
Pediatric Diffuse Midline Glioma H3K27-Altered: From Developmental Origins to Therapeutic Challenges

 ,
,  ,
,  and
and {kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
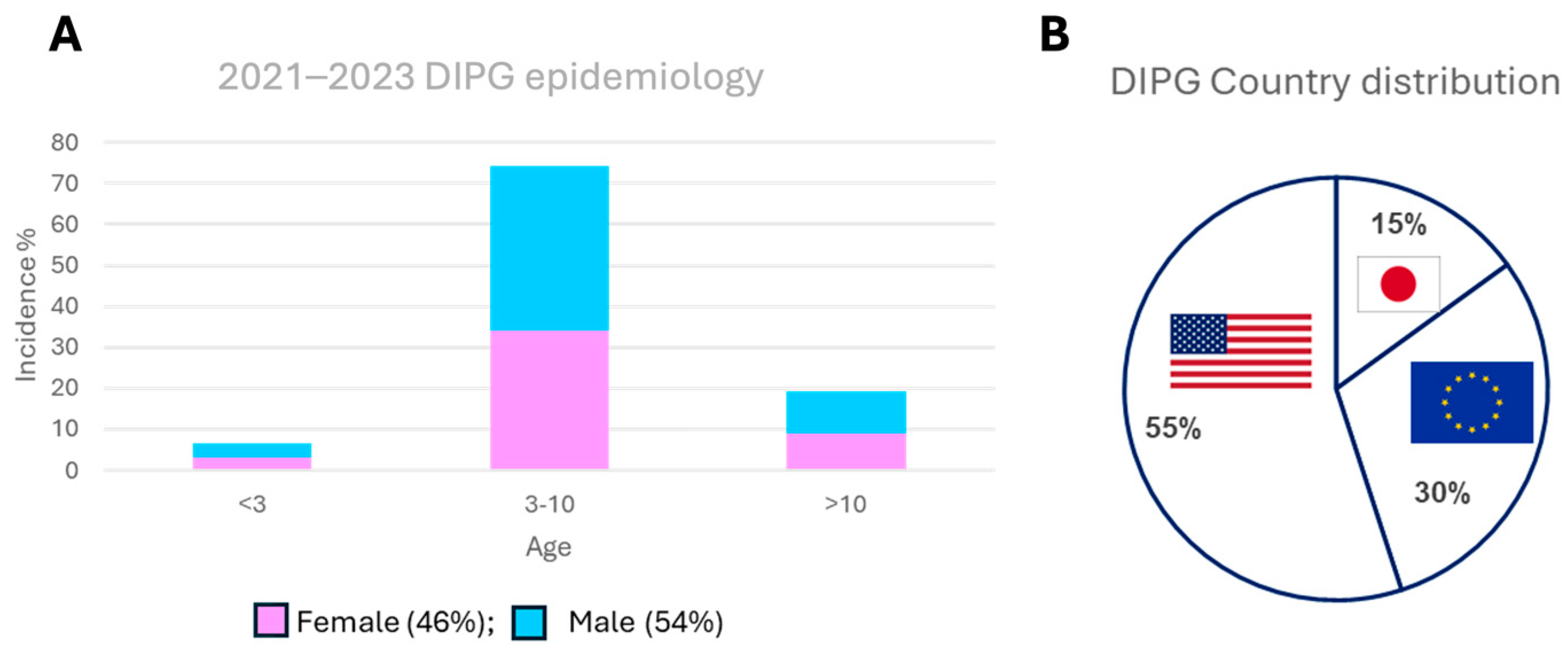
1. Introduction
2. Material and Methods
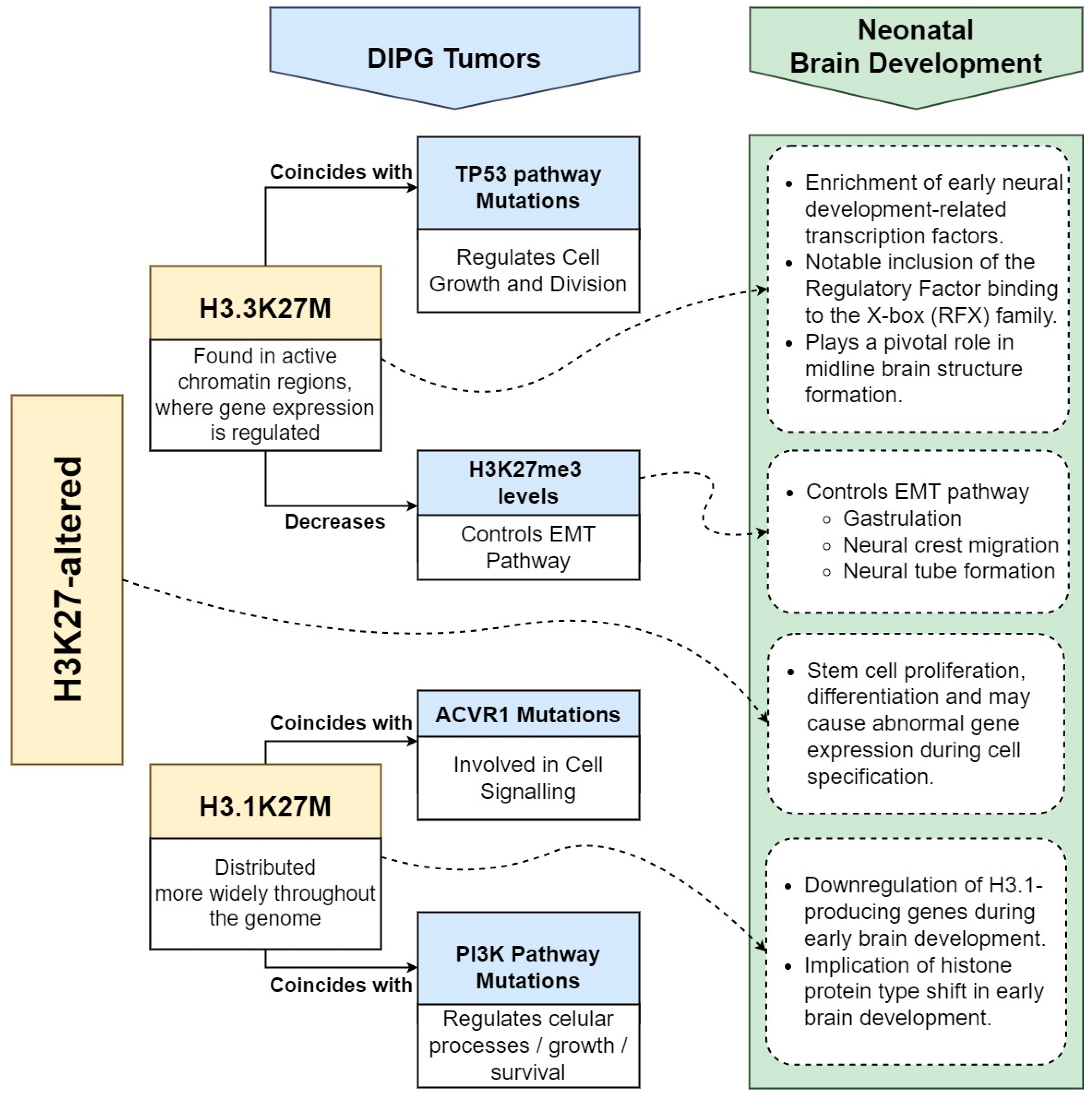
3. H3K27-Altered DIPG and Brain Development
3.1. Histone H3 Dynamics
3.2. Impacts of H3.3K27M and H3.1K27M Oncohistones in DIPG Tumorigenesis
3.3. EMT and H3K27me3 Dysregulation in Gliomagenesis
3.4. H3K27M and Oligodendroglial Precursor Cells (OPCs)
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Perrone, M.G.; Ruggiero, A.; Centonze, A.; Carrieri, A.; Ferorelli, S.; Scilimati, A. Diffuse Intrinsic Pontine Glioma (DIPG): Breakthrough and Clinical Perspective. Curr. Med. Chem. 2021, 28, 3287–3317. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A Summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed]
- Hoogendijk, R.; Van der Lugt, J.; Kranendonk, M.E.G.; Gatta, G.; Capocaccia, R.; Hoving, E.W.; Wesseling, P.; Visser, O.; Van Vuurden, D.G.; Karim-Kos, H. Protocol for investigating data quality and reporting outcomes of pediatric gliomas in population-based cancer registry research. STAR Protoc. 2024, 5, 102905. [Google Scholar] [CrossRef]
- Damodharan, S.; Lara-Velazquez, M.; Williamsen, B.C.; Helgager, J.; Dey, M. Diffuse Intrinsic Pontine Glioma: Molecular Landscape, Evolving Treatment Strategies and Emerging Clinical Trials. J. Pers. Med. 2022, 12, 840. [Google Scholar] [CrossRef]
- Vitanza, N.A.; Monje, M. Diffuse Intrinsic Pontine Glioma: From Diagnosis to Next-Generation Clinical Trials. Curr. Treat. Options Neurol. 2019, 21, 37. [Google Scholar] [CrossRef]
- Freeman, C.R.; Krischer, J.P.; Sanford, R.A.; Cohen, M.E.; Burger, P.C.; del Carpio, R.; Halperin, E.C.; Munoz, L.; Friedman, H.S.; Kun, L.E. Final results of a study of escalating doses of hyperfractionated radiotherapy in brain stem tumors in children: A Pediatric Oncology Group study. Int. J. Radiat. Oncol. Biol. Phys. 1993, 27, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Haas-Kogan, D.A.; Banerjee, A.; Poussaint, T.Y.; Kocak, M.; Prados, M.D.; Geyer, J.R.; Fouladi, M.; Broniscer, A.; Minturn, J.E.; Pollack, I.F.; et al. Phase II trial of tipifarnib and radiation in children with newly diagnosed diffuse intrinsic pontine gliomas. Neuro-oncology 2011, 13, 298–306. [Google Scholar] [CrossRef]
- Mandell, L.R.; Kadota, R.; Freeman, C.; Douglass, E.C.; Fontanesi, J.; Cohen, M.E.; Kovnar, E.; Burger, P.; Sanford, R.A.; Kepner, J.; et al. There is no role for hyperfractionated radiotherapy in the management of children with newly diagnosed diffuse intrinsic brainstem tumors: Results of a Pediatric Oncology Group phase III trial comparing conventional vs. hyperfractionated radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 1999, 43, 959–964. [Google Scholar] [CrossRef] [PubMed]
- Maitra, A.; Mandorino, M.; Armenise, D.; Baldelli, O.M.; Miciaccia, M.; Ferorelli, S.; Papusha, L.; Druy, A.; Perrone, M.G. Decoding Gene Expression Changes in Pediatric Cerebral Tumors: Before and After Radiotherapy. Cancers, 2024; submitted for publication. [Google Scholar]
- Frazier, J.L.; Lee, J.; Thomale, U.W.; Noggle, J.C.; Cohen, K.J.; Jallo, G.I. Treatment of diffuse intrinsic brainstem gliomas: Failed approaches and future strategies. J. Neurosurg. Pediatr. 2009, 3, 259–269. [Google Scholar] [CrossRef]
- Bender, S.; Tang, Y.; Lindroth, A.M.; Hovestadt, V.; Jones, D.T.; Kool, M.; Zapatka, M.; Northcott, P.A.; Sturm, D.; Wang, W.; et al. Reduced H3K27me3 and DNA hypomethylation are major drivers of gene expression in K27M mutant pediatric high-grade gliomas. Cancer Cell 2013, 24, 660–672. [Google Scholar] [CrossRef]
- Chan, K.M.; Fang, D.; Gan, H.; Hashizume, R.; Yu, C.; Schroeder, M.; Gupta, N.; Mueller, S.; James, C.D.; Jenkins, R.; et al. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev. 2013, 27, 985–990. [Google Scholar] [CrossRef]
- Khuong-Quang, D.A.; Buczkowicz, P.; Rakopoulos, P.; Liu, X.Y.; Fontebasso, A.M.; Bouffet, E.; Bartels, U.; Albrecht, S.; Schwartzentruber, J.; Letourneau, L.; et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 2012, 124, 439–447. [Google Scholar] [CrossRef]
- Lewis, P.W.; Müller, M.M.; Koletsky, M.S.; Cordero, F.; Lin, S.; Banaszynski, L.A.; Garcia, B.A.; Muir, T.W.; Becher, O.J.; Allis, C.D. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science 2013, 340, 857–861. [Google Scholar] [CrossRef] [PubMed]
- Schwartzentruber, J.; Korshunov, A.; Liu, X.Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.A.; Tönjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231. [Google Scholar] [CrossRef]
- Taylor, K.R.; Mackay, A.; Truffaux, N.; Butterfield, Y.; Morozova, O.; Philippe, C.; Castel, D.; Grasso, C.S.; Vinci, M.; Carvalho, D.; et al. Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat. Genet. 2014, 46, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Broniscer, A.; McEachron, T.A.; Lu, C.; Paugh, B.S.; Becksfort, J.; Qu, C.; Ding, L.; Huether, R.; Parker, M.; et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 2012, 44, 251–253. [Google Scholar] [CrossRef] [PubMed]
- Audia, J.E.; Campbell, R.M. Histone modifications and cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019521. [Google Scholar] [CrossRef] [PubMed]
- Sievers, P.; Sill, M.; Schrimpf, D.; Stichel, D.; Reuss, D.E.; Sturm, D.; Hench, J.; Frank, S.; Krskova, L.; Vicha, A.; et al. A subset of pediatric-type thalamic gliomas share a distinct DNA methylation profile, H3K27me3 loss and frequent alteration of EGFR. Neuro-Oncology 2021, 23, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Packer, R.J. The 2021 WHO Classification of Tumors of the Central Nervous System: Clinical implications. Neuro-Oncology 2021, 23, 1215–1217. [Google Scholar] [CrossRef] [PubMed]
- Castel, D.; Philippe, C.; Calmon, R.; Le Dret, L.; Truffaux, N.; Boddaert, N.; Pagès, M.; Taylor, K.R.; Saulnier, P.; Lacroix, L.; et al. Histone H3F3A and HIST1H3B K27M mutations define two subgroups of diffuse intrinsic pontine gliomas with different prognosis and phenotypes. Acta Neuropathol. 2015, 130, 815–827. [Google Scholar] [CrossRef]
- Buczkowicz, P.; Hoeman, C.; Rakopoulos, P.; Pajovic, S.; Letourneau, L.; Dzamba, M.; Morrison, A.; Lewis, P.; Bouffet, E.; Bartels, U.; et al. Genomic analysis of diffuse intrinsic pontine gliomas identifies three molecular subgroups and recurrent activating ACVR1 mutations. Nat. Genet. 2014, 46, 451–456. [Google Scholar] [CrossRef]
- Fontebasso, A.M.; Papillon-Cavanagh, S.; Schwartzentruber, J.; Nikbakht, H.; Gerges, N.; Fiset, P.O.; Bechet, D.; Faury, D.; De Jay, N.; Ramkissoon, L.A.; et al. Recurrent somatic mutations in ACVR1 in pediatric midline high-grade astrocytoma. Nat. Genet. 2014, 46, 462–466. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Diaz, A.K.; Paugh, B.S.; Rankin, S.L.; Ju, B.; Li, Y.; Zhu, X.; Qu, C.; Chen, X.; Zhang, J.; et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat. Genet. 2014, 46, 444–450. [Google Scholar] [CrossRef]
- Mackay, A.; Burford, A.; Carvalho, D.; Izquierdo, E.; Fazal-Salom, J.; Taylor, K.R.; Bjerke, L.; Clarke, M.; Vinci, M.; Nandhabalan, M.; et al. Integrated molecular meta-analysis of 1,000 pediatric high-grade and diffuse intrinsic pontine glioma. Cancer Cell 2017, 32, 520–537.e5. [Google Scholar] [CrossRef]
- Insel, T.R. Brain somatic mutations: The dark matter of psychiatric genetics? Mol. Psychiatry 2014, 19, 156–158. [Google Scholar] [CrossRef]
- Sanai, N.; Nguyen, T.; Ihrie, R.A.; Mirzadeh, Z.; Tsai, H.H.; Wong, M.; Gupta, N.; Berger, M.S.; Huang, E.; Garcia-Verdugo, J.M.; et al. Corridors of migrating neurons in the human brain and their decline during infancy. Nature 2011, 478, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Evrony, G.D.; Lehmann, H.S.; Elhosary, P.C.; Mehta, B.K.; Poduri, A.; Walsh, C.A. Single-cell, genome-wide sequencing identifies clonal somatic copy-number variation in the human brain. Cell Rep. 2014, 8, 1280–1289. [Google Scholar] [CrossRef]
- McConnell, M.J.; Lindberg, M.R.; Brennand, K.J.; Piper, J.C.; Voet, T.; Cowing-Zitron, C.; Shumilina, S.; Lasken, R.S.; Vermeesch, J.R.; Hall, I.M.; et al. Mosaic copy number variation in human neurons. Science 2013, 342, 632–637. [Google Scholar] [CrossRef] [PubMed]
- Ren, M.; van Nocker, S. In silico analysis of histone H3 gene expression during human brain development. Int. J. Dev. Biol. 2016, 60, 167–173. [Google Scholar] [CrossRef]
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.A.; Jones, D.T.; Konermann, C.; Pfaff, E.; Tönjes, M.; Sill, M.; Bender, S.; et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 2012, 22, 425–437. [Google Scholar] [CrossRef]
- Castel, D.; Philippe, C.; Kergrohen, T.; Sill, M.; Merlevede, J.; Barret, E.; Puget, S.; Sainte-Rose, C.; Kramm, C.M.; Jones, C.; et al. Transcriptomic and epigenetic profiling of ‘diffuse midline gliomas, H3 K27M-mutant’ discriminate two subgroups based on the type of histone H3 mutated and not supratentorial or infratentorial location. Acta Neuropathol. Commun. 2018, 6, 117. [Google Scholar] [CrossRef] [PubMed]
- Nagaraja, S.; Quezada, M.A.; Gillespie, S.M.; Arzt, M.; Lennon, J.J.; Woo, P.J.; Hovestadt, V.; Kambhampati, M.; Filbin, M.G.; Suva, M.L.; et al. Histone variant and cell context determine H3K27M reprogramming of the enhancer landscape and oncogenic state. Mol. Cell 2019, 76, 965–980.e12. [Google Scholar] [CrossRef]
- Sedykh, I.; Keller, A.N.; Yoon, B.; Roberson, L.; Moskvin, O.V.; Grinblat, Y. Zebrafish Rfx4 controls dorsal and ventral midline formation in the neural tube. Dev. Dyn. 2018, 247, 650–659. [Google Scholar] [CrossRef]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the hallmarks of cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef] [PubMed]
- Piunti, A.; Hashizume, R.; Morgan, M.A.; Bartom, E.T.; Horbinski, C.M.; Marshall, S.A.; Rendleman, E.J.; Ma, Q.; Takahashi, Y.H.; Woodfin, A.R.; et al. Therapeutic targeting of polycomb and BET bromodomain proteins in diffuse intrinsic pontine gliomas. Nat. Med. 2017, 23, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Justin, N.; Zhang, Y.; Tarricone, C.; Martin, S.R.; Chen, S.; Underwood, E.; De Marco, V.; Haire, L.F.; Walker, P.A.; Reinberg, D.; et al. Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nat. Commun. 2016, 7, 11316. [Google Scholar] [CrossRef] [PubMed]
- Diehl, K.L.; Ge, E.J.; Weinberg, D.N.; Jani, K.S.; Allis, C.D.; Muir, T.W. PRC2 engages a bivalent H3K27M-H3K27me3 dinucleosome inhibitor. Proc. Natl. Acad. Sci. USA 2019, 116, 22152–22157. [Google Scholar] [CrossRef] [PubMed]
- Stafford, J.M.; Lee, C.H.; Voigt, P.; Descostes, N.; Saldaña-Meyer, R.; Yu, J.R.; Leroy, G.; Oksuz, O.; Chapman, J.R.; Suarez, F.; et al. Multiple modes of PRC2 inhibition elicit global chromatin alterations in H3K27M pediatric glioma. Sci. Adv. 2018, 4, eaau5935. [Google Scholar] [CrossRef] [PubMed]
- Harutyunyan, A.S.; Krug, B.; Chen, H.; Papillon-Cavanagh, S.; Zeinieh, M.; De Jay, N.; Deshmukh, S.; Chen CC, L.; Belle, J.; Mikael, L.G.; et al. H3K27M induces defective chromatin spread of PRC2-mediated repressive H3K27me2/me3 and is essential for glioma tumorigenesis. Nat. Commun. 2019, 10, 1262. [Google Scholar] [CrossRef]
- Venneti, S.; Garimella, M.T.; Sullivan, L.M.; Martinez, D.; Huse, J.T.; Heguy, A.; Santi, M.; Thompson, C.B.; Judkins, A.R. Evaluation of histone 3 lysine 27 trimethylation (H3K27me3) and enhancer of Zest 2 (EZH2) in pediatric glial and glioneuronal tumors shows decreased H3K27me3 in H3F3A K27M mutant glioblastomas. Brain Pathol. 2013, 23, 558–564. [Google Scholar] [CrossRef]
- Funato, K.; Major, T.; Lewis, P.W.; Allis, C.D.; Tabar, V. Use of human embryonic stem cells to model pediatric gliomas with H3.3K27M histone mutation. Science 2014, 346, 1529–1533. [Google Scholar] [CrossRef] [PubMed]
- Viebahn, C. Epithelio-mesenchymal transformation during formation of the mesoderm in the mammalian embryo. Acta Anat. 1995, 154, 79–97. [Google Scholar] [CrossRef] [PubMed]
- Duband, J.L. Diversity in the molecular and cellular strategies of epithelium-to-mesenchyme transitions: Insights from the neural crest. Cell Adhes. Migr. 2010, 4, 458–482. [Google Scholar] [CrossRef]
- Zou, S.; Zhang, D.; Xu, Z.; Wen, X.; Zhang, Y. JMJD3 promotes the epithelial-mesenchymal transition and migration of glioma cells via the CXCL12/CXCR4 axis. Oncol. Lett. 2019, 18, 5930–5940. [Google Scholar] [CrossRef]
- Sanders, L.M.; Cheney, A.; Seninge, L.; van den Bout, A.; Chen, M.; Beale, H.C.; Kephart, E.T.; Pfeil, J.; Learned, K.; Lyle, A.G.; et al. Identification of a differentiation stall in epithelial mesenchymal transition in histone H3-mutant diffuse midline glioma. GigaScience 2020, 9, giaa136. [Google Scholar] [CrossRef] [PubMed]
- Pun, M.; Pratt, D.; Nano, P.R.; Joshi, P.K.; Jiang, L.; Englinger, B.; Rao, A.; Cieslik, M.; Chinnaiyan, A.M.; Aldape, K.; et al. Common molecular features of H3K27M DMGs and PFA ependymomas map to hindbrain developmental pathways. Acta Neuropathol. Commun. 2023, 11, 25. [Google Scholar] [CrossRef]
- Calver, A.R.; Hall, A.C.; Yu, W.P.; Walsh, F.S.; Heath, J.K.; Betsholtz, C.; Richardson, W.D. Oligodendrocyte population dynamics and the role of PDGF in vivo. Neuron 1998, 20, 869–882. [Google Scholar] [CrossRef]
- Cardona, H.J.; Somasundaram, A.; Crabtree, D.M.; Gadd, S.L.; Becher, O.J. Prenatal overexpression of platelet-derived growth factor receptor A results in central nervous system hypomyelination. Brain Behav. 2021, 11, e2332. [Google Scholar] [CrossRef]
- Pathania, M.; De Jay, N.; Maestro, N.; Harutyunyan, A.S.; Nitarska, J.; Pahlavan, P.; Henderson, S.; Mikael, L.G.; Richard-Londt, A.; Zhang, Y.; et al. H3.3K27M Cooperates with Trp53 Loss and PDGFRA Gain in Mouse Embryonic Neural Progenitor Cells to Induce Invasive High-Grade Gliomas. Cancer Cell 2017, 32, 684–700.e9. [Google Scholar] [CrossRef]
- Filbin, M.G.; Tirosh, I.; Hovestadt, V.; Shaw, M.L.; Escalante, L.E.; Mathewson, N.D.; Neftel, C.; Frank, N.; Pelton, K.; Hebert, C.M.; et al. Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science 2018, 360, 331–335. [Google Scholar] [CrossRef]
- Tomita, Y.; Shimazu, Y.; Somasundaram, A.; Tanaka, Y.; Takata, N.; Ishi, Y.; Gadd, S.; Hashizume, R.; Angione, A.; Pinero, G. A novel mouse model of diffuse midline glioma initiated in neonatal oligodendrocyte progenitor cells highlights cell-of-origin dependent effects of H3K27M. Glia 2022, 70, 1681–1698. [Google Scholar] [CrossRef] [PubMed]
- Monje, M.; Mitra, S.S.; Freret, M.E.; Raveh, T.B.; Kim, J.; Masek, M.; Attema, J.L.; Li, G.; Haddix, T.; Edwards, M.S.; et al. Hedgehog-responsive candidate cell of origin for diffuse intrinsic pontine glioma. Proc. Natl. Acad. Sci. USA 2011, 108, 4453–4458. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Esfahani, D.R.; Huang, T.; Ozark, P.; Bartom, E.; Hashizume, R.; Bonner, E.R.; An, S.; Horbinski, C.M.; James, C.D.; et al. Tenascin-C expression contributes to pediatric brainstem glioma tumor phenotype and represents a novel biomarker of disease. Acta Neuropathol. Commun. 2019, 7, 75. [Google Scholar] [CrossRef]
- Larson, J.D.; Kasper, L.H.; Paugh, B.S.; Jin, H.; Wu, G.; Kwon, C.H.; Fan, Y.; Shaw, T.I.; Silveira, A.B.; Qu, C.; et al. Histone H3.3 K27M accelerates spontaneous brainstem glioma and drives restricted changes in bivalent gene expression. Cancer Cell 2019, 35, 140–155.e7. [Google Scholar] [CrossRef] [PubMed]
- Lange, K.; Kammerer, M.; Saupe, F.; Hegi, M.E.; Grotegut, S.; Fluri, E.; Orend, G. Combined lysophosphatidic acid/platelet-derived growth factor signaling triggers glioma cell migration in a tenascin-C microenvironment. Cancer Res. 2008, 68, 6942–6952. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Sage, J.C.; Miller, M.R.; Verhaak, R.G.; Hippenmeyer, S.; Vogel, H.; Foreman, O.; Bronson, R.T.; Nishiyama, A.; Luo, L.; et al. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell 2011, 146, 209–221. [Google Scholar] [CrossRef]
- Alcantara Llaguno, S.R.; Wang, Z.; Sun, D.; Chen, J.; Xu, J.; Kim, E.; Hatanpaa, K.J.; Raisanen, J.M.; Burns, D.K.; Johnson, J.E.; et al. Adult lineage-restricted CNS progenitors specify distinct glioblastoma subtypes. Cancer Cell 2015, 28, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Silveira, A.B.; Kasper, L.H.; Fan, Y.; Jin, H.; Wu, G.; Shaw, T.I.; Zhu, X.; Larson, J.D.; Easton, J.; Shao, Y.; et al. H3.3 K27M depletion increases differentiation and extends latency of diffuse intrinsic pontine glioma growth in vivo. Acta Neuropathol. 2019, 137, 637–655. [Google Scholar] [CrossRef]
- Haag, D.; Mack, N.; Benites Goncalves da Silva, P.; Statz, B.; Clark, J.; Tanabe, K.; Sharma, T.; Jäger, N.; Jones DT, W.; Kawauchi, D.; et al. H3.3-K27M drives neural stem cell-specific gliomagenesis in a human iPSC-derived model. Cancer Cell 2021, 39, 407–422.e13. [Google Scholar] [CrossRef]
- Kfoury-Beaumont, N.; Prakasam, R.; Pondugula, S.; Lagas, J.S.; Matkovich, S.; Gontarz, P.; Yang, L.; Yano, H.; Kim, A.H.; Rubin, J.B.; et al. The H3K27M mutation alters stem cell growth, epigenetic regulation, and differentiation potential. BMC Biol. 2022, 20, 124. [Google Scholar] [CrossRef]
- Miciaccia, M.; Rizzo, F.; Centonze, A.; Cavallaro, G.; Contino, M.; Armenise, D.; Baldelli, O.M.; Solidoro, R.; Ferorelli, S.; Scarcia, P.; et al. Harmaline to Human Mitochondrial Caseinolytic Serine Protease Activation for Pediatric Diffuse Intrinsic Pontine Glioma Treatment. Pharmaceuticals 2024, 17, 135. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mandorino, M.; Maitra, A.; Armenise, D.; Baldelli, O.M.; Miciaccia, M.; Ferorelli, S.; Perrone, M.G.; Scilimati, A. Pediatric Diffuse Midline Glioma H3K27-Altered: From Developmental Origins to Therapeutic Challenges. Cancers 2024, 16, 1814. https://doi.org/10.3390/cancers16101814
Mandorino M, Maitra A, Armenise D, Baldelli OM, Miciaccia M, Ferorelli S, Perrone MG, Scilimati A. Pediatric Diffuse Midline Glioma H3K27-Altered: From Developmental Origins to Therapeutic Challenges. Cancers. 2024; 16(10):1814. https://doi.org/10.3390/cancers16101814
Chicago/Turabian StyleMandorino, Manuela, Ahana Maitra, Domenico Armenise, Olga Maria Baldelli, Morena Miciaccia, Savina Ferorelli, Maria Grazia Perrone, and Antonio Scilimati. 2024. "Pediatric Diffuse Midline Glioma H3K27-Altered: From Developmental Origins to Therapeutic Challenges" Cancers 16, no. 10: 1814. https://doi.org/10.3390/cancers16101814
APA StyleMandorino, M., Maitra, A., Armenise, D., Baldelli, O. M., Miciaccia, M., Ferorelli, S., Perrone, M. G., & Scilimati, A. (2024). Pediatric Diffuse Midline Glioma H3K27-Altered: From Developmental Origins to Therapeutic Challenges. Cancers, 16(10), 1814. https://doi.org/10.3390/cancers16101814