
Driver Mutations in Pancreatic Cancer and Opportunities for Targeted Therapy

 , , , ,
, , , ,
Abstract
Simple Summary
Abstract
1. Introduction
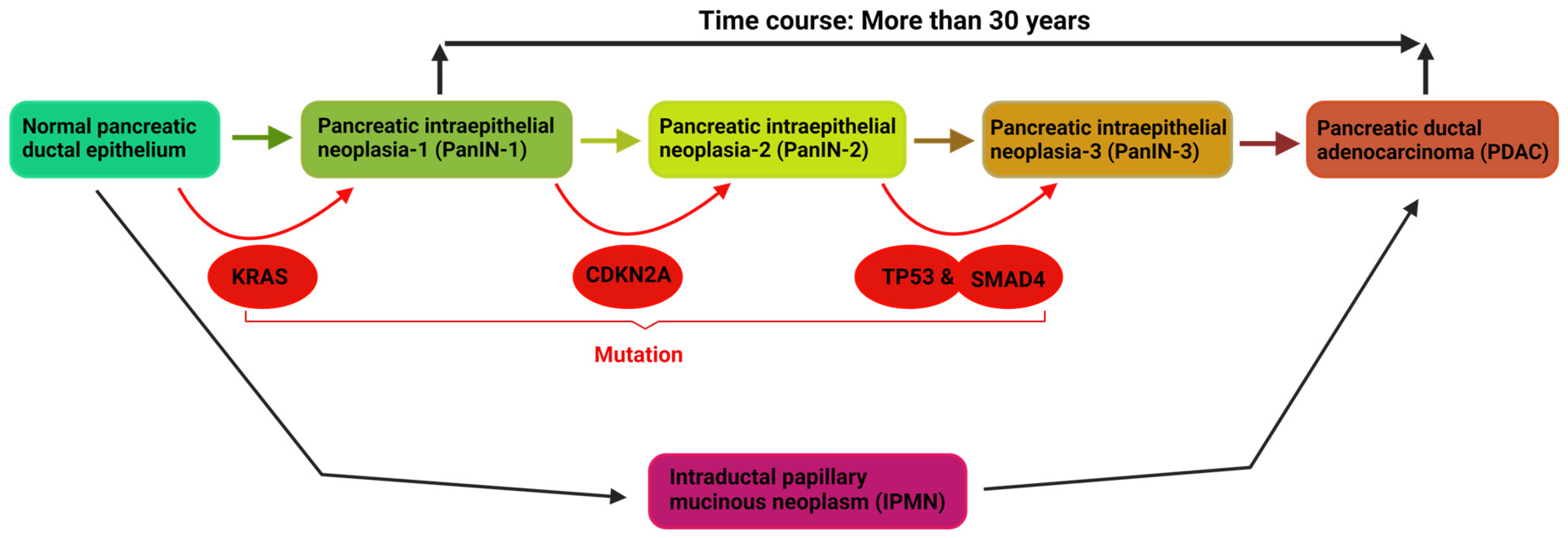
2. Driver Mutations, and Mechanisms of PDAC
3. Targeting Oncogenic Signaling Pathways for PDAC Treatment: KRAS
Targeting KRAS: Clinical and Therapeutic Investigations
4. Targeting Tumor Suppressor Mutations for PDAC Treatment
4.1. TP53
4.1.1. Targeting Negative Regulators of wt-p53
4.1.2. Targeting Mutant p53
4.2. SMAD4
4.3. CDKN2A

{kind=link}
{kind=link}
| Target | Agent(s) | Phase | Disease(s) | Mechanism | Reference(s) |
|---|---|---|---|---|---|
| MDM2 | RG7112 | I | WDLPS/DDLPS | Occupy p53 binding pocket of MDM2 for p53 stabilization/activation | [94,95,96,97,98] |
| MDM2 | Idasanutlin | I | Polycythemia Vera and Essential Thrombocythemia | Promote TP53 expression and prevent p53 degradation | [99] |
| MDM2 | Venetoclax-Idasanutlin | Ib | Relapsed/Refractory Acute Myeloid Leukemia | p53 activation to inactivate MCL-1 and BCL-xL | [100] |
| MDM2 | RO6839921 | I | Acute Myeloid Leukemia | Plasma esterase cleaves inactive prodrug to release active Idasanutlin to restore p53 activity | [101,102] |
| MDM2 and CDK4/6 | Siremadlin + Ribociclib | Ib | WDLPS/DDLPS | p53 pathway activation and inhibition of CDK enzymes | [103] |
| MDM2 | AMG-232 | I | P53WT Solid Tumors or Multiple Myeloma | Strong binding blocking MDM2-p53 interaction | [105,106,107] |
| MDM2 | KRT-232 | I | Solid Tumors or Multiple Myeloma and Acute Myeloid Leukemia | Binds to MDM2 and inhibits interaction with p53 for p53 activation | [104] |
| Mutant p53 | APR-246 + azacytidine | Ib/II | Myelodysplastic Syndrome | Induce apoptosis and reprogram TAMS to enhance immune checkpoint inhibitors | [113] |
| Mutant p53 | COTI-2 | I | Head and Neck Squamous Cell Carcinoma and Gynecologic Malignancies | Restore functionality to mutated p53 | [118] |
| SMAD4 | DTLL | Pre-Clinical | PDAC | Block ATR/mTOR pathway and restore SMAD4 mediated activation of Nf-кβ shunt | [129] |
| SMAD4 | Qianlongtong | Randomized Control | Benign Prostate Hyperplasia | Increase expression of SMAD4 | [131] |
| SMAD4 | Duvelisib | I | T-cell Lymphoma | PI3K-δ/γ inhibition in TGFβ-PI3/AKT Axis | [134] |
| SMAD4 | Perifosine and MK-2206 | II | Recurrent Glioblastoma and Breast Cancer | MSP/RON pathway in TGFβ-PI3/AKT Axis | [135,136] |
| CDK4/6 | Abemaciclib | Open label III | HR+, HER2-, node-positive, Breast Cancer | Inhibition of cell cycle progression through CDK4/6 inhibitor | [148] |
| CDK4/6 | Abemaciclib + Bevacizumab | I | Recurrent GBM with loss of CDKN2A | Inhibition of cell cycle progression through CDK4/6 inhibitor and anti-angiogenic therapy | NCT04074785 |
| CDK4/6 | Palbociclib + Fulvestrant | Placebo Controlled Randomized Trial | HR+, HER2-, Breast Cancer | Inhibition cell cycle progression through CDK4/6 inhibitor | NCT01942135 |
| CDK4/6 | Trilaciclib | Ib/II | Naïve Extensive Stage Small Cell Lung Cancer | Chemotherapy damage prevention by HSPC remaining in G1 arrest | [150] |
| CDNK2A | Ilorasertinib | II | Advanced Solid Tumors | Multikinase Inhibition to induce cell cycle arrest | NCT02478320 |
| CDK4/6 | Palbociclib | Non-randomized Open Label II | Metastatic Grade 1 and 2 Pancreatic Neuroendocrine Tumors | Inhibition of cell cycle progression in RB+ cells through CDK4/6 inhibitor | [153] |
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Panchal, K.; Sahoo, R.K.; Gupta, U.; Chaurasiya, A. Role of targeted immunotherapy for pancreatic ductal adenocarcinoma (PDAC) treatment: An overview. Int. Immunopharmacol. 2021, 95, 107508. [Google Scholar] [CrossRef]
- Siegel, R.L.; Giaquinto, A.N.; Jemal, A. Cancer statistics, 2024. CA Cancer J. Clin. 2024, 74, 12–49. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Olaoba, O.T.; Ligali, F.C.; Alabi, Z.O.; Akinyemi, A.O.; Ayinde, K.S. Of immune checkpoint maladies and remedies: The throwing of jabs in the oncogenic ring of PDAC. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2021, 1875, 188483. [Google Scholar] [CrossRef]
- Collisson, E.A.; Bailey, P.; Chang, D.K.; Biankin, A.V. Molecular subtypes of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 207–220. [Google Scholar] [CrossRef]
- Zhao, Z.; Liu, W. Pancreatic Cancer: A Review of Risk Factors, Diagnosis, and Treatment. Technol. Cancer Res. Treat. 2020, 19, 1533033820962117. [Google Scholar] [CrossRef]
- McGuigan, A.; Kelly, P.; Turkington, R.C.; Jones, C.; Coleman, H.G.; McCain, R.S. Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World J. Gastroenterol. 2018, 24, 4846–4861. [Google Scholar] [CrossRef]
- McMenamin, Ú.; McCain, S.; Kunzmann, A.T. Do smoking and alcohol behaviours influence GI cancer survival? Best Pract. Res. Clin. Gastroenterol. 2017, 31, 569–577. [Google Scholar] [CrossRef]
- Midha, S.; Chawla, S.; Garg, P.K. Modifiable and non-modifiable risk factors for pancreatic cancer: A review. Cancer Lett. 2016, 381, 269–277. [Google Scholar] [CrossRef]
- Yeo, T.P. Demographics, epidemiology, and inheritance of pancreatic ductal adenocarcinoma. Semin. Oncol. 2015, 42, 8–18. [Google Scholar] [CrossRef]
- Brown, D.G.; Rao, S.; Weir, T.L.; O’Malia, J.; Bazan, M.; Brown, R.J.; Ryan, E.P. Metabolomics and metabolic pathway networks from human colorectal cancers, adjacent mucosa, and stool. Cancer Metab. 2016, 4, 11. [Google Scholar] [CrossRef]
- Meng, C.; Bai, C.; Brown, T.D.; Hood, L.E.; Tian, Q. Human Gut Microbiota and Gastrointestinal Cancer. Genom. Proteom. Bioinform. 2018, 16, 33–49. [Google Scholar] [CrossRef]
- Downes, D.P.; Daurio, N.A.; McLaren, D.G.; Carrington, P.; Previs, S.F.; Williams, K.B. Impact of Extracellular Fatty Acids and Oxygen Tension on Lipid Synthesis and Assembly in Pancreatic Cancer Cells. ACS Chem. Biol. 2020, 15, 1892–1900. [Google Scholar] [CrossRef]
- Genkinger, J.M.; Spiegelman, D.; Anderson, K.E.; Bergkvist, L.; Bernstein, L.; Brandt, P.A.v.D.; English, D.R.; Freudenheim, J.L.; Fuchs, C.S.; Giles, G.G.; et al. Alcohol intake and pancreatic cancer risk: A pooled analysis of fourteen cohort studies. Cancer Epidemiol. Biomark. Prev. 2009, 18, 765–776. [Google Scholar] [CrossRef]
- Molina-Montes, E.; Van Hoogstraten, L.; Gomez-Rubio, P.; Löhr, M.; Sharp, L.; Molero, X.; Márquez, M.; Michalski, C.W.; Farré, A.; Perea, J.; et al. Pancreatic Cancer Risk in Relation to Lifetime Smoking Patterns, Tobacco Type, and Dose–Response Relationships. Cancer Epidemiol. Biomark. Prev. 2020, 29, 1009–1018. [Google Scholar] [CrossRef]
- Samokhvalov, A.V.; Rehm, J.; Roerecke, M. Alcohol Consumption as a Risk Factor for Acute and Chronic Pancreatitis: A Systematic Review and a Series of Meta-analyses. EBioMedicine 2015, 2, 1996–2002. [Google Scholar] [CrossRef]
- Kolbeinsson, H.M.; Chandana, S.; Wright, G.P.; Chung, M. Pancreatic Cancer: A Review of Current Treatment and Novel Therapies. J. Investig. Surg. 2023, 36, 2129884. [Google Scholar] [CrossRef]
- Ostroverkhova, D.; Przytycka, T.M.; Panchenko, A.R. Cancer driver mutations: Predictions and reality. Trends Mol. Med. 2023, 29, 554–566. [Google Scholar] [CrossRef]
- Hu, H.-F.; Ye, Z.; Qin, Y.; Xu, X.-W.; Yu, X.-J.; Zhuo, Q.-F.; Ji, S.-R. Mutations in key driver genes of pancreatic cancer: Molecularly targeted therapies and other clinical implications. Acta Pharmacol. Sin. 2021, 42, 1725–1741. [Google Scholar] [CrossRef]
- Vitellius, C.; Griveaux, O.; Morvant, B.; Pedrono, E.; Venara, A.; Ingster, O.; Baize, N.; Dincuff, E.; Rousselet, M.-C.; Guardiola, P.; et al. Impact of Driver Mutations on the Evolution of Isolated Metachronous Lung Metastasis of Pancreatic Ductal adenocarcinoma. Mol. Diagn. Ther. 2020, 24, 443–449. [Google Scholar] [CrossRef]
- Luo, J. KRAS mutation in pancreatic cancer. Semin. Oncol. 2021, 48, 10–18. [Google Scholar] [CrossRef]
- Peters, M.L.B.; Eckel, A.; Mueller, P.P.; Tramontano, A.C.; Weaver, D.T.; Lietz, A.; Hur, C.; Kong, C.Y.; Pandharipande, P.V. Progression to pancreatic ductal adenocarcinoma from pancreatic intraepithelial neoplasia: Results of a simulation model. Pancreatology 2018, 18, 928–934. [Google Scholar] [CrossRef]
- Huang, L.; Guo, Z.; Wang, F.; Fu, L. KRAS mutation: From undruggable to druggable in cancer. Signal Transduct. Target. Ther. 2021, 6, 386. [Google Scholar] [CrossRef]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef]
- Wood, K.; Hensing, T.; Malik, R.; Salgia, R. Prognostic and Predictive Value in KRAS in Non-Small-Cell Lung Cancer: A Review. JAMA Oncol. 2016, 2, 805–812. [Google Scholar] [CrossRef]
- Dearden, S.; Stevens, J.; Wu, Y.-L.; Blowers, D. Mutation incidence and coincidence in non small-cell lung cancer: Meta-analyses by ethnicity and histology (mutMap). Ann. Oncol. 2013, 24, 2371–2376. [Google Scholar] [CrossRef]
- Shen, H.; Lundy, J.; Strickland, A.H.; Harris, M.; Swan, M.; Desmond, C.; Jenkins, B.J.; Croagh, D. KRAS G12D Mutation Subtype in Pancreatic Ductal Adenocarcinoma: Does It Influence Prognosis or Stage of Disease at Presentation? Cells 2022, 11, 3175. [Google Scholar] [CrossRef]
- Bryant, K.L.; Mancias, J.D.; Kimmelman, A.C.; Der, C.J. KRAS: Feeding pancreatic cancer proliferation. Trends Biochem. Sci. 2014, 39, 91–100. [Google Scholar] [CrossRef]
- Neumann, J.; Zeindl-Eberhart, E.; Kirchner, T.; Jung, A. Frequency and type of KRAS mutations in routine diagnostic analysis of metastatic colorectal cancer. Pathol.-Res. Pract. 2009, 205, 858–862. [Google Scholar] [CrossRef]
- Waters, A.M.; Der, C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a031435. [Google Scholar] [CrossRef]
- Saiki, A.Y.; Gaida, K.; Rex, K.; Achanta, P.; Miguel, T.S.; Koppada, N.; Bagal, D.; Lanman, B.A.; Foti, R.S.; McCarter, J.D.; et al. Abstract 4484: Discovery and in vitro characterization of AMG 510–a potent and selective covalent small-molecule inhibitor of KRASG12C. Cancer Res. 2019, 79 (Suppl. S13), 4484. [Google Scholar] [CrossRef]
- Sikdar, N.; Saha, G.; Dutta, A.; Ghosh, S.; Shreekhande, S.V.; Banerjee, S. Genetic Alterations of Periampullary and Pancreatic Ductal Adenocarcinoma: An Overview. Curr. Genom. 2018, 19, 444–463. [Google Scholar] [CrossRef]
- Mohamadkhani, A.; Naderi, E.; Sharafkhah, M.; Fazli, H.R.; Moradzadeh, M.; Pourshams, A. Detection of TP53 R249 Mutation in Iranian Patients with Pancreatic Cancer. J. Oncol. 2013, 2013, 738915. [Google Scholar] [CrossRef]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef]
- Bos, J.L.; Rehmann, H.; Wittinghofer, A. GEFs and GAPs: Critical elements in the control of small G proteins. Cell 2007, 129, 865–877. [Google Scholar] [CrossRef]
- Warne, P.H.; Vician, P.R.; Downward, J. Direct interaction of Ras and the amino-terminal region of Raf-1 in vitro. Nature 1993, 364, 352–355. [Google Scholar] [CrossRef]
- Roux, P.P.; Blenis, J. ERK and p38 MAPK-activated protein kinases: A family of protein kinases with diverse biological functions. Microbiol. Mol. Biol. Rev. 2004, 68, 320–344. [Google Scholar] [CrossRef]
- Zhang, X.-F.; Settleman, J.; Kyriakis, J.; Takeuchi-Suzuki, E.; Elledge, S.J.; Marshall, M.S.; Bruder, J.T.; Rapp, U.R.; Avruch, J. Normal and oncogenic p21ras proteins bind to the amino-terminal regulatory domain of c-Raf-1. Nature 1993, 364, 308–313. [Google Scholar] [CrossRef]
- Molina, J.R.; Adjei, A.A. The Ras/Raf/MAPK pathway. J. Thorac. Oncol. 2006, 1, 7–9. [Google Scholar] [CrossRef]
- Oyedele, A.-Q.K.; Ogunlana, A.T.; Boyenle, I.D.; Ibrahim, N.O.; Gbadebo, I.O.; Owolabi, N.A.; Ayoola, A.M.; Francis, A.C.; Eyinade, O.H.; Adelusi, T.I. Pharmacophoric analogs of sotorasib-entrapped KRAS G12C in its inactive GDP-bound conformation: Covalent docking and molecular dynamics investigations. Mol. Divers. 2022, 27, 1795–1807. [Google Scholar] [CrossRef]
- Scheffzek, K.; Ahmadian, M.R.; Kabsch, W.; Wiesmuller, L.; Lautwein, A.; Schmitz, F.; Wittinghofer, A. The Ras-RasGAP complex: Structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science 1997, 277, 333–338. [Google Scholar] [CrossRef]
- Haigis, K.M. KRAS Alleles: The Devil Is in the Detail. Trends Cancer 2017, 3, 686–697. [Google Scholar] [CrossRef]
- Wennerberg, K.; Rossman, K.L.; Der, C.J. The Ras superfamily at a glance. J. Cell Sci. 2005, 118 Pt 5, 843–846. [Google Scholar] [CrossRef]
- Astrain, G.B.; Nikolova, M.; Smith, M.J. Functional diversity in the RAS subfamily of small GTPases. Biochem. Soc. Trans. 2022, 50, 921–933. [Google Scholar] [CrossRef]
- Lu, Z.; Luo, R.Z.; Lu, Y.; Zhang, X.; Yu, Q.; Khare, S.; Kondo, S.; Kondo, Y.; Yu, Y.; Mills, G.B.; et al. The tumor suppressor gene ARHI regulates autophagy and tumor dormancy in human ovarian cancer cells. J. Clin. Investig. 2008, 118, 3917–3929. [Google Scholar] [CrossRef]
- Sutton, M.N.; Lu, Z.; Li, Y.-C.; Zhou, Y.; Huang, T.; Reger, A.S.; Hurwitz, A.M.; Palzkill, T.; Logsdon, C.; Liang, X.; et al. DIRAS3 (ARHI) Blocks RAS/MAPK Signaling by Binding Directly to RAS and Disrupting RAS Clusters. Cell Rep. 2019, 29, 3448–3459.e6. [Google Scholar] [CrossRef]
- Sutton, M.N.; Yang, H.; Huang, G.Y.; Fu, C.; Pontikos, M.; Wang, Y.; Mao, W.; Pang, L.; Yang, M.; Liu, J.; et al. RAS-related GTPases DIRAS1 and DIRAS2 induce autophagic cancer cell death and are required for autophagy in murine ovarian cancer cells. Autophagy 2018, 14, 637–653. [Google Scholar] [CrossRef]
- Castellano, E.; Downward, J. RAS Interaction with PI3K: More Than Just Another Effector Pathway. Genes. Cancer 2011, 2, 261–274. [Google Scholar] [CrossRef]
- Pawson, T. Specificity in signal transduction: From phosphotyrosine-SH2 domain interactions to complex cellular systems. Cell 2004, 116, 191–203. [Google Scholar] [CrossRef]
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov. 2009, 8, 627–644. [Google Scholar] [CrossRef]
- Castellano, E.; Downward, J. Role of RAS in the regulation of PI 3-kinase. Curr. Top. Microbiol. Immunol. 2010, 346, 143–169. [Google Scholar]
- He, Q.; Liu, Z.; Wang, J. Targeting KRAS in PDAC: A New Way to Cure It? Cancers 2022, 14, 4982. [Google Scholar] [CrossRef]
- Pantsar, T. The current understanding of KRAS protein structure and dynamics. Comput. Struct. Biotechnol. J. 2020, 18, 189–198. [Google Scholar] [CrossRef]
- Tang, D.; Kroemer, G.; Kang, R. Oncogenic KRAS blockade therapy: Renewed enthusiasm and persistent challenges. Mol. Cancer 2021, 20, 128. [Google Scholar] [CrossRef]
- Cruz-Migoni, A.; Canning, P.; Quevedo, C.E.; Bataille, C.J.R.; Bery, N.; Miller, A.; Russell, A.J.; Phillips, S.E.V.; Carr, S.B.; Rabbitts, T.H. Structure-based development of new RAS-effector inhibitors from a combination of active and inactive RAS-binding compounds. Proc. Natl. Acad. Sci. USA 2019, 116, 2545–2550. [Google Scholar] [CrossRef]
- Kessler, D.; Gmachl, M.; Mantoulidis, A.; Martin, L.J.; Zoephel, A.; Mayer, M.; Gollner, A.; Covini, D.; Fischer, S.; Gerstberger, T.; et al. Drugging an undruggable pocket on KRAS. Proc. Natl. Acad. Sci. USA 2019, 116, 15823–15829. [Google Scholar] [CrossRef]
- McCarthy, M.J.; Pagba, C.V.; Prakash, P.; Naji, A.K.; van der Hoeven, D.; Liang, H.; Gupta, A.K.; Zhou, Y.; Cho, K.-J.; Hancock, J.F.; et al. Discovery of High-Affinity Noncovalent Allosteric KRAS Inhibitors That Disrupt Effector Binding. ACS Omega 2019, 4, 2921–2930. [Google Scholar] [CrossRef]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef]
- Welsch, M.E.; Kaplan, A.; Chambers, J.M.; Stokes, M.E.; Bos, P.H.; Zask, A.; Zhang, Y.; Sanchez-Martin, M.; Badgley, M.A.; Huang, C.S.; et al. Multivalent Small-Molecule Pan-RAS Inhibitors. Cell 2017, 168, 878–889.e29. [Google Scholar] [CrossRef]
- Liu, J.; Kang, R.; Tang, D. The KRAS-G12C inhibitor: Activity and resistance. Cancer Gene Ther. 2022, 29, 875–878. [Google Scholar] [CrossRef]
- Lee, A. Sotorasib: A Review in KRAS G12C Mutation-Positive Non-small Cell Lung Cancer. Target. Oncol. 2022, 17, 727–733. [Google Scholar] [CrossRef]
- Kwan, A.K.; Piazza, G.A.; Keeton, A.B.; Leite, C.A. The path to the clinic: A comprehensive review on direct KRAS(G12C) inhibitors. J. Exp. Clin. Cancer Res. 2022, 41, 27. [Google Scholar] [CrossRef]
- Nagasaka, M.; Li, Y.; Sukari, A.; Ou, S.H.; Al-Hallak, M.N.; Azmi, A.S. KRAS G12C Game of Thrones, which direct KRAS inhibitor will claim the iron throne? Cancer Treat. Rev. 2020, 84, 101974. [Google Scholar] [CrossRef]
- Fakih, M.; O’Neil, B.; Price, T.J.; Falchook, G.S.; Desai, J.; Kuo, J.; Govindan, R.; Rasmussen, E.; Morrow, P.K.; Ngang, J.; et al. Phase 1 Study Evaluating the Safety, Tolerability, Pharmacokinetics (PK), and Efficacy of AMG 510, a Novel Small Molecule KRASG12C Inhibitor, in Advanced Solid Tumors; American Society of Clinical Oncology: Alexandria, VA, USA, 2019. [Google Scholar]
- Papadopoulos, K.P.; Ou, S.-H.I.; Johnson, M.L.; Christensen, J.; Velastegui, K.; Potvin, D.; Faltaos, D.; Chao, R.C. A phase I/II multiple expansion cohort trial of MRTX849 in patients with advanced solid tumors with KRAS G12C mutation. J. Clin. Oncol. 2019, 37 (Suppl. S15), TPS3161. [Google Scholar] [CrossRef]
- Janes, M.R.; Zhang, J.; Li, L.-S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L.; et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018, 172, 578–589.e17. [Google Scholar] [CrossRef]
- Gentile, D.R.; Rathinaswamy, M.K.; Jenkins, M.L.; Moss, S.M.; Siempelkamp, B.D.; Renslo, A.R.; Burke, J.E.; Shokat, K.M. Ras Binder Induces a Modified Switch-II Pocket in GTP and GDP States. Cell Chem. Biol. 2017, 24, 1455–1466.e14. [Google Scholar] [CrossRef]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Robert, C.; Grob, J.J.; Stroyakovskiy, D.; Karaszewska, B.; Hauschild, A.; Levchenko, E.; Chiarion Sileni, V.; Schachter, J.; Garbe, C.; Bondarenko, I.; et al. Five-Year Outcomes with Dabrafenib plus Trametinib in Metastatic Melanoma. N. Engl. J. Med. 2019, 381, 626–636. [Google Scholar] [CrossRef]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRAS(G12C) Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef]
- Karp, J.E.; Flatten, K.; Feldman, E.J.; Greer, J.M.; Loegering, D.A.; Ricklis, R.M.; Morris, L.E.; Ritchie, E.; Smith, B.D.; Ironside, V.; et al. Active oral regimen for elderly adults with newly diagnosed acute myelogenous leukemia: A preclinical and phase 1 trial of the farnesyltransferase inhibitor tipifarnib (R115777, Zarnestra) combined with etoposide. Blood 2009, 113, 4841–4852. [Google Scholar] [CrossRef]
- Youssef, M.E.; Cavalu, S.; Hasan, A.M.; Yahya, G.; Abd-Eldayem, M.A.; Saber, S. Role of Ganetespib, an HSP90 Inhibitor, in Cancer Therapy: From Molecular Mechanisms to Clinical Practice. Int. J. Mol. Sci. 2023, 24, 5014. [Google Scholar] [CrossRef]
- Kim, M.J.; Lee, S.J.; Ryu, J.H.; Kim, S.H.; Kwon, I.C.; Roberts, T.M. Combination of KRAS gene silencing and PI3K inhibition for ovarian cancer treatment. J. Control. Release 2020, 318, 98–108. [Google Scholar] [CrossRef]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef]
- Wu, Y.; Walker, J.R.; Westberg, M.; Ning, L.; Monje, M.; Kirkland, T.A.; Lin, M.Z.; Su, Y. Kinase-Modulated Bioluminescent Indicators Enable Noninvasive Imaging of Drug Activity in the Brain. ACS Central Sci. 2023, 9, 719–732. [Google Scholar] [CrossRef]
- Sakamoto, K.; Lin, B.; Nunomura, K.; Izawa, T.; Nakagawa, S. The K-Ras(G12D)-inhibitory peptide KS-58 suppresses growth of murine CT26 colorectal cancer cell-derived tumors. Sci. Rep. 2022, 12, 8121. [Google Scholar] [CrossRef]
- Wang, A.; Liu, J.; Li, X.; Zou, F.; Qi, Z.; Qi, S.; Liu, Q.; Wang, Z.; Cao, J.; Jiang, Z.; et al. Discovery of a highly potent pan-RAF inhibitor IHMT-RAF-128 for cancer treatment. Eur. J. Pharmacol. 2023, 952, 175752. [Google Scholar] [CrossRef]
- Ghosh, S.; Fan, F.; Powell, R.T.; Roszik, J.; Park, Y.S.; Stephan, C.; Sebastian, M.; Tan, L.; Sorokin, A.V.; Lorenzi, P.L.; et al. Vincristine Enhances the Efficacy of MEK Inhibitors in Preclinical Models of KRAS-mutant Colorectal Cancer. Mol. Cancer Ther. 2023, 22, 962–975. [Google Scholar] [CrossRef]
- Carbone, D.; De Franco, M.; Pecoraro, C.; Bassani, D.; Pavan, M.; Cascioferro, S.; Parrino, B.; Cirrincione, G.; Dall’acqua, S.; Moro, S.; et al. Discovery of the 3-Amino-1,2,4-triazine-Based Library as Selective PDK1 Inhibitors with Therapeutic Potential in Highly Aggressive Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 2023, 24, 3679. [Google Scholar] [CrossRef]
- Hassin, O.; Oren, M. Drugging p53 in cancer: One protein, many targets. Nat. Rev. Drug Discov. 2023, 22, 127–144. [Google Scholar] [CrossRef]
- Hu, J.; Cao, J.; Topatana, W.; Juengpanich, S.; Li, S.; Zhang, B.; Shen, J.; Cai, L.; Cai, X.; Chen, M. Targeting mutant p53 for cancer therapy: Direct and indirect strategies. J. Hematol. Oncol. 2021, 14, 157. [Google Scholar] [CrossRef] [PubMed]
- Maddalena, M.; Mallel, G.; Nataraj, N.B.; Shreberk-Shaked, M.; Hassin, O.; Mukherjee, S.; Arandkar, S.; Rotkopf, R.; Kapsack, A.; Lambiase, G.; et al. TP53 missense mutations in PDAC are associated with enhanced fibrosis and an immunosuppressive microenvironment. Proc. Natl. Acad. Sci. USA 2021, 118, e2025631118. [Google Scholar] [CrossRef]
- McLornan, D.P.; List, A.; Mufti, G.J. Applying synthetic lethality for the selective targeting of cancer. N. Engl. J. Med. 2014, 371, 1725–1735. [Google Scholar] [CrossRef] [PubMed]
- Cooks, T.; Pateras, I.S.; Jenkins, L.M.; Patel, K.M.; Robles, A.I.; Morris, J.; Forshew, T.; Appella, E.; Gorgoulis, V.G.; Harris, C.C. Mutant p53 cancers reprogram macrophages to tumor supporting macrophages via exosomal miR-1246. Nat. Commun. 2018, 9, 771. [Google Scholar] [CrossRef] [PubMed]
- Wellenstein, M.D.; Coffelt, S.B.; Duits, D.E.M.; Van Miltenburg, M.H.; Slagter, M.; De Rink, I.; Henneman, L.; Kas, S.M.; Prekovic, S.; Hau, C.-S.; et al. Loss of p53 triggers WNT-dependent systemic inflammation to drive breast cancer metastasis. Nature 2019, 572, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.P.; Lozano, G. Mutant p53 partners in crime. Cell Death Differ. 2018, 25, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Pilley, S.; Rodriguez, T.A.; Vousden, K.H. Mutant p53 in cell-cell interactions. Genes Dev. 2021, 35, 433–448. [Google Scholar] [CrossRef] [PubMed]
- Sabapathy, K.; Lane, D.P. Therapeutic targeting of p53: All mutants are equal, but some mutants are more equal than others. Nat. Rev. Clin. Oncol. 2018, 15, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Stein, Y.; Rotter, V.; Aloni-Grinstein, R. Gain-of-function mutant p53: All the roads lead to tumorigenesis. Int. J. Mol. Sci. 2019, 20, 6197. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, J.; Xu, D.; Zhang, T.; Hu, W.; Feng, Z. Gain-of-function mutant p53 in cancer progression and therapy. J. Mol. Cell Biol. 2020, 12, 674–687. [Google Scholar] [CrossRef]
- Zhu, G.; Pan, C.; Bei, J.-X.; Li, B.; Liang, C.; Xu, Y.; Fu, X. Mutant p53 in cancer progression and targeted therapies. Front. Oncol. 2020, 10, 595187. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, A.; Tolcher, A.; Beeram, M.; Nemunaitis, J.; Weiss, G.J.; Bhalla, K.; Agrawal, M.; Nichols, G.; Middleton, S.; Beryozkina, A.; et al. Clinical pharmacology characterization of RG7112, an MDM2 antagonist, in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2015, 76, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Andreeff, M.; Kelly, K.R.; Yee, K.; Assouline, S.; Strair, R.; Popplewell, L.; Bowen, D.; Martinelli, G.; Drummond, M.W.; Vyas, P.; et al. Results of the Phase I Trial of RG7112, a Small-Molecule MDM2 Antagonist in Leukemia. Clin. Cancer Res. 2016, 22, 868–876. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Chen, G.; Jukofsky, L.; Geho, D.; Han, S.W.; Birzele, F.; Bader, S.; Himmelein, L.; Cai, J.; Albertyn, Z.; et al. MDM2 antagonist clinical response association with a gene expression signature in acute myeloid leukaemia. Br. J. Haematol. 2015, 171, 432–435. [Google Scholar] [CrossRef] [PubMed]
- Ray-Coquard, I.; Blay, J.-Y.; Italiano, A.; Le Cesne, A.; Penel, N.; Zhi, J.; Heil, F.; Rueger, R.; Graves, B.; Ding, M.; et al. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: An exploratory proof-of-mechanism study. Lancet Oncol. 2012, 13, 1133–1140. [Google Scholar] [CrossRef] [PubMed]
- Vu, B.; Wovkulich, P.; Pizzolato, G.; Lovey, A.; Ding, Q.; Jiang, N.; Liu, J.-J.; Zhao, C.; Glenn, K.; Wen, Y.; et al. Discovery of RG7112: A Small-Molecule MDM2 Inhibitor in Clinical Development. ACS Med. Chem. Lett. 2013, 4, 466–469. [Google Scholar] [CrossRef]
- Mascarenhas, J.; Lu, M.; Kosiorek, H.; Virtgaym, E.; Xia, L.; Sandy, L.; Mesa, R.; Petersen, B.; Farnoud, N.; Najfeld, V.; et al. Oral idasanutlin in patients with polycythemia vera. Blood 2019, 134, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.G.; Dail, M.; Garcia, J.S.; Jonas, B.A.; Yee, K.W.L.; Kelly, K.R.; Vey, N.; Assouline, S.; Roboz, G.J.; Paolini, S.; et al. Venetoclax and idasanutlin in relapsed/refractory AML: A nonrandomized, open-label phase 1b trial. Blood 2023, 141, 1265–1276. [Google Scholar] [CrossRef]
- Uy, G.L.; Assouline, S.; Young, A.-M.; Blotner, S.; Higgins, B.; Chen, L.-C.; Yee, K. Phase 1 study of the MDM2 antagonist RO6839921 in patients with acute myeloid leukemia. Investig. New Drugs 2020, 38, 1430–1441. [Google Scholar] [CrossRef]
- Razak, A.R.A.; Miller, W.H.; Uy, G.L.; Blotner, S.; Young, A.-M.; Higgins, B.; Chen, L.-C.; Gore, L. A phase 1 study of the MDM2 antagonist RO6839921, a pegylated prodrug of idasanutlin, in patients with advanced solid tumors. Investig. New Drugs 2020, 38, 1156–1165. [Google Scholar] [CrossRef] [PubMed]
- Abdul Razak, A.R.; Bauer, S.; Suarez, C.; Lin, C.C.; Quek, R.; Hütter-Krönke, M.L.; Cubedo, R.; Ferretti, S.; Guerreiro, N.; Jullion, A.; et al. Co-Targeting of MDM2 and CDK4/6 with Siremadlin and Ribociclib for the Treatment of Patients with Well-Differentiated or Dedifferentiated Liposarcoma: Results from a Proof-of-Concept, Phase Ib Study. Clin. Cancer Res. 2022, 28, 1087–1097. [Google Scholar] [CrossRef]
- Taylor, A.; Lee, D.; Allard, M.; Poland, B.; Greg Slatter, J. Phase 1 Concentration-QTc and Cardiac Safety Analysis of the MDM2 Antagonist KRT-232 in Patients with Advanced Solid Tumors, Multiple Myeloma, or Acute Myeloid Leukemia. Clin. Pharmacol. Drug Dev. 2021, 10, 918–926. [Google Scholar] [CrossRef]
- Gluck, W.L.; Gounder, M.M.; Frank, R.; Eskens, F.; Blay, J.Y.; Cassier, P.A.; Soria, J.-C.; Chawla, S.; De Weger, V.; Wagner, A.J.; et al. Phase 1 study of the MDM2 inhibitor AMG 232 in patients with advanced P53 wild-type solid tumors or multiple myeloma. Investig. New Drugs 2020, 38, 831–843. [Google Scholar] [CrossRef]
- Erba, H.P.; Becker, P.S.; Shami, P.J.; Grunwald, M.R.; Flesher, D.L.; Zhu, M.; Rasmussen, E.; Henary, H.A.; Anderson, A.A.; Wang, E.S. Phase 1b study of the MDM2 inhibitor AMG 232 with or without trametinib in relapsed/refractory acute myeloid leukemia. Blood Adv. 2019, 3, 1939–1949. [Google Scholar] [CrossRef] [PubMed]
- Jeay, S.; Ferretti, S.; Holzer, P.; Fuchs, J.; Chapeau, E.A.; Wartmann, M.; Sterker, D.; Romanet, V.; Murakami, M.; Kerr, G.; et al. Dose and Schedule Determine Distinct Molecular Mechanisms Underlying the Efficacy of the p53–MDM2 Inhibitor HDM201. Cancer Res. 2018, 78, 6257–6267. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Fujiwara, Y.; Nakano, K.; Shimizu, T.; Tomomatsu, J.; Koyama, T.; Ogura, M.; Tachibana, M.; Kakurai, Y.; Yamashita, T.; et al. Safety and pharmacokinetics of milademetan, a MDM2 inhibitor, in Japanese patients with solid tumors: A phase I study. Cancer Sci. 2021, 112, 2361–2370. [Google Scholar] [CrossRef] [PubMed]
- De Weger, V.A.; de Jonge, M.; Langenberg, M.H.; Schellens, J.H.; Lolkema, M.; Varga, A.; Demers, B.; Thomas, K.; Hsu, K.; Tuffal, G.; et al. A phase I study of the HDM2 antagonist SAR405838 combined with the MEK inhibitor pimasertib in patients with advanced solid tumours. Br. J. Cancer 2019, 120, 286–293. [Google Scholar] [CrossRef]
- Bidou, L.; Bugaud, O.; Belakhov, V.; Baasov, T.; Namy, O. Characterization of new-generation aminoglycoside promoting premature termination codon readthrough in cancer cells. RNA Biol. 2017, 14, 378–388. [Google Scholar] [CrossRef]
- Xie, X.; Fan, C.; Luo, B.; Zhang, J.; Jensen, L.D.; Burman, J.; Jönsson, C.; Ljusberg, A.; Larsson, P.; Zhao, Z.; et al. APR-246 enhances colorectal cancer sensitivity to radiotherapy. Mol. Cancer Ther. 2023, 22, 947–961. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Abrams, S.L.; Steelman, L.S.; Cocco, L.; Ratti, S.; Martelli, A.M.; Lombardi, P.; Gizak, A.; Duda, P. APR-246—The Mutant TP53 Reactivator—Increases the Effectiveness of Berberine and Modified Berberines to Inhibit the Proliferation of Pancreatic Cancer Cells. Biomolecules 2022, 12, 276. [Google Scholar] [CrossRef]
- Sallman, D.A.; DeZern, A.E.; Garcia-Manero, G.; Steensma, D.P.; Roboz, G.J.; Sekeres, M.A.; Cluzeau, T.; Sweet, K.L.; McLemore, A.; McGraw, K.L.; et al. Eprenetapopt (APR-246) and Azacitidine in TP53-Mutant Myelodysplastic Syndromes. J. Clin. Oncol. 2021, 39, 1584–1594. [Google Scholar] [CrossRef] [PubMed]
- Zatloukalová, P.; Galoczová, M.; Vojtěšek, B. Prima-1 and APR-246 in Cancer Therapy. Klin. Onkol. 2018, 31 (Suppl. S2), 71–76. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Michels, J.; Mezzadra, R.; Venkatesh, D.; Dong, L.; Gomez, R.; Samaan, F.; Ho, Y.-J.; Campesato, L.F.; Mangarin, L.; et al. Increased p53 expression induced by APR-246 reprograms tumor-associated macrophages to augment immune checkpoint blockade. J. Clin. Investig. 2022, 132. [Google Scholar] [CrossRef] [PubMed]
- Lindemann, A.; Patel, A.A.; Silver, N.L.; Tang, L.; Liu, Z.; Wang, L.; Tanaka, N.; Rao, X.; Takahashi, H.; Maduka, N.K.; et al. COTI-2, A Novel Thiosemicarbazone Derivative, Exhibits Antitumor Activity in HNSCC through p53-dependent and -independent Mechanisms. Clin. Cancer Res. 2019, 25, 5650–5662. [Google Scholar] [CrossRef] [PubMed]
- Salim, K.Y.; Vareki, S.M.; Danter, W.R.; San-Marina, S.; Koropatnick, J. COTI-2, a novel small molecule that is active against multiple human cancer cell lines in vitro and in vivo. Oncotarget 2016, 7, 41363–41379. [Google Scholar] [CrossRef] [PubMed]
- Nunes, J.H.B.; Hager, S.; Mathuber, M.; Pósa, V.; Roller, A.; Enyedy, É.A.; Stefanelli, A.; Berger, W.; Keppler, B.K.; Heffeter, P.; et al. Cancer Cell Resistance Against the Clinically Investigated Thiosemicarbazone COTI-2 Is Based on Formation of Intracellular Copper Complex Glutathione Adducts and ABCC1-Mediated Efflux. J. Med. Chem. 2020, 63, 13719–13732. [Google Scholar] [CrossRef] [PubMed]
- Hahn, S.A.; Schutte, M.; Hoque, A.T.M.S.; Moskaluk, C.A.; da Costa, L.T.; Rozenblum, E.; Weinstein, C.L.; Fischer, A.; Yeo, C.J.; Hruban, R.H.; et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 1996, 271, 350–353. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, T.; Sunamura, M.; Horii, A. Molecular mechanisms of pancreatic carcinogenesis. Cancer Sci. 2006, 97, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Waddell, N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef]
- Xia, X.; Wu, W.; Huang, C.; Cen, G.; Jiang, T.; Cao, J.; Huang, K.; Qiu, Z. SMAD4 and its role in pancreatic cancer. Tumor Biol. 2015, 36, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Saiki, Y.; Horii, A. Molecular pathology of pancreatic cancer. Pathol. Int. 2014, 64, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, A.; Heldin, C.H. Non-Smad TGF-beta signals. J. Cell Sci. 2005, 118 Pt 16, 3573–3584. [Google Scholar] [CrossRef] [PubMed]
- Alhopuro, P.; Alazzouzi, H.; Sammalkorpi, H.; Dávalos, V.; Salovaara, R.; Hemminki, A.; Järvinen, H.; Mecklin, J.-P.; Schwartz, S.; Aaltonen, L.A.; et al. SMAD4 levels and response to 5-fluorouracil in colorectal cancer. Clin. Cancer Res. 2005, 11, 6311–6316. [Google Scholar] [CrossRef] [PubMed]
- Kozak, M.M.; von Eyben, R.; Pai, J.; Vossler, S.R.; Limaye, M.; Jayachandran, P.; Anderson, E.M.; Shaffer, J.L.; Longacre, T.; Pai, R.K.; et al. Smad4 inactivation predicts for worse prognosis and response to fluorouracil-based treatment in colorectal cancer. J. Clin. Pathol. 2015, 68, 341–345. [Google Scholar] [CrossRef]
- Cheng, H.; Fertig, E.J.; Ozawa, H.; Hatakeyama, H.; Howard, J.D.; Perez, J.; Considine, M.; Thakar, M.; Ranaweera, R.; Krigsfeld, G.; et al. Decreased SMAD4 expression is associated with induction of epithelial-to-mesenchymal transition and cetuximab resistance in head and neck squamous cell carcinoma. Cancer Biol. Ther. 2015, 16, 1252–1258. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, K.; Hablesreiter, R.; Inoue, Y.; Yoshida, K.; Briest, F.; Christen, F.; Kakiuchi, N.; Yoshizato, T.; Shiozawa, Y.; Shiraishi, Y.; et al. A genetically defined signature of responsiveness to erlotinib in early-stage pancreatic cancer patients: Results from the CONKO-005 trial. EBioMedicine 2021, 66, 103327. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Song, W.; Cao, R.; Ye, C.; Zhang, L.; Chen, H.; Wang, J.; Shi, Y.; Li, R.; Li, Y.; et al. An EGFR/HER2-targeted conjugate sensitizes gemcitabine-sensitive and resistant pancreatic cancer through different SMAD4-mediated mechanisms. Nat. Commun. 2022, 13, 5506. [Google Scholar] [CrossRef] [PubMed]
- Hufford, D.J. Folk medicine and health culture in contemporary society. Prim. Care Clin. Off. Pract. 1997, 24, 723–741. [Google Scholar] [CrossRef]
- Ling, Z.; Liu, H.; Yang, J.; He, J.-Q.; Yang, S.; Chen, Q.-H. Qianlongtong capsule elevates the Smad4 gene expression in prostate stromal cells. Zhonghua Nan Ke Xue Natl. J. Androl. 2014, 20, 730–733. [Google Scholar]
- Cappuccio, G.; Caiazza, M.; Roca, A.; Melis, D.; Iuliano, A.; Matyas, G.; Rubino, M.; Limongelli, G.; Brunetti-Pierri, N. A pilot clinical trial with losartan in Myhre syndrome. Am. J. Med. Genet. Part A 2021, 185, 702–709. [Google Scholar] [CrossRef] [PubMed]
- Hou, M.-M.; Liu, X.; Wheler, J.; Naing, A.; Hong, D.; Coleman, R.L.; Tsimberidou, A.; Janku, F.; Zinner, R.; Lu, K.; et al. Targeted PI3K/AKT/mTOR therapy for metastatic carcinomas of the cervix: A phase I clinical experience. Oncotarget 2014, 5, 11168–11179. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, S.M.; Koch, R.; Porcu, P.; Oki, Y.; Moskowitz, A.; Perez, M.; Myskowski, P.; Officer, A.; Jaffe, J.D.; Morrow, S.N.; et al. Activity of the PI3K-δ,γ inhibitor duvelisib in a phase 1 trial and preclinical models of T-cell lymphoma. Blood 2018, 131, 888–898. [Google Scholar] [CrossRef] [PubMed]
- Kaley, T.J.; Panageas, K.S.; Mellinghoff, I.K.; Nolan, C.; Gavrilovic, I.T.; DeAngelis, L.M.; Abrey, L.E.; Holland, E.C.; Lassman, A.B. Phase II trial of an AKT inhibitor (perifosine) for recurrent glioblastoma. J. Neuro-Oncol. 2019, 144, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Lin, N.U.; Maurer, M.A.; Chen, H.; Mahvash, A.; Sahin, A.; Akcakanat, A.; Li, Y.; Abramson, V.; Litton, J.; et al. Phase II trial of AKT inhibitor MK-2206 in patients with advanced breast cancer who have tumors with PIK3CA or AKT mutations, and/or PTEN loss/PTEN mutation. Breast Cancer Res. 2019, 21, 78. [Google Scholar] [CrossRef]
- LoRusso, P.M.; Gounder, M.; Jalal, S.I.; André, V.; Kambhampati, S.R.P.; Loizos, N.; Hall, J.; Holzer, T.R.; Nasir, A.; Cosaert, J.; et al. Phase 1 study of narnatumab, an anti-RON receptor monoclonal antibody, in patients with advanced solid tumors. Investig. New Drugs 2017, 35, 442–450. [Google Scholar] [CrossRef]
- Kreuger, I.Z.; Slieker, R.C.; van Groningen, T.; van Doorn, R. Therapeutic Strategies for Targeting CDKN2A Loss in Melanoma. J. Investig. Dermatol. 2023, 143, 18–25.e1. [Google Scholar] [CrossRef]
- Liggett, W.H.; Sidransky, D. Role of the p16 tumor suppressor gene in cancer. J. Clin. Oncol. 1998, 16, 1197–1206. [Google Scholar] [CrossRef]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Stott, F.J.; Bates, S.; James, M.C.; McConnell, B.B.; Starborg, M.; Brookes, S.; Palmero, I.; Ryan, K.; Hara, E.; Vousden, K.H.; et al. The alternative product from the human CDKN2A locus, p14ARF, participates in a regulatory feedback loop with p53 and MDM2. EMBO J. 1998, 17, 5001–5014. [Google Scholar] [CrossRef]
- Xing, X.; Cai, W.; Shi, H.; Wang, Y.; Li, M.; Jiao, J.; Chen, M. The prognostic value of CDKN2A hypermethylation in colorectal cancer: A meta-analysis. Br. J. Cancer 2013, 108, 2542–2548. [Google Scholar] [CrossRef]
- Liu, M.T.; Liu, J.Y.; Su, J. CDKN2A gene in melanoma. Zhonghua Bing Li Xue Za Zhi 2019, 48, 909–912. [Google Scholar]
- Tsuriel, S.; Hannes, V.; Hasona, A.; Raz, M.; Hershkovitz, D. Digital PCR-Based Method for Detecting CDKN2A Loss in Brain Tumours. Mol. Diagn. Ther. 2022, 26, 689–698. [Google Scholar] [CrossRef]
- Ladanyi, M. Implications of P16/CDKN2A deletion in pleural mesotheliomas. Lung Cancer 2005, 49 (Suppl. S1), S95–S98. [Google Scholar] [CrossRef]
- Wang, J.; Patil, V.; Liu, J.; Dogan, H.; Tabatabai, G.S.; Yefet, L.; Behling, F.; Hoffman, E.; Bunda, S.; Yakubov, R.; et al. Increased mRNA expression of CDKN2A is a transcriptomic marker of clinically aggressive meningiomas. Acta Neuropathol. 2023, 146, 145–162. [Google Scholar] [CrossRef]
- Kimura, H.; Klein, A.P.M.; Hruban, R.H.; Roberts, N.J.V. The Role of Inherited Pathogenic CDKN2A Variants in Susceptibility to Pancreatic Cancer. Pancreas 2021, 50, 1123–1130. [Google Scholar] [CrossRef]
- Johnston, S.R.D.; Harbeck, N.; Hegg, R.; Toi, M.; Martin, M.; Shao, Z.M.; Zhang, Q.Y.; Rodriguez, J.L.M.; Campone, M.; Hamilton, E.; et al. Abemaciclib Combined With Endocrine Therapy for the Adjuvant Treatment of HR+, HER2−, Node-Positive, High-Risk, Early Breast Cancer (monarchE). J. Clin. Oncol. 2020, 38, 3987–3998. [Google Scholar] [CrossRef]
- Turner, N.C.; Slamon, D.J.; Ro, J.; Bondarenko, I.; Im, S.-A.; Masuda, N.; Colleoni, M.; DeMichele, A.; Loi, S.; Verma, S.; et al. Overall Survival with Palbociclib and Fulvestrant in Advanced Breast Cancer. N. Engl. J. Med. 2018, 379, 1926–1936. [Google Scholar] [CrossRef]
- Weiss, J.M.; Csoszi, T.; Maglakelidze, M.; Hoyer, R.J.; Beck, J.T.; Gomez, M.D.; Lowczak, A.; Aljumaily, R.; Lima, C.R.; Boccia, R.V.; et al. Myelopreservation with the CDK4/6 inhibitor trilaciclib in patients with small-cell lung cancer receiving first-line chemotherapy: A phase Ib/randomized phase II trial. Ann. Oncol. 2019, 30, 1613–1621. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Tibes, R.; Kadia, T.; Kantarjian, H.; Arellano, M.; Knight, E.A.; Xiong, H.; Qin, Q.; Munasinghe, W.; Roberts-Rapp, L.; et al. Phase 1 dose escalation trial of ilorasertib, a dual Aurora/VEGF receptor kinase inhibitor, in patients with hematologic malignancies. Investig. New Drugs 2015, 33, 870–880. [Google Scholar] [CrossRef]
- Maitland, M.L.; Piha-Paul, S.; Falchook, G.; Kurzrock, R.; Nguyen, L.; Janisch, L.; Karovic, S.; McKee, M.; Hoening, E.; Wong, S.; et al. Clinical pharmacodynamic/exposure characterisation of the multikinase inhibitor ilorasertib (ABT-348) in a phase 1 dose-escalation trial. Br. J. Cancer 2018, 118, 1042–1050. [Google Scholar] [CrossRef] [PubMed]
- Grande, E.; Teulé, A.; Alonso-Gordoa, T.; Jiménez-Fonseca, P.; Benavent, M.; Capdevila, J.; Custodio, A.; Vera, R.; Munarriz, J.; La Casta, A.; et al. The PALBONET Trial: A Phase II Study of Palbociclib in Metastatic Grade 1 and 2 Pancreatic Neuroendocrine Tumors (GETNE-1407). Oncology 2020, 25, 745-e1265. [Google Scholar] [CrossRef] [PubMed]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 16022. [Google Scholar] [CrossRef] [PubMed]
- Singhi, A.D.; Koay, E.J.; Chari, S.T.; Maitra, A. Early Detection of Pancreatic Cancer: Opportunities and Challenges. Gastroenterology 2019, 156, 2024–2040. [Google Scholar] [CrossRef]

| S/No | Gene | Frequency of Mutation in PDACs | Types of Mutation | Amino Acid Residues | References |
|---|---|---|---|---|---|
| 1 | KRAS | 90% | Missense point | G12C, G12D, and G12V | [28,29,30,31] |
| 2 | CDNK2A | 30–40% | Point mutation, deletion and loss of heterozygousity, insertion and frame shift mutation, promoter methylation, splice site mutation | P16INK4A (p16-Leu148) and P14ARF mutations | [32,33] |
| 3 | DPC4/SMAD4 | 50% | Nonsense mutation, missense mutation, frameshift mutation, splice site mutation, deletion and insertion, point mutation, promoter methylation, large rearrangement, silent mutation | MH1 domain: R361C, R361H, R361S, and R361G MH2 domain: R100C, D351N, L384P, P529L. Linker region: E249K, G253V C-terminal region: Q408P and G437E | [19,32] |
| 4 | TP53 | ≥50% | Missense, nonsense, frameshift, splice site, deletion and insertion, promoter methylation, hotspot, wild-type p53. | R17H, R28Q, R273H, R282W, and Y220C | [19,34] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olaoba, O.T.; Adelusi, T.I.; Yang, M.; Maidens, T.; Kimchi, E.T.; Staveley-O’Carroll, K.F.; Li, G. Driver Mutations in Pancreatic Cancer and Opportunities for Targeted Therapy. Cancers 2024, 16, 1808. https://doi.org/10.3390/cancers16101808
Olaoba OT, Adelusi TI, Yang M, Maidens T, Kimchi ET, Staveley-O’Carroll KF, Li G. Driver Mutations in Pancreatic Cancer and Opportunities for Targeted Therapy. Cancers. 2024; 16(10):1808. https://doi.org/10.3390/cancers16101808
Chicago/Turabian StyleOlaoba, Olamide T., Temitope I. Adelusi, Ming Yang, Tessa Maidens, Eric T. Kimchi, Kevin F. Staveley-O’Carroll, and Guangfu Li. 2024. "Driver Mutations in Pancreatic Cancer and Opportunities for Targeted Therapy" Cancers 16, no. 10: 1808. https://doi.org/10.3390/cancers16101808
APA StyleOlaoba, O. T., Adelusi, T. I., Yang, M., Maidens, T., Kimchi, E. T., Staveley-O’Carroll, K. F., & Li, G. (2024). Driver Mutations in Pancreatic Cancer and Opportunities for Targeted Therapy. Cancers, 16(10), 1808. https://doi.org/10.3390/cancers16101808