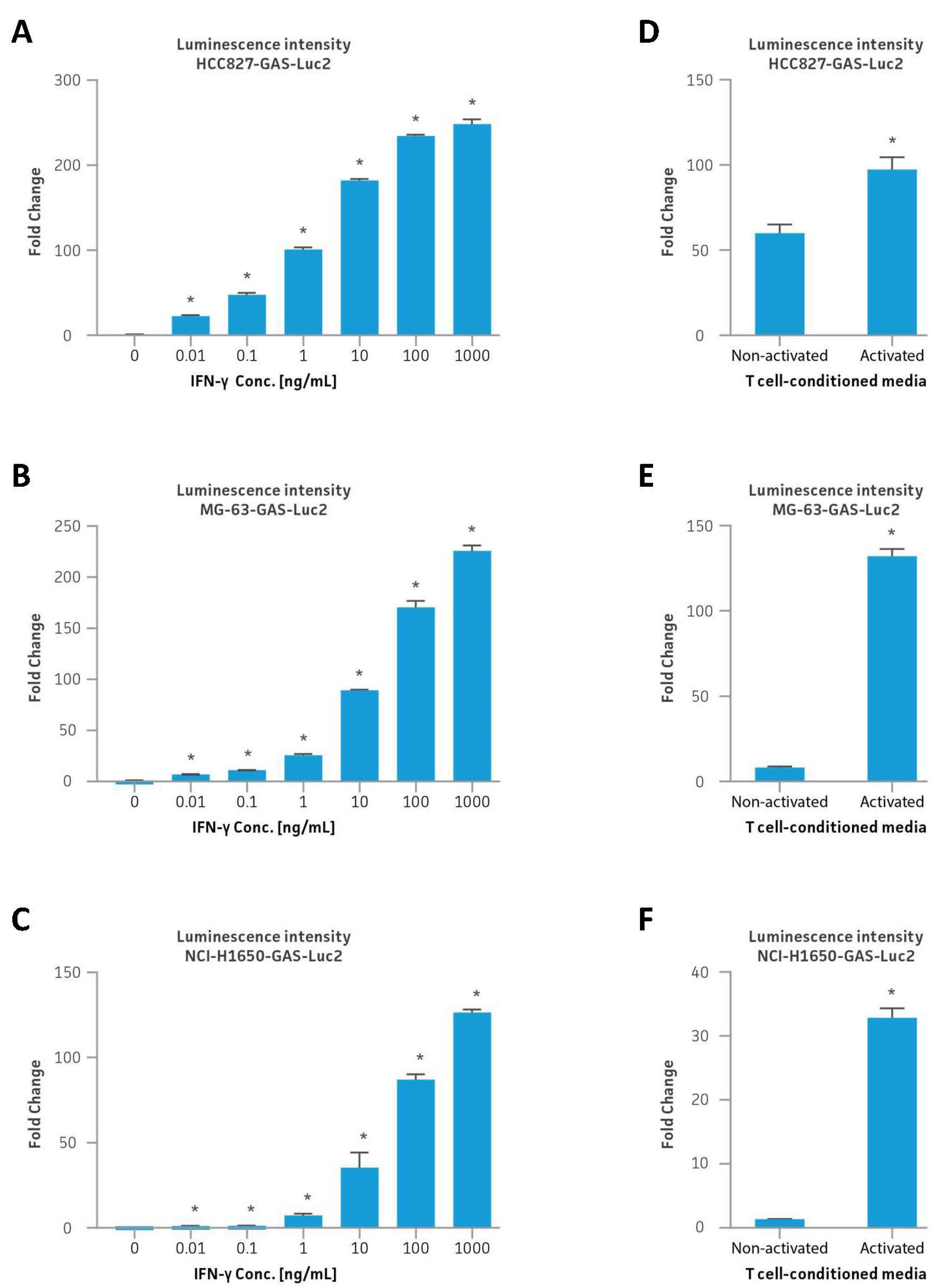
2.4. Interferon Gamma Stimulation
2.5. T Cell-Conditioned Media Stimulation
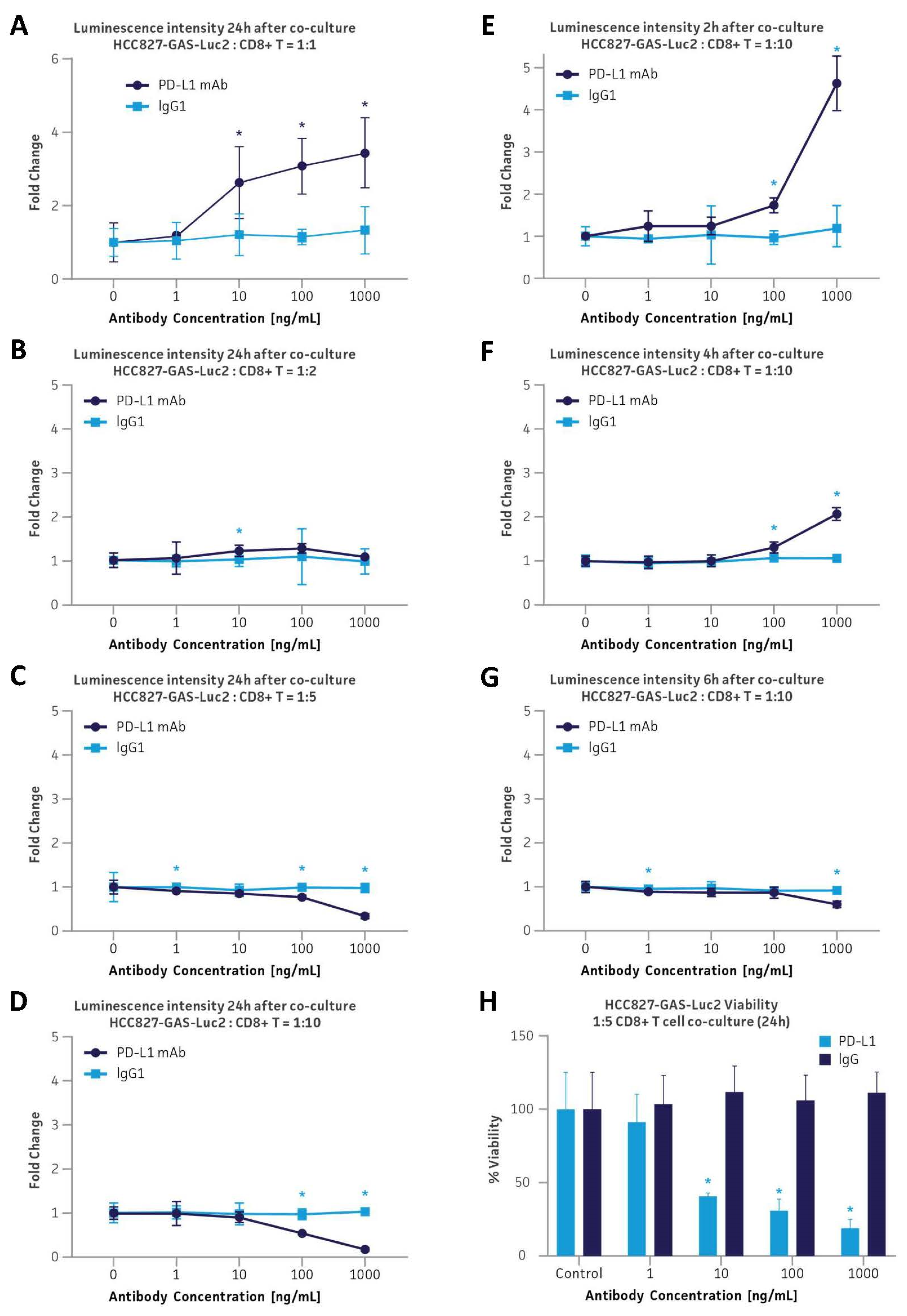
2.6. Cancer Cell and Immune Cell Co-Culture
2.7. CD8+ Cytotoxic T Cell Co-Culture on Cancer Cell Viability
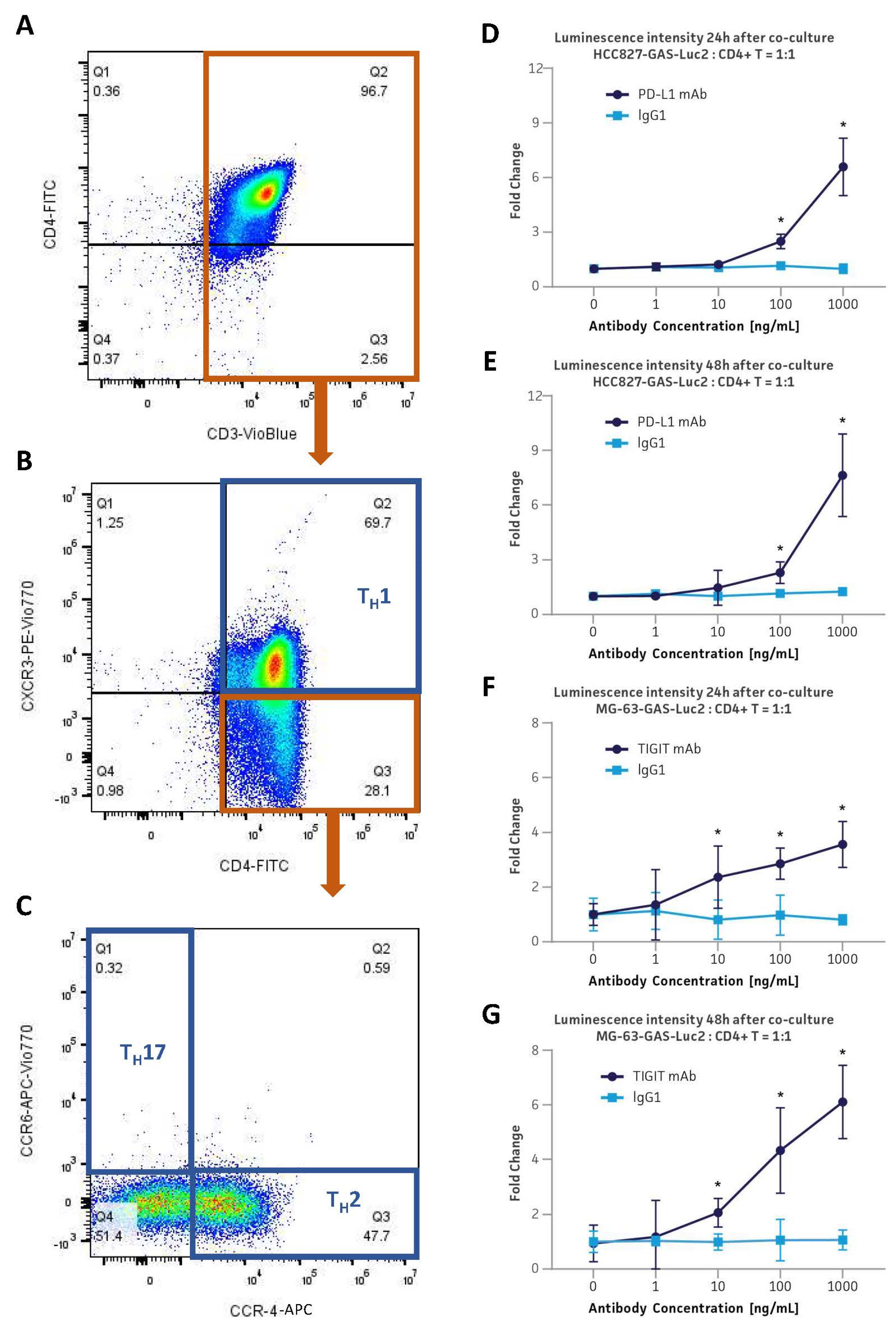
2.8. CD4+ Helper T Cell Subset Phenotyping
2.9. Generation of Artificial Antigen-Presenting Cell Line
2.10. Quantification and Statistical Analysis
3. Results
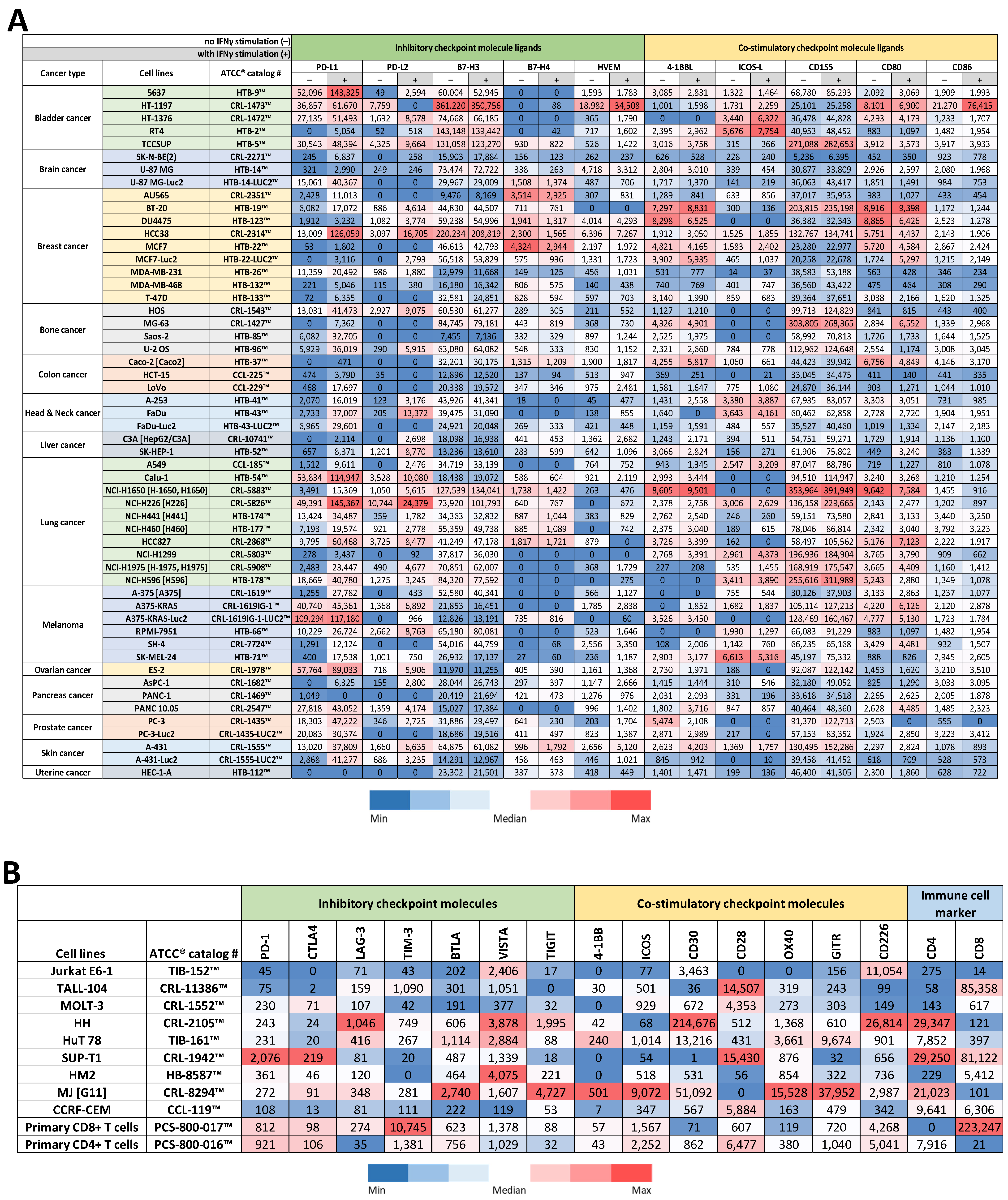
3.1. Protein Profiling of Cancer Cell Lines for Immune Checkpoint Molecule Ligand Expression
3.2. Protein Profiling of T Cell Lines & Primary T Cells for Immune Checkpoint Molecule Expression
3.3. Selection of Candidate Reporter Cell Lines
3.4. Comparisons of Physical Properties of Reporter Cell Lines to Parental Cell Lines
3.5. IFNγ and T Cell-Conditioned Media Stimulation of Reporter Cell Lines
3.6. Application in the PD-L1 Immune Checkpoint Inhibitor Platform via Co-Culture with Primary Human CD8+ Cytotoxic T Cells
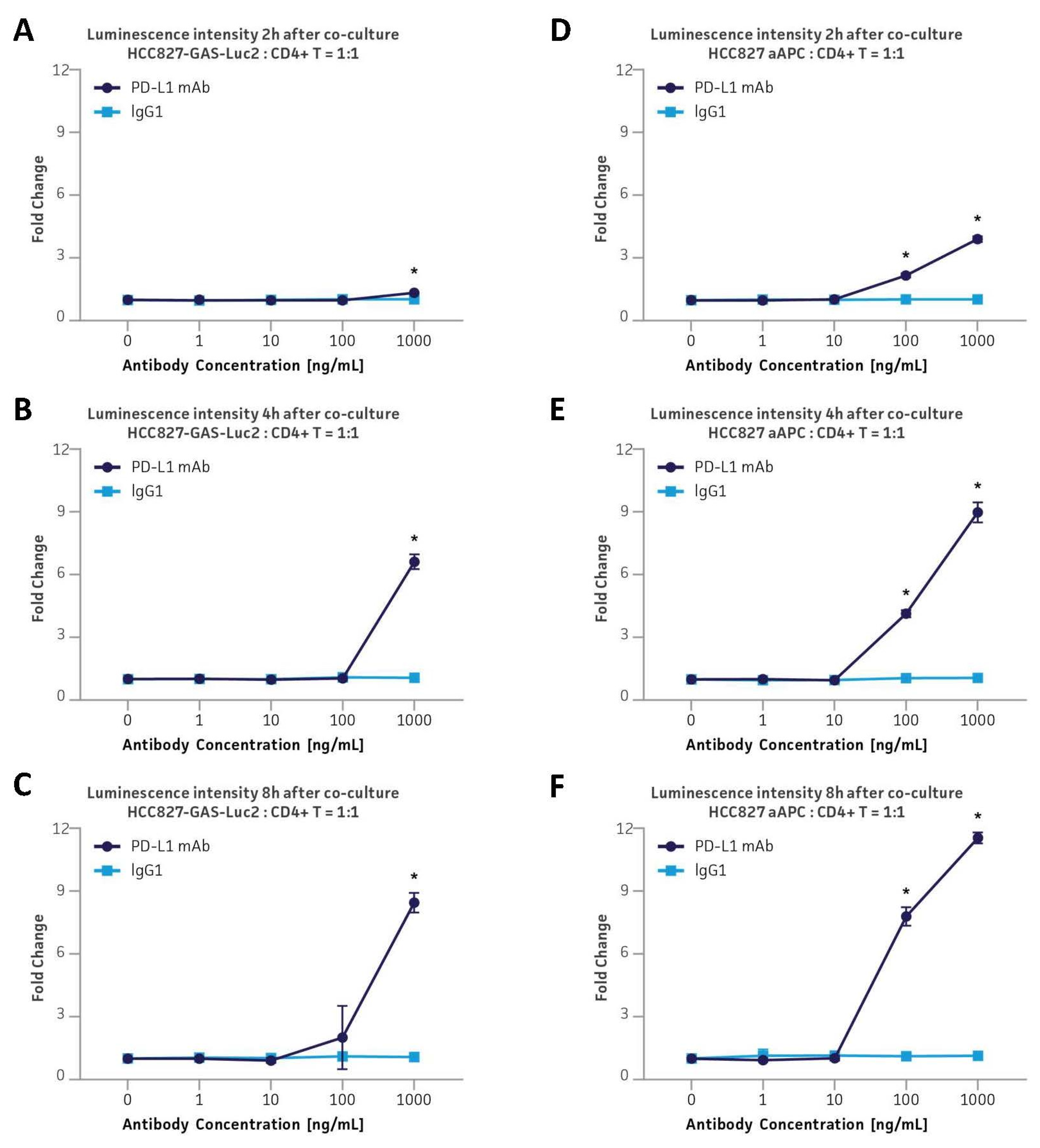
3.7. Application in the PD-L1 & TIGIT Immune Checkpoint Inhibitor Platforms via Co-Culture with Primary Human CD4+ Helper T Cells
3.8. Application in the PD-L1 Immune Checkpoint Inhibitor Platform via Co-Culture with Primary Human CD4+ Helper T Cells by Employing an Anti-CD3/CD28-Expressing aAPC
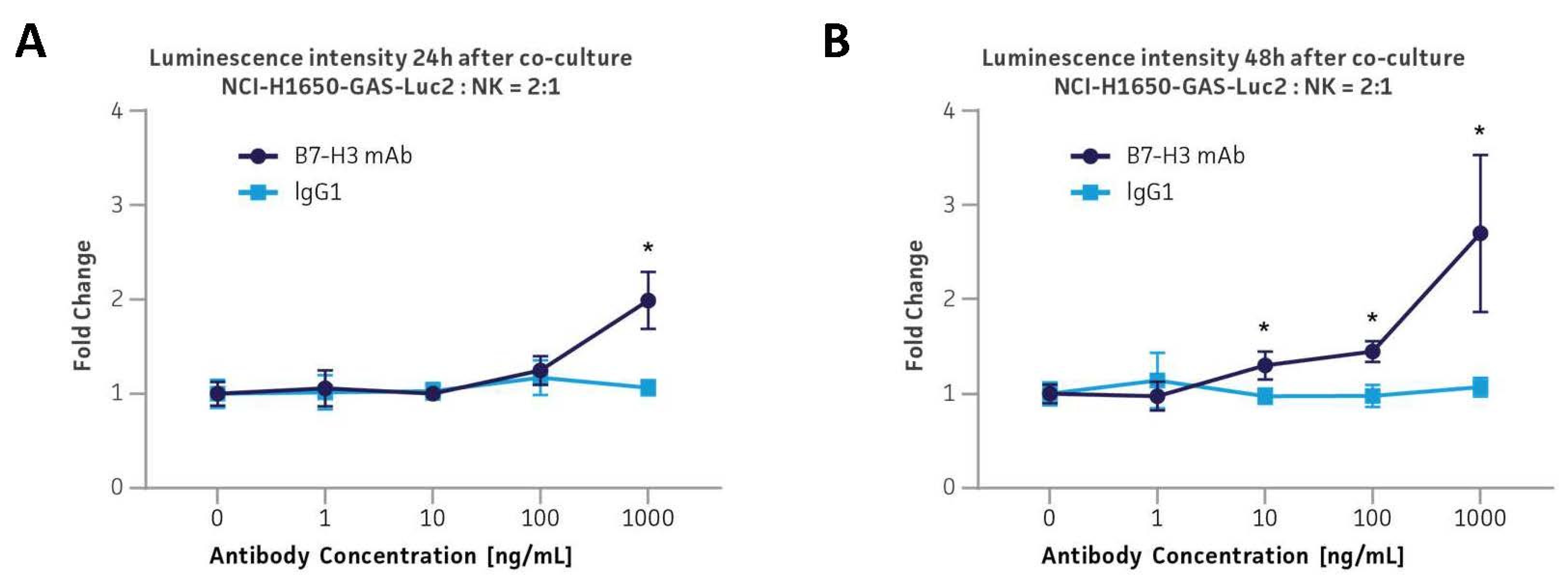
3.9. Application in the B7-H3 Immune Checkpoint ADCC Platform via Co-Culture with Primary Human CD56+ NK Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Korman, A.J.; Garrett-Thomson, S.C.; Lonberg, N. The foundations of immune checkpoint blockade and the ipilimumab approval decennial. Nat. Rev. Drug Discov. 2022, 21, 509–528. [Google Scholar] [CrossRef]
- Kubli, S.P.; Berger, T.; Araujo, D.V.; Siu, L.L.; Mak, T.W. Beyond immune checkpoint blockade: Emerging immunological strategies. Nat. Rev. Drug Discov. 2021, 20, 899–919. [Google Scholar] [CrossRef]
- Olson, B.; Li, Y.; Lin, Y.; Liu, E.T.; Patnaik, A. Mouse Models for Cancer Immunotherapy Research. Cancer Discov. 2018, 8, 1358–1365. [Google Scholar] [CrossRef]
- Neal, J.T.; Li, X.; Zhu, J.; Giangarra, V.; Grzeskowiak, C.L.; Ju, J.; Liu, I.H.; Chiou, S.H.; Salahudeen, A.A.; Smith, A.R.; et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell 2018, 175, 1972–1988 e1916. [Google Scholar] [CrossRef]
- Dijkstra, K.K.; Cattaneo, C.M.; Weeber, F.; Chalabi, M.; van de Haar, J.; Fanchi, L.F.; Slagter, M.; van der Velden, D.L.; Kaing, S.; Kelderman, S.; et al. Generation of Tumor-Reactive T Cells by Co-culture of Peripheral Blood Lymphocytes and Tumor Organoids. Cell 2018, 174, 1586–1598 e1512. [Google Scholar] [CrossRef]
- Versteven, M.; Van den Bergh, J.M.J.; Broos, K.; Fujiki, F.; Campillo-Davo, D.; De Reu, H.; Morimoto, S.; Lecocq, Q.; Keyaerts, M.; Berneman, Z.; et al. A versatile T cell-based assay to assess therapeutic antigen-specific PD-1-targeted approaches. Oncotarget 2018, 9, 27797–27808. [Google Scholar] [CrossRef]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef]
- Yang, S.; Wei, W.; Zhao, Q. B7-H3, a checkpoint molecule, as a target for cancer immunotherapy. Int. J. Biol. Sci. 2020, 16, 1767–1773. [Google Scholar] [CrossRef]
- Zhou, W.-T.; Jin, W.-L. B7-H3/CD276: An Emerging Cancer Immunotherapy. Front. Immunol. 2021, 12, 701006. [Google Scholar] [CrossRef]
- Kučan Brlić, P.; Lenac Roviš, T.; Cinamon, G.; Tsukerman, P.; Mandelboim, O.; Jonjić, S. Targeting PVR (CD155) and its receptors in anti-tumor therapy. Cell. Mol. Immunol. 2019, 16, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Braun, M.; Aguilera, A.R.; Sundarrajan, A.; Corvino, D.; Stannard, K.; Krumeich, S.; Das, I.; Lima, L.G.; Meza Guzman, L.G.; Li, K.; et al. CD155 on Tumor Cells Drives Resistance to Immunotherapy by Inducing the Degradation of the Activating Receptor CD226 in CD8(+) T Cells. Immunity 2020, 53, 805–823.e815. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Dall’Olio, F.G.; Marabelle, A.; Caramella, C.; Garcia, C.; Aldea, M.; Chaput, N.; Robert, C.; Besse, B. Tumour burden and efficacy of immune-checkpoint inhibitors. Nat. Rev. Clin. Oncol. 2022, 19, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Larson, R.C.; Kann, M.C.; Bailey, S.R.; Haradhvala, N.J.; Llopis, P.M.; Bouffard, A.A.; Scarfo, I.; Leick, M.B.; Grauwet, K.; Berger, T.R.; et al. CAR T cell killing requires the IFNgammaR pathway in solid but not liquid tumours. Nature 2022, 604, 563–570. [Google Scholar] [CrossRef]
- Chen, D.; Varanasi, S.K.; Hara, T.; Traina, K.; Sun, M.; McDonald, B.; Farsakoglu, Y.; Clanton, J.; Xu, S.; Garcia-Rivera, L.; et al. CTLA-4 blockade induces CD4(+) T cell IFNgamma-driven microglial phagocytosis and anti-tumor function in glioblastoma. Immunity 2023, 56, 2086–2104.e8. [Google Scholar] [CrossRef] [PubMed]
- Platanias, L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Brasier, A.R.; Tate, J.E.; Habener, J.F. Optimized use of the firefly luciferase assay as a reporter gene in mammalian cell lines. Biotechniques 1989, 7, 1116–1122. [Google Scholar] [PubMed]
- Grewal, T.; Priceputu, E.; Davignon, J.; Bernier, L. Identification of a gamma-interferon-responsive element in the promoter of the human macrophage scavenger receptor A gene. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 825–831. [Google Scholar] [CrossRef]
- Schmidts, A.; Marsh, L.C.; Srivastava, A.A.; Bouffard, A.A.; Boroughs, A.C.; Scarfo, I.; Larson, R.C.; Bedoya, F.; Choi, B.D.; Frigault, M.J.; et al. Cell-based artificial APC resistant to lentiviral transduction for efficient generation of CAR-T cells from various cell sources. J. Immunother. Cancer 2020, 8, e000990. [Google Scholar] [CrossRef]
- Shrestha, B.; Zhang, Y.; Yu, B.; Li, G.; Boucher, J.C.; Beatty, N.J.; Tsai, H.C.; Wang, X.; Mishra, A.; Sweet, K.; et al. Generation of Antitumor T Cells For Adoptive Cell Therapy With Artificial Antigen Presenting Cells. J. Immunother. 2020, 43, 79–88. [Google Scholar] [CrossRef]
- Ghandi, M.; Huang, F.W.; Jane-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R., 3rd; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019, 569, 503–508. [Google Scholar] [CrossRef]
- Nusinow, D.P.; Szpyt, J.; Ghandi, M.; Rose, C.M.; McDonald, E.R., 3rd; Kalocsay, M.; Jane-Valbuena, J.; Gelfand, E.; Schweppe, D.K.; Jedrychowski, M.; et al. Quantitative Proteomics of the Cancer Cell Line Encyclopedia. Cell 2020, 180, 387–402 e316. [Google Scholar] [CrossRef]
- Upadhaya, S.; Hubbard-Lucey, V.M.; Yu, J.X. Immuno-oncology drug development forges on despite COVID-19. Nat. Rev. Drug Discov. 2020, 19, 751–752. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Cancer Cell Line Encyclopedia, C.; Genomics of Drug Sensitivity in Cancer, C. Pharmacogenomic agreement between two cancer cell line data sets. Nature 2015, 528, 84–87. [Google Scholar] [CrossRef]
- Li, H.; Ning, S.; Ghandi, M.; Kryukov, G.V.; Gopal, S.; Deik, A.; Souza, A.; Pierce, K.; Keskula, P.; Hernandez, D.; et al. The landscape of cancer cell line metabolism. Nat. Med. 2019, 25, 850–860. [Google Scholar] [CrossRef]
- Jaffe, J.D.; Wang, Y.; Chan, H.M.; Zhang, J.; Huether, R.; Kryukov, G.V.; Bhang, H.E.; Taylor, J.E.; Hu, M.; Englund, N.P.; et al. Global chromatin profiling reveals NSD2 mutations in pediatric acute lymphoblastic leukemia. Nat. Genet. 2013, 45, 1386–1391. [Google Scholar] [CrossRef]
- Guo, Y.; Xiao, P.; Lei, S.; Deng, F.; Xiao, G.G.; Liu, Y.; Chen, X.; Li, L.; Wu, S.; Chen, Y.; et al. How is mRNA expression predictive for protein expression? A correlation study on human circulating monocytes. Acta Biochim. Biophys. Sin. 2008, 40, 426–436. [Google Scholar] [CrossRef]
- Koussounadis, A.; Langdon, S.P.; Um, I.H.; Harrison, D.J.; Smith, V.A. Relationship between differentially expressed mRNA and mRNA-protein correlations in a xenograft model system. Sci. Rep. 2015, 5, 10775. [Google Scholar] [CrossRef]
- Liu, B.; Hu, X.; Feng, K.; Gao, R.; Xue, Z.; Zhang, S.; Zhang, Y.; Corse, E.; Hu, Y.; Han, W.; et al. Temporal single-cell tracing reveals clonal revival and expansion of precursor exhausted T cells during anti-PD-1 therapy in lung cancer. Nat. Cancer 2022, 3, 108–121. [Google Scholar] [CrossRef]
- Melenhorst, J.J.; Chen, G.M.; Wang, M.; Porter, D.L.; Chen, C.; Collins, M.A.; Gao, P.; Bandyopadhyay, S.; Sun, H.; Zhao, Z.; et al. Decade-long leukaemia remissions with persistence of CD4(+) CAR T cells. Nature 2022, 602, 503–509. [Google Scholar] [CrossRef]
- Xu, Y.; Wei, Z.; Feng, M.; Zhu, D.; Mei, S.; Wu, Z.; Feng, Q.; Chang, W.; Ji, M.; Liu, C.; et al. Tumor-infiltrated activated B cells suppress liver metastasis of colorectal cancers. Cell Rep. 2022, 40, 111295. [Google Scholar] [CrossRef]
- Borriello, L.; Coste, A.; Traub, B.; Sharma, V.P.; Karagiannis, G.S.; Lin, Y.; Wang, Y.; Ye, X.; Duran, C.L.; Chen, X.; et al. Primary tumor associated macrophages activate programs of invasion and dormancy in disseminating tumor cells. Nat. Commun. 2022, 13, 626. [Google Scholar] [CrossRef]
- Buechler, M.B.; Pradhan, R.N.; Krishnamurty, A.T.; Cox, C.; Calviello, A.K.; Wang, A.W.; Yang, Y.A.; Tam, L.; Caothien, R.; Roose-Girma, M.; et al. Cross-tissue organization of the fibroblast lineage. Nature 2021, 593, 575–579. [Google Scholar] [CrossRef]
- Xie, Y.; He, L.; Lugano, R.; Zhang, Y.; Cao, H.; He, Q.; Chao, M.; Liu, B.; Cao, Q.; Wang, J.; et al. Key molecular alterations in endothelial cells in human glioblastoma uncovered through single-cell RNA sequencing. JCI Insight 2021, 6, 15. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Gibson, J.; Ou, K.; Lopez, L.S.; Ng, R.H.; Leggett, N.; Jonsson, V.D.; Zarif, J.C.; Lee, P.P.; Wang, X.; et al. PD-L1 blockade restores CAR T cell activity through IFN-gamma-regulation of CD163+ M2 macrophages. J. Immunother. Cancer 2022, 10, e004400. [Google Scholar] [CrossRef]
- Eggermont, L.J.; Paulis, L.E.; Tel, J.; Figdor, C.G. Towards efficient cancer immunotherapy: Advances in developing artificial antigen-presenting cells. Trends Biotechnol. 2014, 32, 456–465. [Google Scholar] [CrossRef]
- Sunshine, J.C.; Green, J.J. Nanoengineering approaches to the design of artificial antigen-presenting cells. Nanomedicine 2013, 8, 1173–1189. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, H.; Foulke, J.G.; Chen, L.; Tian, F.; Gu, Z. GAS-Luc2 Reporter Cell Lines for Immune Checkpoint Drug Screening in Solid Tumors. Cancers 2024, 16, 1965. https://doi.org/10.3390/cancers16111965
Chang H, Foulke JG, Chen L, Tian F, Gu Z. GAS-Luc2 Reporter Cell Lines for Immune Checkpoint Drug Screening in Solid Tumors. Cancers. 2024; 16(11):1965. https://doi.org/10.3390/cancers16111965
Chicago/Turabian StyleChang, Hyeyoun, John G. Foulke, Luping Chen, Fang Tian, and Zhizhan Gu. 2024. "GAS-Luc2 Reporter Cell Lines for Immune Checkpoint Drug Screening in Solid Tumors" Cancers 16, no. 11: 1965. https://doi.org/10.3390/cancers16111965
APA StyleChang, H., Foulke, J. G., Chen, L., Tian, F., & Gu, Z. (2024). GAS-Luc2 Reporter Cell Lines for Immune Checkpoint Drug Screening in Solid Tumors. Cancers, 16(11), 1965. https://doi.org/10.3390/cancers16111965