


Genome-Wide Methylation Analysis in Two Wild-Type Non-Small Cell Lung Cancer Subgroups with Negative and High PD-L1 Expression


Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
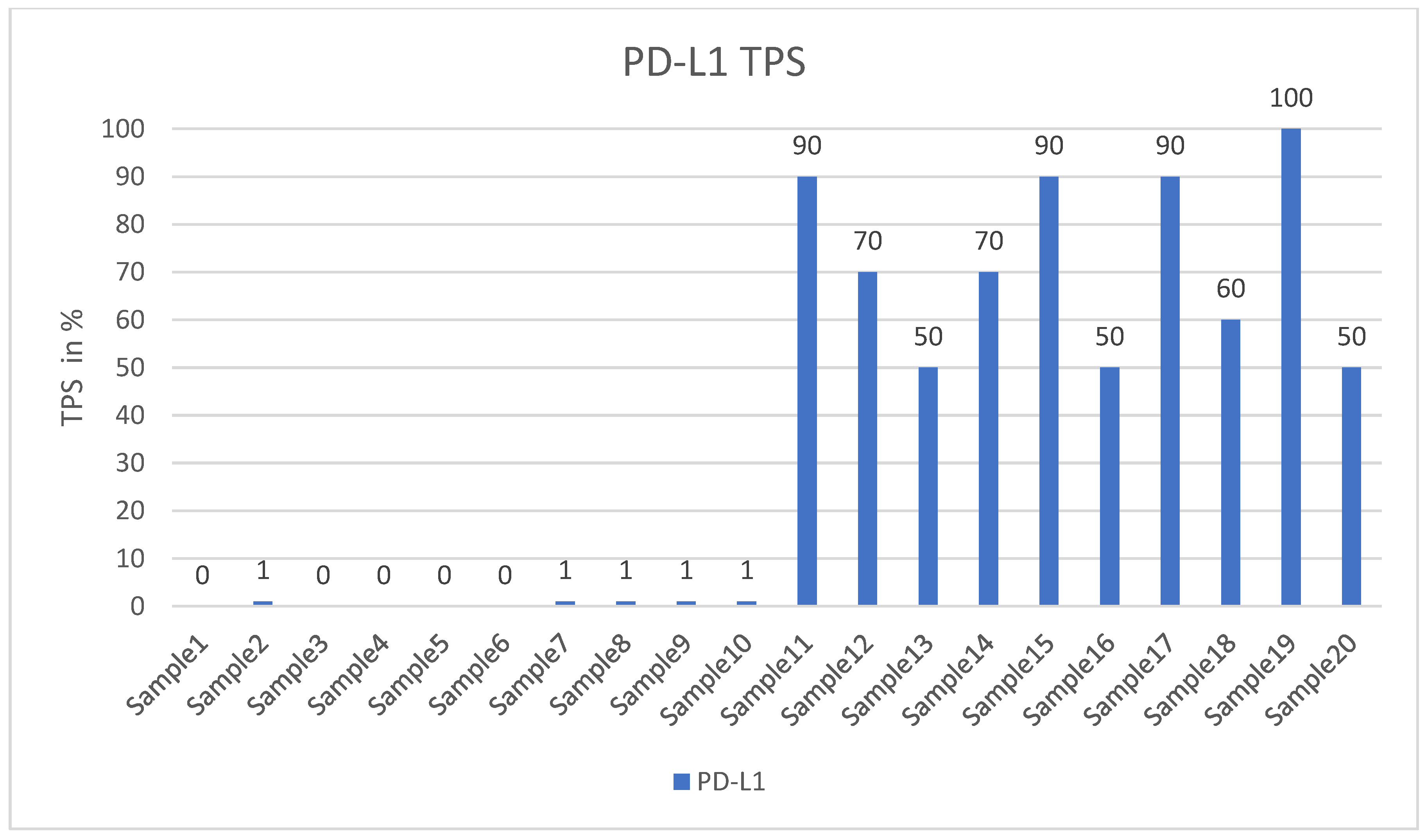
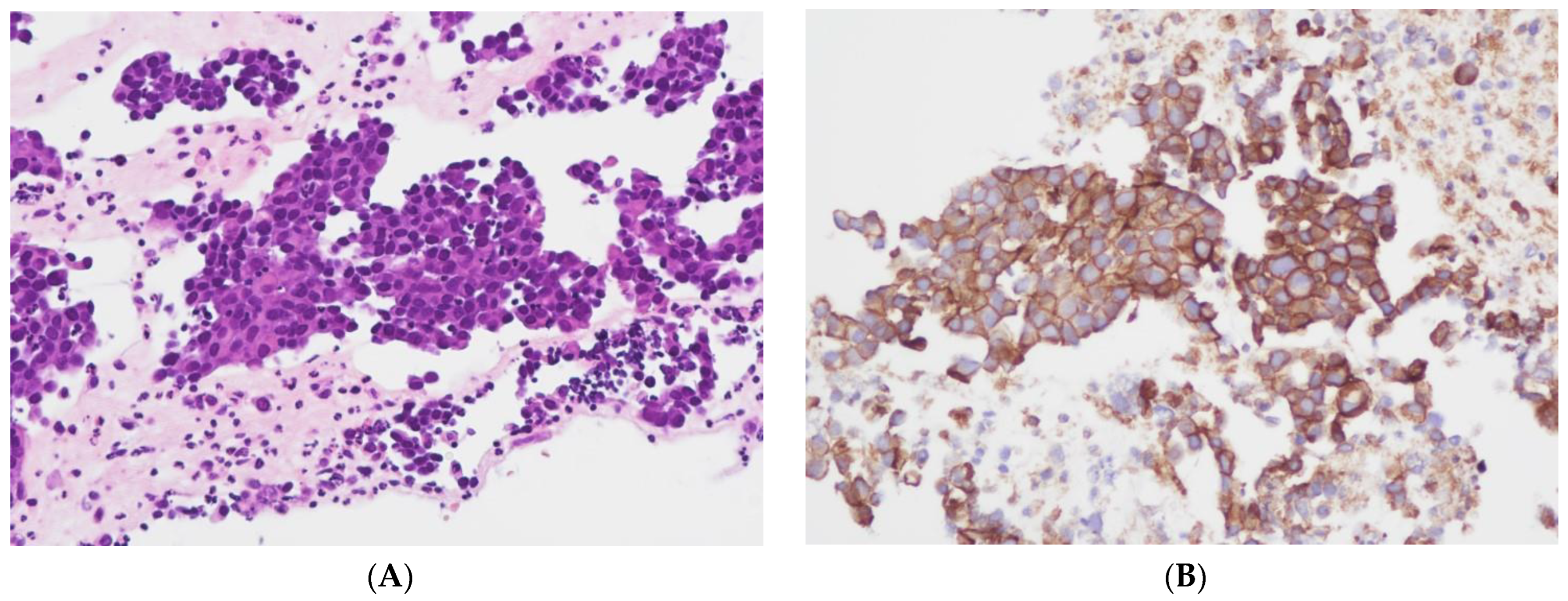
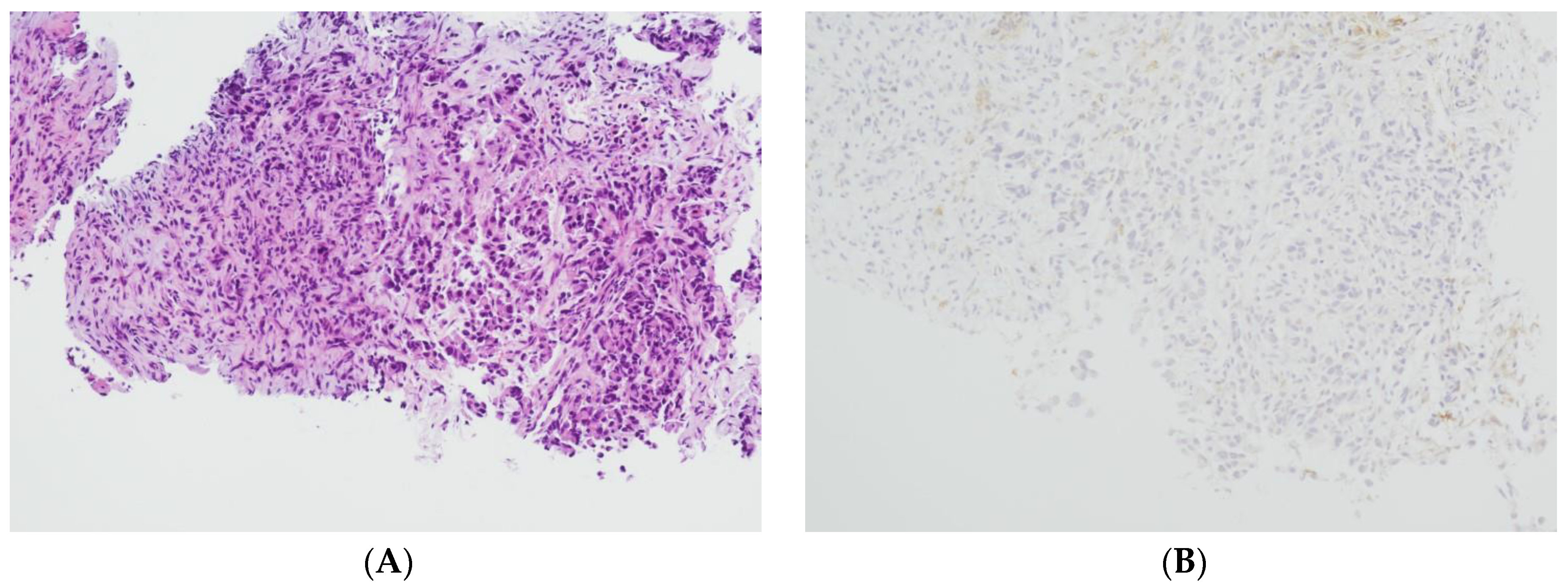
2.1. Tissue Collection and Immunohistochemical Analysis
2.2. Molecular and Genetic Analysis
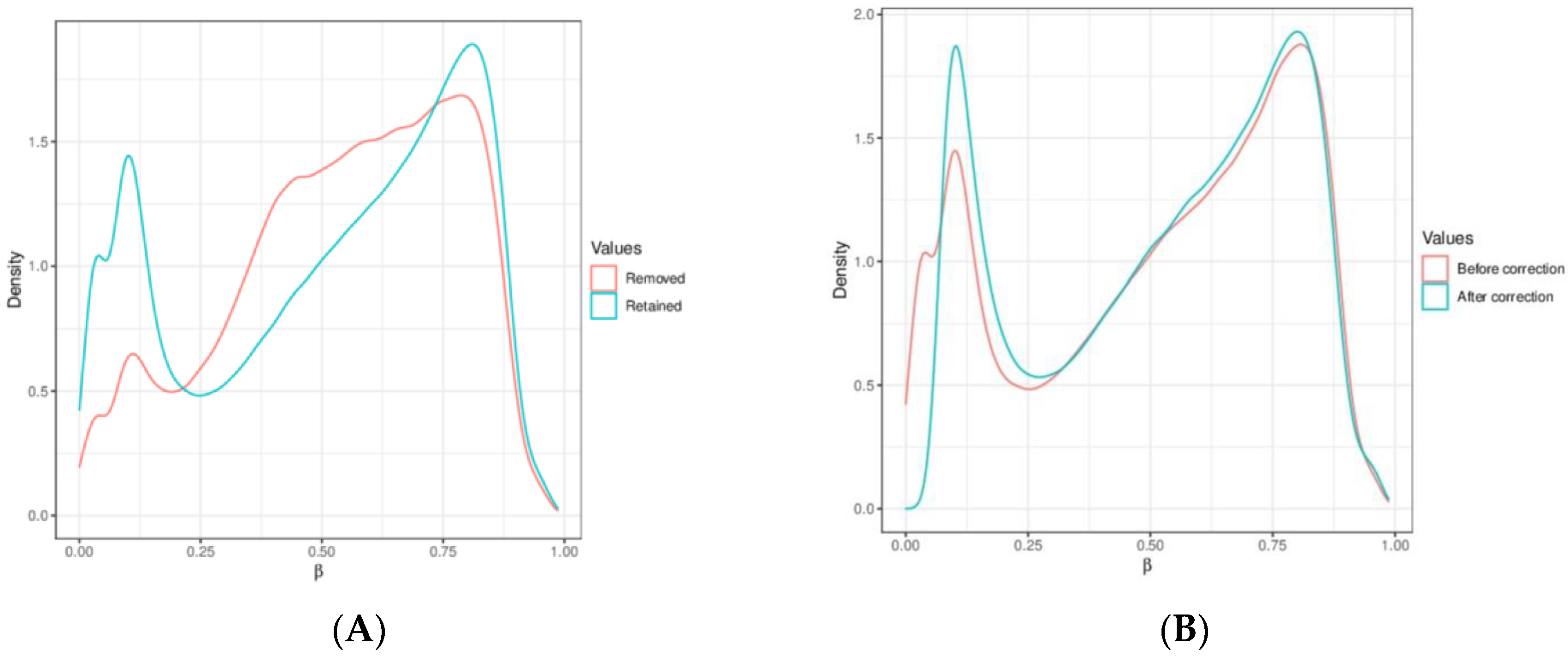
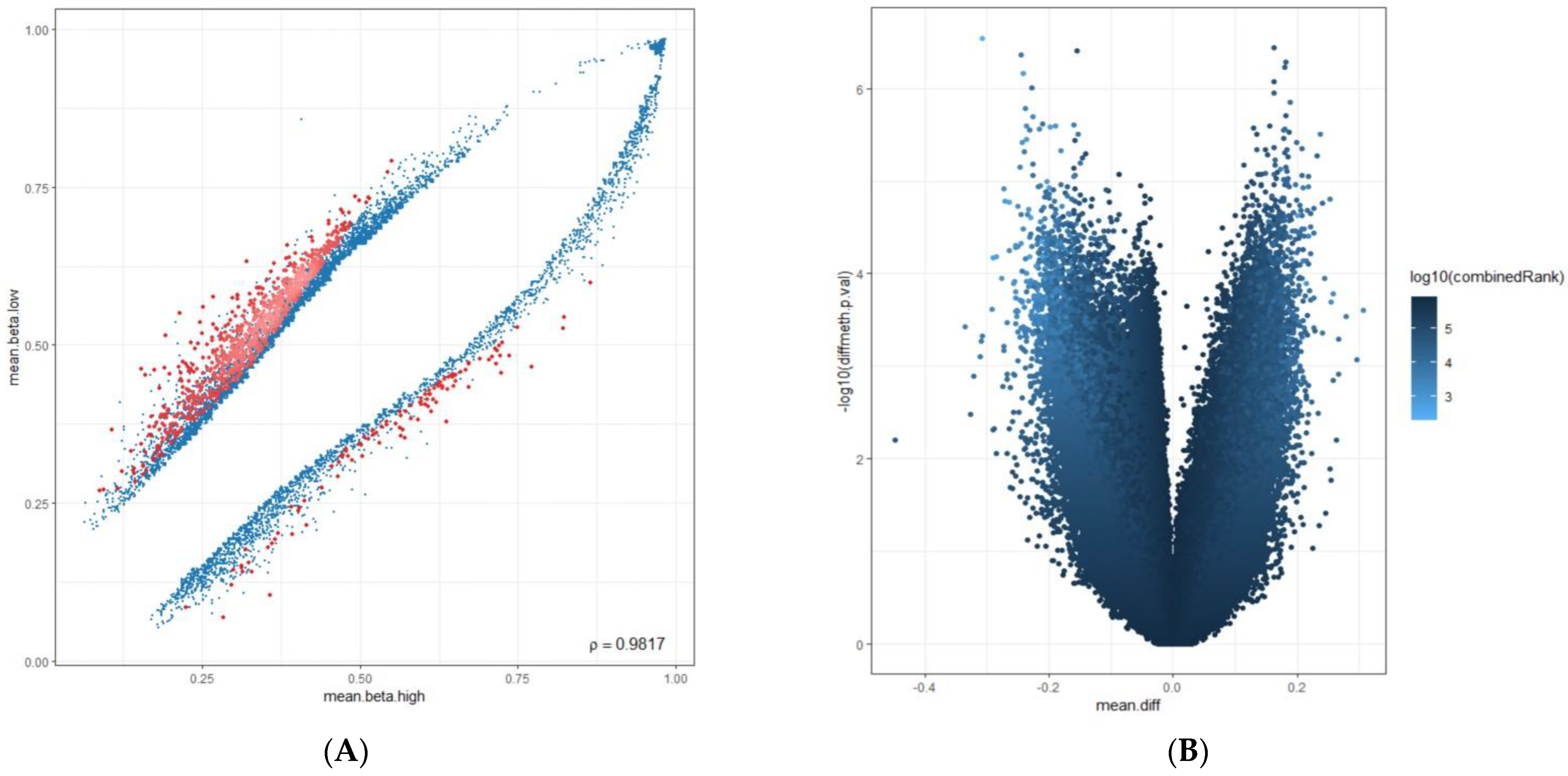
2.3. Computational Data Analysis
3. Results
3.1. Hypermethylated Genes and Promoters in PD-L1 High-Expressing Cases
3.2. Hypomethylated Genes and Promoters in PD-L1 High-Expressing Cases
3.3. Hypermethylated Genes and Promoters in PD-L1 Negative-Expressing Cases
3.4. Hypomethylated Genes and Promoters in PD-L1 Negative-Expressing Cases
4. Discussion
4.1. Pathobiological Mechanisms of Hypermethylated Genes and Promoters in the PD-L1 High-Expressing Tumors
4.2. Pathobiological Mechanisms of Hypomethylated Genes and Promoters in the PD-L1 High-Expression Group
4.3. Pathobiological Mechanisms of Hypermethylated Genes and Promoters in PD-L1 Negative-Expressing Cases
4.4. Pathobiological Mechanisms of Hypomethylated Genes and Promoters in the PD-L1 Negative-Expressing Group
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chi, S.A.; Yu, H.; Choi, Y.L.; Park, S.; Sun, J.M.; Lee, S.H.; Ahn, J.S.; Ahn, M.J.; Choi, D.H.; Kim, K.; et al. Trends in Survival Rates of Non-Small Cell Lung Cancer with Use of Molecular Testing and Targeted Therapy in Korea, 2010–2020. JAMA Netw. Open 2023, 6, e232002. [Google Scholar] [CrossRef] [PubMed]
- Paci, E.; Puliti, D.; Lopes Pegna, A.; Carrozzi, L.; Picozzi, G.; Falaschi, F.; Pistelli, F.; Aquilini, F.; Ocello, C.; Zappa, M.; et al. Mortality, survival and incidence rates in the ITALUNG randomised lung cancer screening trial. Thorax 2017, 72, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Bruno, R.; Fontanini, G. Next Generation Sequencing for Gene Fusion Analysis in Lung Cancer: A Literature Review. Diagnostics 2020, 10, 521. [Google Scholar] [CrossRef]
- Zacharias, M.; Konjic, S.; Kratochwill, N.; Absenger, G.; Terbuch, A.; Jost, P.J.; Wurm, R.; Lindenmann, J.; Kashofer, K.; Gollowitsch, F.; et al. Expanding Broad Molecular Reflex Testing in Non-Small Cell Lung Cancer to Squamous Histology. Cancers 2024, 16, 903. [Google Scholar] [CrossRef] [PubMed]
- Hardtstock, F.; Myers, D.; Li, T.; Cizova, D.; Maywald, U.; Wilke, T.; Griesinger, F. Real-world treatment and survival of patients with advanced non-small cell lung Cancer: A German retrospective data analysis. BMC Cancer 2020, 20, 260. [Google Scholar] [CrossRef]
- Yu, Z.; Xu, F.; Zou, J. Feasibility and safety of EGFR-TKI neoadjuvant therapy for EGFR-mutated NSCLC: A meta-analysis. Eur. J. Clin. Pharmacol. 2024, 80, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Choi, J.H.; Ahn, M.S.; Lee, H.W.; Kang, S.Y.; Choi, Y.W.; Koh, Y.W.; Sheen, S.S. Differential efficacy of tyrosine kinase inhibitors according to the types of EGFR mutations and agents in non-small cell lung cancer: A real-world study. BMC Cancer 2024, 24, 70. [Google Scholar] [CrossRef]
- Paliogiannis, P.; Colombino, M.; Sini, M.C.; Manca, A.; Casula, M.; Palomba, G.; Pisano, M.; Doneddu, V.; Zinellu, A.; Santeufemia, D.; et al. Global prognostic impact of driver genetic alterations in patients with lung adenocarcinoma: A real-life study. BMC Pulm. Med. 2022, 22, 32. [Google Scholar] [CrossRef]
- Godoy, L.A.; Chen, J.; Ma, W.; Lally, J.; Toomey, K.A.; Rajappa, P.; Sheridan, R.; Mahajan, S.; Stollenwerk, N.; Phan, C.T.; et al. Emerging precision neoadjuvant systemic therapy for patients with resectable non-small cell lung cancer: Current status and perspectives. Biomark. Res. 2023, 11, 7. [Google Scholar] [CrossRef]
- Araghi, M.; Mannani, R.; Heidarnejad Maleki, A.; Hamidi, A.; Rostami, S.; Safa, S.H.; Faramarzi, F.; Khorasani, S.; Alimohammadi, M.; Tahmasebi, S.; et al. Recent advances in non-small cell lung cancer targeted therapy; an update review. Cancer Cell Int. 2023, 23, 162. [Google Scholar] [CrossRef]
- Majeed, U.; Manochakian, R.; Zhao, Y.; Lou, Y. Targeted therapy in advanced non-small cell lung cancer: Current advances and future trends. J. Hematol. Oncol. 2021, 14, 108. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Chen, Y.; Li, X.; Long, S.; Shi, Y.; Yu, Y.; Wu, W.; Han, L.; Wang, S. The role of PD-1/PD-L1 and application of immune-checkpoint inhibitors in human cancers. Front. Immunol. 2022, 13, 964442. [Google Scholar] [CrossRef] [PubMed]
- Wojtukiewicz, M.Z.; Rek, M.M.; Karpowicz, K.; Gorska, M.; Politynska, B.; Wojtukiewicz, A.M.; Moniuszko, M.; Radziwon, P.; Tucker, S.C.; Honn, K.V. Inhibitors of immune checkpoints-PD-1, PD-L1, CTLA-4-new opportunities for cancer patients and a new challenge for internists and general practitioners. Cancer Metastasis Rev. 2021, 40, 949–982. [Google Scholar] [CrossRef] [PubMed]
- Turchan, W.T.; Pitroda, S.P.; Weichselbaum, R.R. Radiotherapy and Immunotherapy Combinations in the Treatment of Patients with Metastatic Disease: Current Status and Future Focus. Clin. Cancer Res. 2021, 27, 5188–5194. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Zheng, T.; Zhang, X.; Peng, Y.; Jiang, X.; Huang, Y.; Tan, B.; Yang, Z. First-line treatment options for advanced non-small cell lung cancer patients with PD-L1 ≥ 50%: A systematic review and network meta-analysis. Cancer Immunol. Immunother. 2022, 71, 1345–1355. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Chen, J.; Zhang, L.; Cheng, S.; Yu, J. Network meta-analysis of first-line immune checkpoint inhibitor therapy in advanced non-squamous non-small cell lung cancer patients with PD-L1 expression ≥ 50. BMC Cancer 2023, 23, 791. [Google Scholar] [CrossRef] [PubMed]
- Jabbour, S.K.; Lee, K.H.; Frost, N.; Breder, V.; Kowalski, D.M.; Pollock, T.; Levchenko, E.; Reguart, N.; Martinez-Marti, A.; Houghton, B.; et al. Pembrolizumab Plus Concurrent Chemoradiation Therapy in Patients with Unresectable, Locally Advanced, Stage III Non-Small Cell Lung Cancer: The Phase 2 KEYNOTE-799 Nonrandomized Trial. JAMA Oncol. 2021, 7, 1351–1359. [Google Scholar] [CrossRef]
- Ma, W.; Xue, R.; Zhu, Z.; Farrukh, H.; Song, W.; Li, T.; Zheng, L.; Pan, C.X. Increasing cure rates of solid tumors by immune checkpoint inhibitors. Exp. Hematol. Oncol. 2023, 12, 10. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, J.; Liu, P.; Wang, J.; Zhao, K.; Zhu, Z.; Gu, K.; Zhao, W. Immunotherapy progress and clinical strategy of unresectable locally advanced non-small cell lung cancer. Front. Oncol. 2023, 13, 1022042. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef]
- Czajka-Francuz, P.; Prendes, M.J.; Mankan, A.; Quintana, A.; Pabla, S.; Ramkissoon, S.; Jensen, T.J.; Peiro, S.; Severson, E.A.; Achyut, B.R.; et al. Mechanisms of immune modulation in the tumor microenvironment and implications for targeted therapy. Front. Oncol. 2023, 13, 1200646. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Niu, M.; Xu, L.; Luo, S.; Wu, K. Regulation of PD-L1 expression in the tumor microenvironment. J. Hematol. Oncol. 2021, 14, 10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Huang, Y.; Yang, X. The complex role of PD-L1 in antitumor immunity: A recent update. Cell Mol. Immunol. 2021, 18, 2067–2068. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Sharma, A.; Jasrotia, S.; Kumar, A. Effects of Chemotherapy on the Immune System: Implications for Cancer Treatment and Patient Outcomes. Naunyn Schmiedebergs Arch. Pharmacol. 2024, 397, 2551–2566. [Google Scholar] [CrossRef]
- Bergholz, J.S.; Wang, Q.; Kabraji, S.; Zhao, J.J. Integrating Immunotherapy and Targeted Therapy in Cancer Treatment: Mechanistic Insights and Clinical Implications. Clin. Cancer Res. 2020, 26, 5557–5566. [Google Scholar] [CrossRef]
- Romero-Garcia, S.; Prado-Garcia, H.; Carlos-Reyes, A. Role of DNA Methylation in the Resistance to Therapy in Solid Tumors. Front. Oncol. 2020, 10, 1152. [Google Scholar] [CrossRef] [PubMed]
- Hoang, P.H.; Landi, M.T. DNA Methylation in Lung Cancer: Mechanisms and Associations with Histological Subtypes, Molecular Alterations, and Major Epidemiological Factors. Cancers 2022, 14, 961. [Google Scholar] [CrossRef]
- De Marchi, P.; Leal, L.F.; Duval da Silva, V.; da Silva, E.C.A.; Cordeiro de Lima, V.C.; Reis, R.M. PD-L1 expression by Tumor Proportion Score (TPS) and Combined Positive Score (CPS) are similar in non-small cell lung cancer (NSCLC). J. Clin. Pathol. 2021, 74, 735–740. [Google Scholar] [CrossRef]
- Schildhaus, H.U. [Predictive value of PD-L1 diagnostics]. Pathologe 2018, 39, 498–519. [Google Scholar] [CrossRef]
- Ruschoff, J.; Schildhaus, H.U.; Ruschoff, J.H.; Johrens, K.; Bocker-Edmonston, T.; Dietmaier, W.; Blaker, H.; Baretton, G.; Horst, D.; Dietel, M.; et al. Erratum zu: Testung auf Mismatch-Reparatur-Defizienz und Mikrosatelliteninstabilität. Pathologie 2023, 44, 71–72. [Google Scholar] [CrossRef] [PubMed]
- Twomey, J.D.; Zhang, B. Cancer Immunotherapy Update: FDA-Approved Checkpoint Inhibitors and Companion Diagnostics. AAPS J. 2021, 23, 39. [Google Scholar] [CrossRef] [PubMed]
- Gosney, J.R.; Peake, M.D.; Kerr, K.M. Improving practice in PD-L1 testing of non-small cell lung cancer in the UK: Current problems and potential solutions. J. Clin. Pathol. 2024, 77, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Kaur, D.; Lee, S.M.; Goldberg, D.; Spix, N.J.; Hinoue, T.; Li, H.-T.; Dwaraka, V.B.; Smith, R.; Shen, H.; Liang, G.; et al. Comprehensive evaluation of the Infinium human MethylationEPIC v2 BeadChip. Epigenetics Commun. 2023, 3, 6. [Google Scholar] [CrossRef] [PubMed]
- Bock, C. Analysing and interpreting DNA methylation data. Nat. Rev. Genet. 2012, 13, 705–719. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gorrie-Stone, T.J.; Grant, O.A.; Andrayas, A.D.; Zhai, X.; McDonald-Maier, K.D.; Schalkwyk, L.C. InterpolatedXY: A two-step strategy to normalize DNA methylation microarray data avoiding sex bias. Bioinformatics 2022, 38, 3950–3957. [Google Scholar] [CrossRef] [PubMed]
- Gardiner-Garden, M.; Frommer, M. CpG islands in vertebrate genomes. J. Mol. Biol. 1987, 196, 261–282. [Google Scholar] [CrossRef]
- Smyth, G.K. limma: Linear Models for Microarray Data. In Bioinformatics and Computational Biology Solutions Using R and Bioconductor; Gentleman, R., Carey, V.J., Huber, W., Irizarry, R.A., Dudoit, S., Eds.; Springer: New York, NY, USA, 2005; pp. 397–420. [Google Scholar]
- Flanagan, J.M. Epigenome-wide association studies (EWAS): Past, present, and future. Methods Mol. Biol. 2015, 1238, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Campagna, M.P.; Xavier, A.; Lechner-Scott, J.; Maltby, V.; Scott, R.J.; Butzkueven, H.; Jokubaitis, V.G.; Lea, R.A. Epigenome-wide association studies: Current knowledge, strategies and recommendations. Clin. Epigenetics 2021, 13, 214. [Google Scholar] [CrossRef]
- McPherson, J.W. Gaussian Statistics: An Overview. In Reliability Physics and Engineering: Time-To-Failure Modeling; McPherson, J.W., Ed.; Springer International Publishing: Cham, Switzerland, 2019; pp. 81–91. [Google Scholar]
- Bhootra, S.; Jill, N.; Shanmugam, G.; Rakshit, S.; Sarkar, K. DNA methylation and cancer: Transcriptional regulation, prognostic, and therapeutic perspective. Med. Oncol. 2023, 40, 71. [Google Scholar] [CrossRef]
- Lee, C.J.; Ahn, H.; Jeong, D.; Pak, M.; Moon, J.H.; Kim, S. Impact of mutations in DNA methylation modification genes on genome-wide methylation landscapes and downstream gene activations in pan-cancer. BMC Med. Genom. 2020, 13, 27. [Google Scholar] [CrossRef] [PubMed]
- Frost, H.R. Analyzing cancer gene expression data through the lens of normal tissue-specificity. PLoS Comput. Biol. 2021, 17, e1009085. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Breeze, C.E.; Zhen, S.; Beck, S.; Teschendorff, A.E. Tissue-independent and tissue-specific patterns of DNA methylation alteration in cancer. Epigenetics Chromatin 2016, 9, 10. [Google Scholar] [CrossRef] [PubMed]
- Stahl, M.; Kohrman, N.; Gore, S.D.; Kim, T.K.; Zeidan, A.M.; Prebet, T. Epigenetics in Cancer: A Hematological Perspective. PLoS Genet. 2016, 12, e1006193. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, J.; Deng, X.; Xiong, F.; Zhang, S.; Gong, Z.; Li, X.; Cao, K.; Deng, H.; He, Y.; et al. The role of microenvironment in tumor angiogenesis. J. Exp. Clin. Cancer Res. 2020, 39, 204. [Google Scholar] [CrossRef]
- Surace, A.E.A.; Hedrich, C.M. The Role of Epigenetics in Autoimmune/Inflammatory Disease. Front. Immunol. 2019, 10, 1525. [Google Scholar] [CrossRef]
- de Mendoza, A.; Nguyen, T.V.; Ford, E.; Poppe, D.; Buckberry, S.; Pflueger, J.; Grimmer, M.R.; Stolzenburg, S.; Bogdanovic, O.; Oshlack, A.; et al. Large-scale manipulation of promoter DNA methylation reveals context-specific transcriptional responses and stability. Genome Biol. 2022, 23, 163. [Google Scholar] [CrossRef] [PubMed]
- Larsen, T.V.; Dybdal, N.; Daugaard, T.F.; Lade-Keller, J.; Lin, L.; Sorensen, B.S.; Nielsen, A.L. Examination of the Functional Relationship between PD-L1 DNA Methylation and mRNA Expression in Non-Small-Cell Lung Cancer. Cancers 2023, 15, 1909. [Google Scholar] [CrossRef]
- Huang, C.; Ren, S.; Chen, Y.; Liu, A.; Wu, Q.; Jiang, T.; Lv, P.; Song, D.; Hu, F.; Lan, J.; et al. PD-L1 methylation restricts PD-L1/PD-1 interactions to control cancer immune surveillance. Sci. Adv. 2023, 9, eade4186. [Google Scholar] [CrossRef]
- Mourksi, N.E.; Morin, C.; Fenouil, T.; Diaz, J.J.; Marcel, V. snoRNAs Offer Novel Insight and Promising Perspectives for Lung Cancer Understanding and Management. Cells 2020, 9, 541. [Google Scholar] [CrossRef]
- Han, P.; Liu, Q.; Xiang, J. Monitoring methylation-driven genes as prognostic biomarkers in patients with lung squamous cell cancer. Oncol. Lett. 2020, 19, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Yeung, Y.T.; Fan, S.; Lu, B.; Yin, S.; Yang, S.; Nie, W.; Wang, M.; Zhou, L.; Li, T.; Li, X.; et al. CELF2 suppresses non-small cell lung carcinoma growth by inhibiting the PREX2-PTEN interaction. Carcinogenesis 2020, 41, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Cuttano, R.; Colangelo, T.; Guarize, J.; Dama, E.; Cocomazzi, M.P.; Mazzarelli, F.; Melocchi, V.; Palumbo, O.; Marino, E.; Belloni, E.; et al. miRNome profiling of lung cancer metastases revealed a key role for miRNA-PD-L1 axis in the modulation of chemotherapy response. J. Hematol. Oncol. 2022, 15, 178. [Google Scholar] [CrossRef] [PubMed]
- Guz, M.; Rivero-Muller, A.; Okon, E.; Stenzel-Bembenek, A.; Polberg, K.; Slomka, M.; Stepulak, A. MicroRNAs-role in lung cancer. Dis. Markers 2014, 2014, 218169. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.L.; Zuo, R.; Ou, W.B. The hippo pathway provides novel insights into lung cancer and mesothelioma treatment. J. Cancer. Res. Clin. Oncol. 2018, 144, 2097–2106. [Google Scholar] [CrossRef] [PubMed]
- Padilla, L.; Dakhel, S.; Adan, J.; Masa, M.; Martinez, J.M.; Roque, L.; Coll, T.; Hervas, R.; Calvis, C.; Llinas, L.; et al. S100A7: From mechanism to cancer therapy. Oncogene 2017, 36, 6749–6761. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lu, X.; Zhang, Y.; Zhao, D.; Gong, H.; Du, Y.; Sun, H. The Effect of Extracellular Superoxide Dismutase (SOD3) Gene in Lung Cancer. Front. Oncol. 2022, 12, 722646. [Google Scholar] [CrossRef] [PubMed]
- Xiao-Jie, L.; Ai-Mei, G.; Li-Juan, J.; Jiang, X. Pseudogene in cancer: Real functions and promising signature. J. Med. Genet. 2015, 52, 17–24. [Google Scholar] [CrossRef]
- Dong, P.; Xiong, Y.; Yue, J.; Hanley, S.J.B.; Watari, H. Tumor-Intrinsic PD-L1 Signaling in Cancer Initiation, Development and Treatment: Beyond Immune Evasion. Front. Oncol. 2018, 8, 386. [Google Scholar] [CrossRef]
- Pang, B.; Wu, N.; Guan, R.; Pang, L.; Li, X.; Li, S.; Tang, L.; Guo, Y.; Chen, J.; Sun, D.; et al. Overexpression of RCC2 Enhances Cell Motility and Promotes Tumor Metastasis in Lung Adenocarcinoma by Inducing Epithelial-Mesenchymal Transition. Clin. Cancer Res. 2017, 23, 5598–5610. [Google Scholar] [CrossRef]
- Herath, S.; Sadeghi Rad, H.; Radfar, P.; Ladwa, R.; Warkiani, M.; O’Byrne, K.; Kulasinghe, A. The Role of Circulating Biomarkers in Lung Cancer. Front. Oncol. 2021, 11, 801269. [Google Scholar] [CrossRef] [PubMed]
- Sheng, X.; Bowen, N.; Wang, Z. GLI pathogenesis-related 1 functions as a tumor-suppressor in lung cancer. Mol. Cancer 2016, 15, 25. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wang, H.; He, H.; Niu, H.; Li, Y. Interferon induced transmembrane protein 3 regulates the growth and invasion of human lung adenocarcinoma. Thorac. Cancer 2017, 8, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Lu, Y.; Zhang, F.; Huang, J.; Ren, X.L.; Zhang, R. The Emerging Roles of Long Noncoding RNAs as Hallmarks of Lung Cancer. Front. Oncol. 2021, 11, 761582. [Google Scholar] [CrossRef] [PubMed]
- Gong, S.; Xu, M.; Zhang, Y.; Shan, Y.; Zhang, H. The Prognostic Signature and Potential Target Genes of Six Long Non-coding RNA in Laryngeal Squamous Cell Carcinoma. Front. Genet. 2020, 11, 413. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.L.; Tsai, Y.M.; Lien, C.T.; Kuo, P.L.; Hung, A.J. The Roles of MicroRNA in Lung Cancer. Int. J. Mol. Sci. 2019, 20, 1611. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Zhang, Y.; Li, M.; Zhang, Y.; Zhang, H.; Chen, J.; Liu, Z.; Yuan, P.; Yang, Z.; Wang, X. MiR-124-3p impedes the metastasis of non-small cell lung cancer via extracellular exosome transport and intracellular PI3K/AKT signaling. Biomark. Res. 2023, 11, 1. [Google Scholar] [CrossRef] [PubMed]
- Loedige, I.; Gaidatzis, D.; Sack, R.; Meister, G.; Filipowicz, W. The mammalian TRIM-NHL protein TRIM71/LIN-41 is a repressor of mRNA function. Nucleic Acids Res. 2013, 41, 518–532. [Google Scholar] [CrossRef]
- Treiber, T.; Treiber, N.; Plessmann, U.; Harlander, S.; Daiss, J.L.; Eichner, N.; Lehmann, G.; Schall, K.; Urlaub, H.; Meister, G. A Compendium of RNA-Binding Proteins that Regulate MicroRNA Biogenesis. Mol. Cell 2017, 66, 270–284.e213. [Google Scholar] [CrossRef]
- Jin, J.; Lu, Z.; Wang, X.; Liu, Y.; Han, T.; Wang, Y.; Wang, T.; Gan, M.; Xie, C.; Wang, J.; et al. E3 ubiquitin ligase TRIM7 negatively regulates NF-kappa B signaling pathway by degrading p65 in lung cancer. Cell Signal. 2020, 69, 109543. [Google Scholar] [CrossRef]
- Schuck, S. Microautophagy-distinct molecular mechanisms handle cargoes of many sizes. J. Cell Sci. 2020, 133, jcs246322. [Google Scholar] [CrossRef] [PubMed]
- Kravic, B.; Behrends, C.; Meyer, H. Regulation of lysosome integrity and lysophagy by the ubiquitin-conjugating enzyme UBE2QL1. Autophagy 2020, 16, 179–180. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Heredia, J.M.; Verdugo Sivianes, E.M.; Lucena-Cacace, A.; Molina-Pinelo, S.; Carnero, A. Numb-like (NumbL) downregulation increases tumorigenicity, cancer stem cell-like properties and resistance to chemotherapy. Oncotarget 2016, 7, 63611–63628. [Google Scholar] [CrossRef] [PubMed]
- Haider, T.; Pandey, V.; Banjare, N.; Gupta, P.N.; Soni, V. Drug resistance in cancer: Mechanisms and tackling strategies. Pharmacol. Rep. 2020, 72, 1125–1151. [Google Scholar] [CrossRef]
- Shen, X.; He, Z.; Li, H.; Yao, C.; Zhang, Y.; He, L.; Li, S.; Huang, J.; Guo, Z. Distinct functional patterns of gene promoter hypomethylation and hypermethylation in cancer genomes. PLoS ONE 2012, 7, e44822. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PD-L1 Expression | Methylation Status | Gene | Methylation | p Value | Chr |
|---|---|---|---|---|---|
| PD-L1 high | Highest methylation in gene | SNORD114-14 (C/D Box 114-14). Small Nucleolar RNAs (SnoRNAs) | 0.9 | 0.04 | 14 |
| Highest methylation in promoter | DCAF4L2 (DDB1 Associated Factor 4 Like 2) | 0.82 | 0.02 | 8 | |
| CELF2-AS1 (CELF2 Antisense RNA 1) | 0.8 | 0.02 | 10 | ||
| LINCMD1 (Long Intergenic Non-Protein Coding RNA, Muscle Differentiation1, MIR133BHG) | 0.8 | 0.01 | 6 | ||
| Lowest methylation in gene | GLIPR1L2 (GLIPR1 Like 2) | 0.21 | 0.007 | 12 | |
| Lowest methylation in promoter | CAPS2 (Calcyphosine 2). | 0.14 | 0.005 | 12 | |
| GLIPR1L2 (GLI Pathogenesis Related 1 Like 2) | 0.19 | 0.008 | 12 | ||
| IFITM3 (Interferon Induced Transmembrane Protein 3) | 0.21 | 0.03 | 11 | ||
| PD-L1 negative | Highest methylation in gene | SNORD114-14, (C/D Box 114-14) Small Nucleolar RNAs (SnoRNAs) | 0.75 | 0.04 | 14 |
| LINC00528 (Long Intergenic Non-Protein Coding RNA 528) | 0.73 | 0.001 | 224 | ||
| Lowest methylation in gene | MIR124-3 (MicroRNA124-3) | 0.21 | 0.01 | 20 | |
| Lowest methylation in promoter | TRIM71 (Tripartite Motif Containing 71, LIN41) | 0.15 | 0.02 | 3 | |
| CAPS2 (Calcyphosine 2). | 0.26 | 0.005 | 12 | ||
| UBE2QL1 (Ubiquitin Conjugating Enzyme E2 Q Family Like 1) | 0.3 | 0.007 | 5 | ||
| GLIPR1L2 (GLIPR1 Like 2) | 0.3 | 0.008 | 12 | ||
| PDL1 high/negative | Difference * in gene methylation | S100A7L2 gene, also known as S100 Calcium Binding Protein A7 Like 2 | 0.74/0.57 Delta 17% | 0.002 | 1 |
| Difference * in promoter methylation | SOD1P3 (Superoxide Dismutase 1 Pseudogene 3), | 0.68/0.54 Delta 14% | 0.02 | 3 | |
| PDL1 negative/high | Difference * in gene methylation | LINC00528 (Long Intergenic Non-Protein Coding RNA 528) | 0.73/0.54 Delta 19% | 0.001 | 22 |
| Difference * in promoter methylation | NUMB gene, (NUMB Endocytic Adaptor Protein) | 0.51/0.31 Delta 20% | 0.0005 | 14 | |
| PD-L1 Expression | Methylation status | Gene | Methylation | p value | Chr |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hutarew, G.; Alinger-Scharinger, B.; Sotlar, K.; Kraus, T.F.J. Genome-Wide Methylation Analysis in Two Wild-Type Non-Small Cell Lung Cancer Subgroups with Negative and High PD-L1 Expression. Cancers 2024, 16, 1841. https://doi.org/10.3390/cancers16101841
Hutarew G, Alinger-Scharinger B, Sotlar K, Kraus TFJ. Genome-Wide Methylation Analysis in Two Wild-Type Non-Small Cell Lung Cancer Subgroups with Negative and High PD-L1 Expression. Cancers. 2024; 16(10):1841. https://doi.org/10.3390/cancers16101841
Chicago/Turabian StyleHutarew, Georg, Beate Alinger-Scharinger, Karl Sotlar, and Theo F. J. Kraus. 2024. "Genome-Wide Methylation Analysis in Two Wild-Type Non-Small Cell Lung Cancer Subgroups with Negative and High PD-L1 Expression" Cancers 16, no. 10: 1841. https://doi.org/10.3390/cancers16101841
APA StyleHutarew, G., Alinger-Scharinger, B., Sotlar, K., & Kraus, T. F. J. (2024). Genome-Wide Methylation Analysis in Two Wild-Type Non-Small Cell Lung Cancer Subgroups with Negative and High PD-L1 Expression. Cancers, 16(10), 1841. https://doi.org/10.3390/cancers16101841