
Pathogenicity Prediction of Gene Fusion in Structural Variations: A Knowledge Graph-Infused Explainable Artificial Intelligence (XAI) Framework

 ,
,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Knowledge Graph
2.2. Explainable AI (XAI)
2.3. Our Explainable Learning Model
2.3.1. Correct Answer Set: Dataset 1
- The target text describes approved drugs in Japan for $DISEASENAME.
- The target text describes FDA-approved drugs for $DISEASENAME.
- The target text is referenced by guidelines about $DISEASENAME.
- The target text describes highly statistically reliable clinical trials/meta-analyses and consensus among experts on $DISEASENAME.
- The target text describes FDA-approved drugs for other cancer types.
- The target text describes highly statistically reliable clinical trials/meta-analyses and consensus among experts regarding other cancer types.
- The target text describes small-scale clinical trials that have shown usefulness regardless of cancer type.
- The target text describes the usefulness shown in case reports regardless of cancer type.
- The usefulness of target text has been reported in preclinical studies (in vitro and in vivo).
2.3.2. Features
- The amount of literature regarding the same fusion genes in Mitelman;
- The number of entries about the same fusion genes in COSMIC;
- The sequence of domains registered with Pfam on each gene;
- The lengths of both UTRs;
- TargetScan-registered miRNAs that affect UTR;
- Families of miRNAs;
- Whether each domain is within the breakpoint;
2.4. Benchmark
2.4.1. Existing Methods
2.4.2. Learning Set: Dataset 2 and Dataset 3
2.4.3. Method of Comparing the Performance of the Models
- Cross Validation
- 2.
- Holdout Validation
2.4.4. Our Benchmark Learning Model
- Sequences of domains registered with Pfam for each gene;
- Lengths of both UTRs;
- TargetScan-registered miRNAs that affect UTR;
- Families of miRNAs;
- Whether each domain was within the breakpoint.
3. Results
3.1. Evaluation of Prediction Accuracy
3.1.1. Evaluation using Cross-Validation
3.1.2. Evaluation Using Holdout Validation
3.2. Evaluation of the Explanation of Prediction
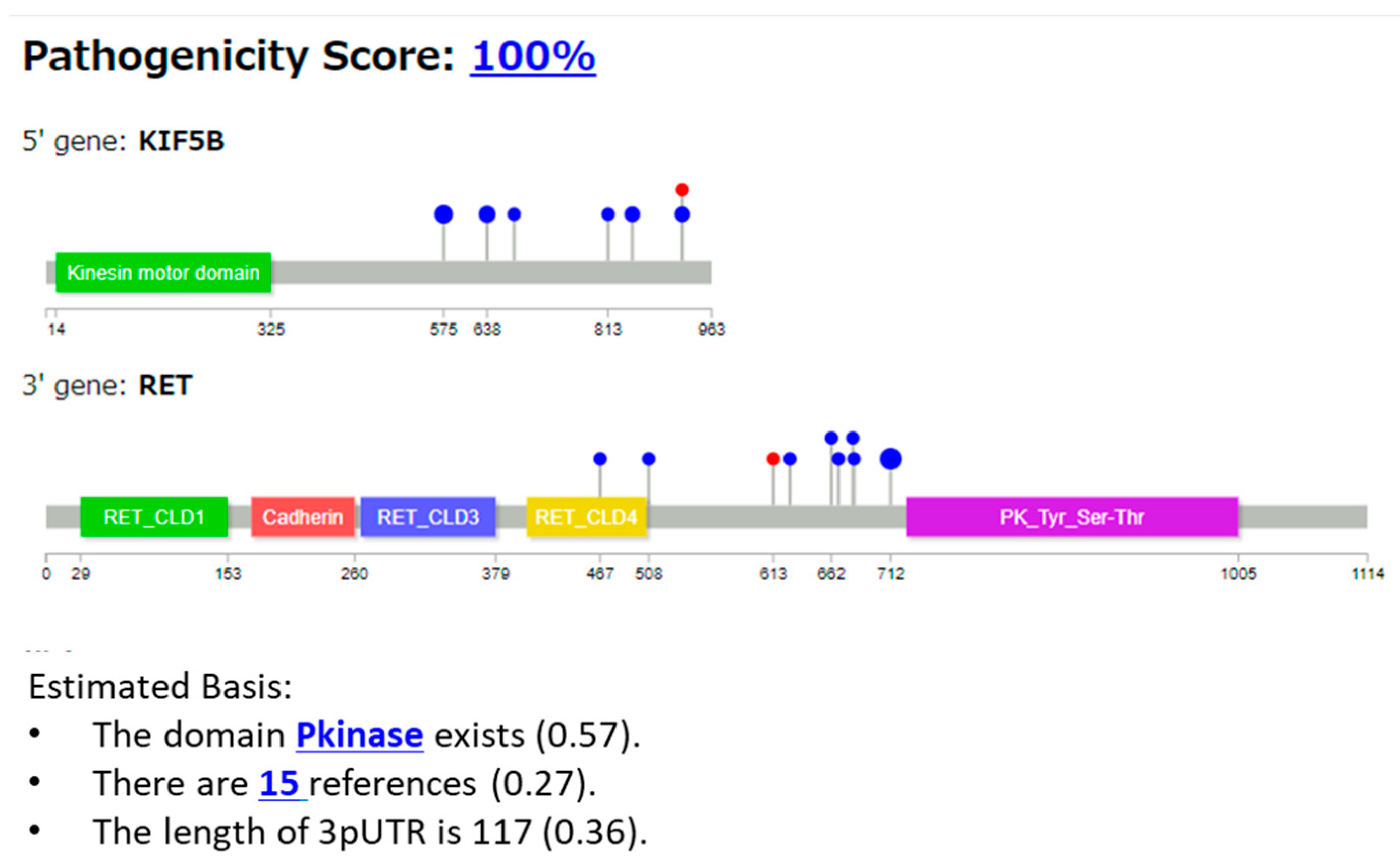
3.2.1. Case 1: KIF5B::RET Fusion
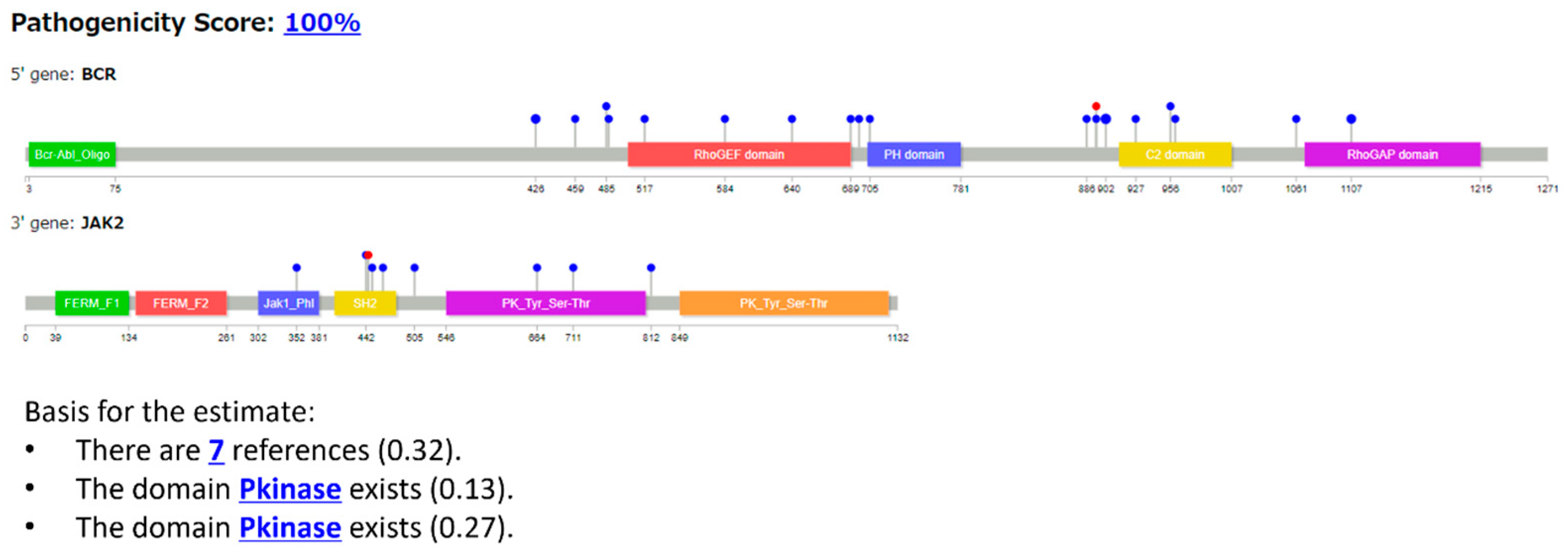
3.2.2. Case 2: BCR::JAK2 T Fusion
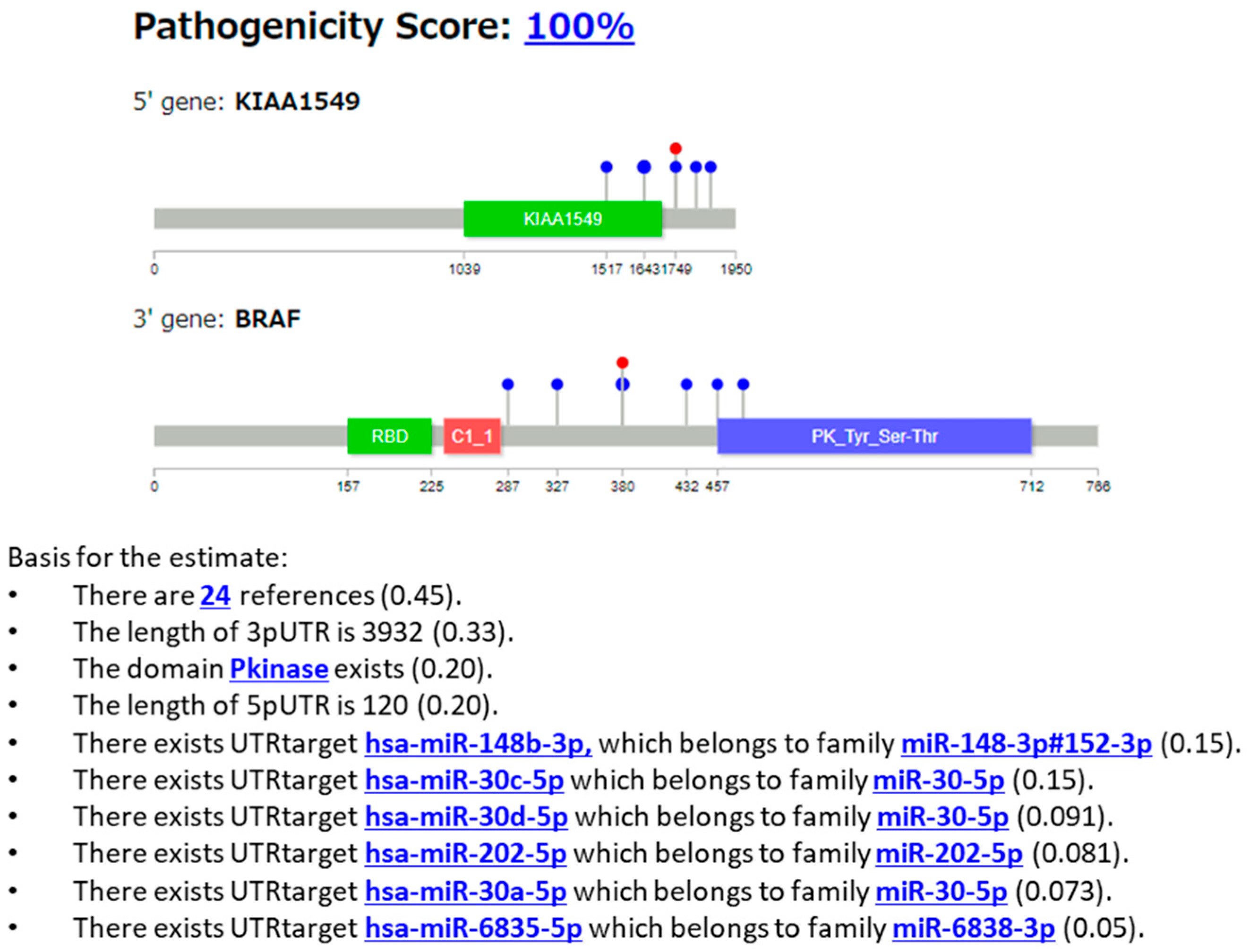
3.2.3. Case 3: KIAA1549::BRAF Fusion
3.2.4. Case 4: IKZF1::LRBA Fusion
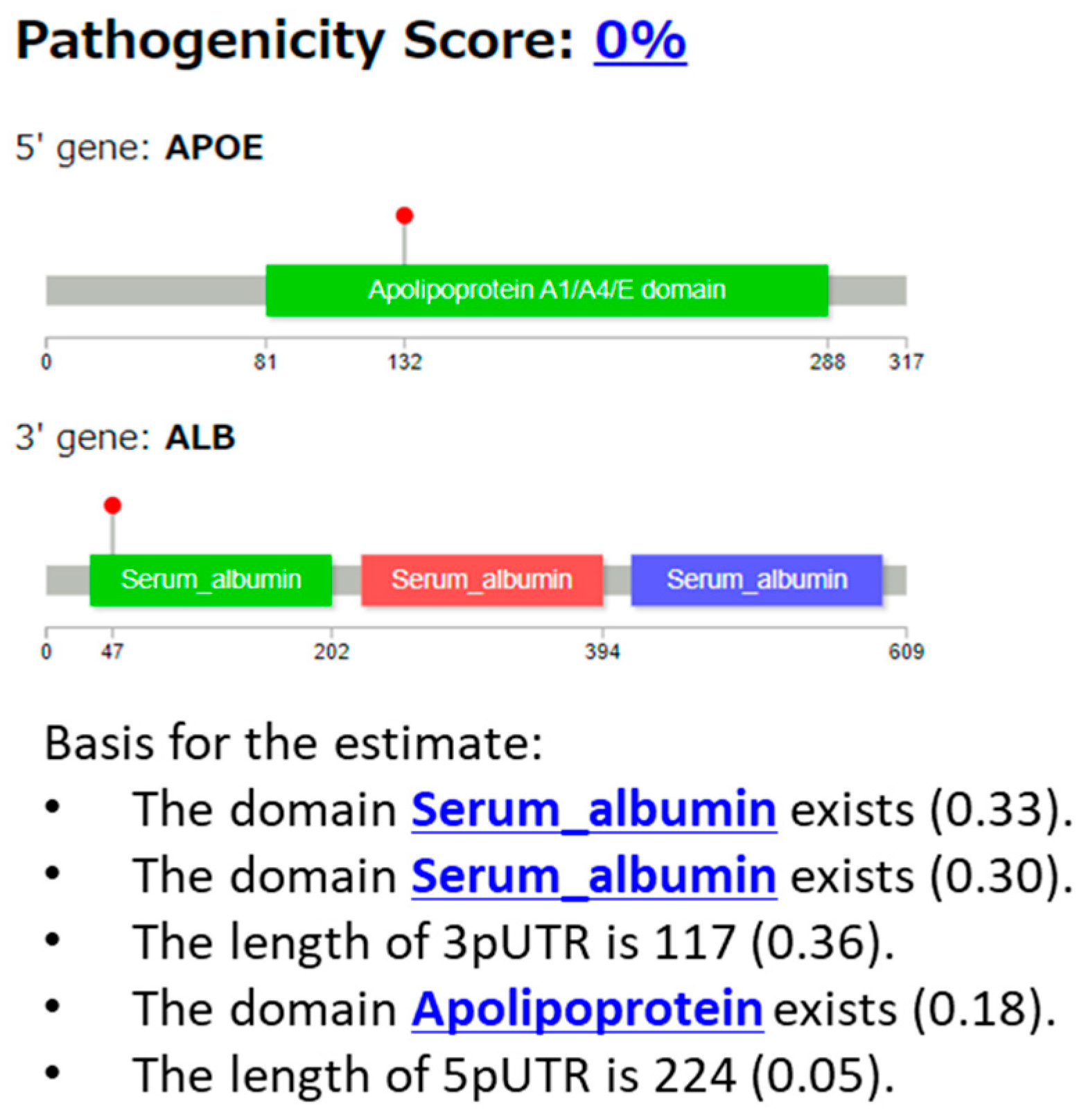
3.2.5. Case 5: APOE::ALB Fusion
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mitelman, F.; Johansson, B.; Mertens, F. The Impact of Translocations and Gene Fusions on Cancer Causation. Nat. Rev. Cancer 2007, 7, 233–245. [Google Scholar] [CrossRef]
- Chen, X.; Schulz-Trieglaff, O.; Shaw, R.; Barnes, B.; Schlesinger, F.; Källberg, M.; Cox, A.J.; Kruglyak, S.; Saunders, C.T. Manta: Rapid Detection of Structural Variants and Indels for Germline and Cancer Sequencing Applications. Bioinformatics 2016, 32, 1220–1222. [Google Scholar] [CrossRef] [PubMed]
- Lovino, M.; Montemurro, M.; Barrese, V.S.; Ficarra, E. Identifying the Oncogenic Potential of Gene Fusions Exploiting MiRNAs. J. Biomed. Inform. 2022, 129, 104057. [Google Scholar] [CrossRef] [PubMed]
- Lovino, M.; Ciaburri, M.S.; Urgese, G.; Di Cataldo, S.; Ficarra, E. DEEPrior: A Deep Learning Tool for the Prioritization of Gene Fusions. Bioinformatics 2020, 36, 3248–3250. [Google Scholar] [CrossRef] [PubMed]
- Shugay, M.; Ortiz de Mendíbil, I.; Vizmanos, J.L.; Novo, F.J. Oncofuse: A Computational Framework for the Prediction of the Oncogenic Potential of Gene Fusions. Bioinformatics 2013, 29, 2539–2546. [Google Scholar] [CrossRef]
- Sheu, R.-K.; Pardeshi, M.S. A Survey on Medical Explainable AI (XAI): Recent Progress, Explainability Approach, Human Interaction and Scoring System. Sensors 2022, 22, 8068. [Google Scholar] [CrossRef]
- Abe, S.; Tago, S.; Yokoyama, K.; Ogawa, M.; Takei, T.; Imoto, S.; Fuji, M. Explainable AI for Estimating Pathogenicity of Genetic Variants Using Large-Scale Knowledge Graphs. Cancers 2023, 15, 1118. [Google Scholar] [CrossRef]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, Ł.; Polosukhin, I. Attention Is All You Need. Adv. Neural Inf. Process. Syst. 2017, 30, 5998–6008. [Google Scholar]
- Resource Description Framework (RDF): Concepts and Abstract Syntax. Available online: https://www.w3.org/TR/rdf-concepts/ (accessed on 28 March 2024).
- Med2RDF. Available online: http://med2rdf.org/ (accessed on 28 March 2024).
- Auer, S.; Bizer, C.; Kobilarov, G.; Lehmann, J.; Cyganiak, R.; Ives, Z. DBpedia: A Nucleus for a Web of Open Data. In Proceedings of the The Semantic Web, Busan, Republic of Korea, 11–15 November 2007; Springer: Berlin/Heidelberg, Germany, 2007; pp. 722–735. [Google Scholar]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef]
- Raney, B.J.; Barber, G.P.; Benet-Pagès, A.; Casper, J.; Clawson, H.; Cline, M.S.; Diekhans, M.; Fischer, C.; Navarro Gonzalez, J.; Hickey, G.; et al. The UCSC Genome Browser Database: 2024 Update. Nucleic Acids Res. 2024, 52, D1082–D1088. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The Protein Families Database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference Sequence (RefSeq) Database at NCBI: Current Status, Taxonomic Expansion, and Functional Annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [PubMed]
- McGeary, S.E.; Lin, K.S.; Shi, C.Y.; Pham, T.M.; Bisaria, N.; Kelley, G.M.; Bartel, D.P. The Biochemical Basis of MicroRNA Targeting Efficacy. Science 2019, 366, eaav1741. [Google Scholar] [CrossRef] [PubMed]
- Johansson, B.; Mertens, F.; Mitelman, F. Geographic Heterogeneity of Neoplasia-Associated Chromosome Aberrations. Genes Chromosomes Cancer 1991, 3, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Mitelman Database Chromosome Aberrations and Gene Fusions in Cancer. Available online: https://mitelmandatabase.isb-cgc.org/about (accessed on 25 March 2024).
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the Unification of Biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Gene Ontology Consortium; Aleksander, S.A.; Balhoff, J.; Carbon, S.; Cherry, J.M.; Drabkin, H.J.; Ebert, D.; Feuermann, M.; Gaudet, P.; Harris, N.L.; et al. The Gene Ontology Knowledgebase in 2023. Genetics 2023, 224, iyad031. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Wang, Q.; Tang, M.; Barthel, F.; Amin, S.; Yoshihara, K.; Lang, F.M.; Martinez-Ledesma, E.; Lee, S.H.; Zheng, S.; et al. TumorFusions: An Integrative Resource for Cancer-Associated Transcript Fusions. Nucleic Acids Res. 2018, 46, D1144–D1149. [Google Scholar] [CrossRef] [PubMed]
- Maruhashi, K.; Todoriki, M.; Ohwa, T.; Goto, K.; Hasegawa, Y.; Inakoshi, H.; Anai, H. Learning Multi-Way Relations via Tensor Decomposition with Neural Networks. AAAI 2018, 32, 3770–3777. [Google Scholar] [CrossRef]
- Ribeiro, M.T.; Singh, S.; Guestrin, C. “Why Should I Trust You?”: Explaining the Predictions of Any Classifier. In Proceedings of the 2016 Conference of the North American Chapter of the Association for Computational Linguistics: Demonstrations, San Diego, CA, USA, 12–17 June 2016. [Google Scholar] [CrossRef]
- Brown, T.B.; Mann, B.; Ryder, N.; Subbiah, M.; Kaplan, J.; Dhariwal, P.; Neelakantan, A.; Shyam, P.; Sastry, G.; Askell, A.; et al. Language Models Are Few-Shot Learners. Adv. Neural Inf. Process. Syst. 2020, 33, 1877–1901. [Google Scholar]
- Azure OpenAI Service. Available online: https://azure.microsoft.com/en-us/products/ai-services/openai-service/ (accessed on 25 March 2024).
- Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer Analysis Project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Liang, W.; Zhang, Y.; Cao, H.; Wang, B.; Ding, D.; Yang, X.; Vodrahalli, K.; He, S.; Smith, D.; Yin, Y.; et al. Can Large Language Models Provide Useful Feedback on Research Papers? A Large-Scale Empirical Analysis. arXiv 2023, arXiv:2310.01783. [Google Scholar]
- JSMO Guideline. Available online: https://www.jsmo.or.jp/about/doc/20200310.pdf (accessed on 22 March 2024).
- Abate, F.; Zairis, S.; Ficarra, E.; Acquaviva, A.; Wiggins, C.H.; Frattini, V.; Lasorella, A.; Iavarone, A.; Inghirami, G.; Rabadan, R. Pegasus: A Comprehensive Annotation and Prediction Tool for Detection of Driver Gene Fusions in Cancer. BMC Syst. Biol. 2014, 8, 97. [Google Scholar] [CrossRef]
- Babiceanu, M.; Qin, F.; Xie, Z.; Jia, Y.; Lopez, K.; Janus, N.; Facemire, L.; Kumar, S.; Pang, Y.; Qi, Y.; et al. Recurrent Chimeric Fusion RNAs in Non-Cancer Tissues and Cells. Nucleic Acids Res. 2016, 44, 2859–2872. [Google Scholar] [CrossRef] [PubMed]
- Kohno, T.; Ichikawa, H.; Totoki, Y.; Yasuda, K.; Hiramoto, M.; Nammo, T.; Sakamoto, H.; Tsuta, K.; Furuta, K.; Shimada, Y.; et al. KIF5B-RET Fusions in Lung Adenocarcinoma. Nat. Med. 2012, 18, 375–377. [Google Scholar] [CrossRef] [PubMed]
- Jay, J.J.; Brouwer, C. Lollipops in the Clinic: Information Dense Mutation Plots for Precision Medicine. PLoS ONE 2016, 11, e0160519. [Google Scholar] [CrossRef] [PubMed]
- Cirmena, G.; Aliano, S.; Fugazza, G.; Bruzzone, R.; Garuti, A.; Bocciardi, R.; Bacigalupo, A.; Ravazzolo, R.; Ballestrero, A.; Sessarego, M. A BCR-JAK2 Fusion Gene as the Result of a t(9;22)(P24;Q11) in a Patient with Acute Myeloid Leukemia. Cancer Genet. Cytogenet. 2008, 183, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Ryall, S.; Krishnatry, R.; Arnoldo, A.; Buczkowicz, P.; Mistry, M.; Siddaway, R.; Ling, C.; Pajovic, S.; Yu, M.; Rubin, J.B.; et al. Targeted Detection of Genetic Alterations Reveal the Prognostic Impact of H3K27M and MAPK Pathway Aberrations in Paediatric Thalamic Glioma. Acta Neuropathol. Commun. 2016, 4, 93. [Google Scholar] [CrossRef] [PubMed]
- Yokota, K.; Sasaki, H.; Okuda, K.; Shimizu, S.; Shitara, M.; Hikosaka, Y.; Moriyama, S.; Yano, M.; Fujii, Y. KIF5B/RET Fusion Gene in Surgically-Treated Adenocarcinoma of the Lung. Oncol. Rep. 2012, 28, 1187–1192. [Google Scholar] [CrossRef]
- Ju, Y.S.; Lee, W.-C.; Shin, J.-Y.; Lee, S.; Bleazard, T.; Won, J.-K.; Kim, Y.T.; Kim, J.-I.; Kang, J.-H.; Seo, J.-S. A Transforming KIF5B and RET Gene Fusion in Lung Adenocarcinoma Revealed from Whole-Genome and Transcriptome Sequencing. Genome Res. 2012, 22, 436–445. [Google Scholar] [CrossRef]
- Cuesta-Domínguez, Á.; Ortega, M.; Ormazábal, C.; Santos-Roncero, M.; Galán-Díez, M.; Steegmann, J.L.; Figuera, Á.; Arranz, E.; Vizmanos, J.L.; Bueren, J.A.; et al. Transforming and Tumorigenic Activity of JAK2 by Fusion to BCR: Molecular Mechanisms of Action of a Novel BCR-JAK2 Tyrosine-Kinase. PLoS ONE 2012, 7, e32451. [Google Scholar] [CrossRef]
- McWhirter, J.R.; Galasso, D.L.; Wang, J.Y. A Coiled-Coil Oligomerization Domain of Bcr Is Essential for the Transforming Function of Bcr-Abl Oncoproteins. Mol. Cell. Biol. 1993, 13, 7587–7595. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.G.; Li, Y.; Payne-Turner, D.; Harvey, R.C.; Yang, Y.-L.; Pei, D.; McCastlain, K.; Ding, L.; Lu, C.; Song, G.; et al. Targetable Kinase-Activating Lesions in Ph-like Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2014, 371, 1005–1015. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, M.; Badiali, M.; Moi, L.; Buttarelli, F.R.; Baldi, C.; Massimino, M.; Sanson, M.; Giangaspero, F. KIAA1549:BRAF Fusion Gene in Pediatric Brain Tumors of Various Histogenesis. Pediatr. Blood Cancer 2015, 62, 724–727. [Google Scholar] [CrossRef] [PubMed]
- Appay, R.; Fina, F.; Macagno, N.; Padovani, L.; Colin, C.; Barets, D.; Ordioni, J.; Scavarda, D.; Giangaspero, F.; Badiali, M.; et al. Duplications of KIAA1549 and BRAF Screening by Droplet Digital PCR from Formalin-Fixed Paraffin-Embedded DNA Is an Accurate Alternative for KIAA1549-BRAF Fusion Detection in Pilocytic Astrocytomas. Mod. Pathol. 2018, 31, 1490–1501. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Deng, X.; Zeng, X.; Peng, X. The Role of Mir-148a in Cancer. J. Cancer 2016, 7, 1233–1241. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, X.; Wang, Y.; Peng, R.; Lin, Z.; Wang, Y.; Hu, B.; Wang, J.; Shi, G. Low Expression of MicroRNA-30c Promotes Prostate Cancer Cells Invasion Involved in Downregulation of KRAS Protein. Oncol. Lett. 2017, 14, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, E.A.; Rajendran, P.; Scherthan, H. The MicroRNA-202 as a Diagnostic Biomarker and a Potential Tumor Suppressor. Int. J. Mol. Sci. 2022, 23, 5870. [Google Scholar] [CrossRef] [PubMed]
- Lind, K.T.; Chatwin, H.V.; DeSisto, J.; Coleman, P.; Sanford, B.; Donson, A.M.; Davies, K.D.; Willard, N.; Ewing, C.A.; Knox, A.J.; et al. Novel RAF Fusions in Pediatric Low-Grade Gliomas Demonstrate MAPK Pathway Activation. J. Neuropathol. Exp. Neurol. 2021, 80, 1099–1107. [Google Scholar] [CrossRef]
- Helgager, J.; Lidov, H.G.; Mahadevan, N.R.; Kieran, M.W.; Ligon, K.L.; Alexandrescu, S. A Novel GIT2-BRAF Fusion in Pilocytic Astrocytoma. Diagn. Pathol. 2017, 12, 82. [Google Scholar] [CrossRef]
- Yan, L.; Ping, N.; Zhu, M.; Sun, A.; Xue, Y.; Ruan, C.; Drexler, H.G.; Macleod, R.A.F.; Wu, D.; Chen, S. Clinical, Immunophenotypic, Cytogenetic, and Molecular Genetic Features in 117 Adult Patients with Mixed-Phenotype Acute Leukemia Defined by WHO-2008 Classification. Haematologica 2012, 97, 1708–1712. [Google Scholar] [CrossRef]
- Mullighan, C.G.; Miller, C.B.; Radtke, I.; Phillips, L.A.; Dalton, J.; Ma, J.; White, D.; Hughes, T.P.; Le Beau, M.M.; Pui, C.-H.; et al. BCR-ABL1 Lymphoblastic Leukaemia Is Characterized by the Deletion of Ikaros. Nature 2008, 453, 110–114. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murakami, K.; Tago, S.-i.; Takishita, S.; Morikawa, H.; Kojima, R.; Yokoyama, K.; Ogawa, M.; Fukushima, H.; Takamori, H.; Nannya, Y.; et al. Pathogenicity Prediction of Gene Fusion in Structural Variations: A Knowledge Graph-Infused Explainable Artificial Intelligence (XAI) Framework. Cancers 2024, 16, 1915. https://doi.org/10.3390/cancers16101915
Murakami K, Tago S-i, Takishita S, Morikawa H, Kojima R, Yokoyama K, Ogawa M, Fukushima H, Takamori H, Nannya Y, et al. Pathogenicity Prediction of Gene Fusion in Structural Variations: A Knowledge Graph-Infused Explainable Artificial Intelligence (XAI) Framework. Cancers. 2024; 16(10):1915. https://doi.org/10.3390/cancers16101915
Chicago/Turabian StyleMurakami, Katsuhiko, Shin-ichiro Tago, Sho Takishita, Hiroaki Morikawa, Rikuhiro Kojima, Kazuaki Yokoyama, Miho Ogawa, Hidehito Fukushima, Hiroyuki Takamori, Yasuhito Nannya, and et al. 2024. "Pathogenicity Prediction of Gene Fusion in Structural Variations: A Knowledge Graph-Infused Explainable Artificial Intelligence (XAI) Framework" Cancers 16, no. 10: 1915. https://doi.org/10.3390/cancers16101915
APA StyleMurakami, K., Tago, S.-i., Takishita, S., Morikawa, H., Kojima, R., Yokoyama, K., Ogawa, M., Fukushima, H., Takamori, H., Nannya, Y., Imoto, S., & Fuji, M. (2024). Pathogenicity Prediction of Gene Fusion in Structural Variations: A Knowledge Graph-Infused Explainable Artificial Intelligence (XAI) Framework. Cancers, 16(10), 1915. https://doi.org/10.3390/cancers16101915