

Extracellular Signal-Regulated Kinases: One Pathway, Multiple Fates


Abstract
Simple Summary
Abstract
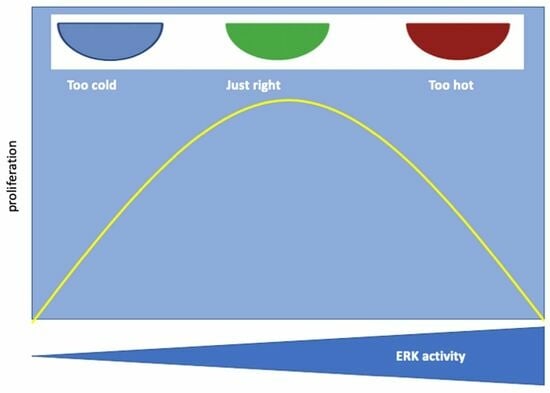
{kind=link}
{kind=link}
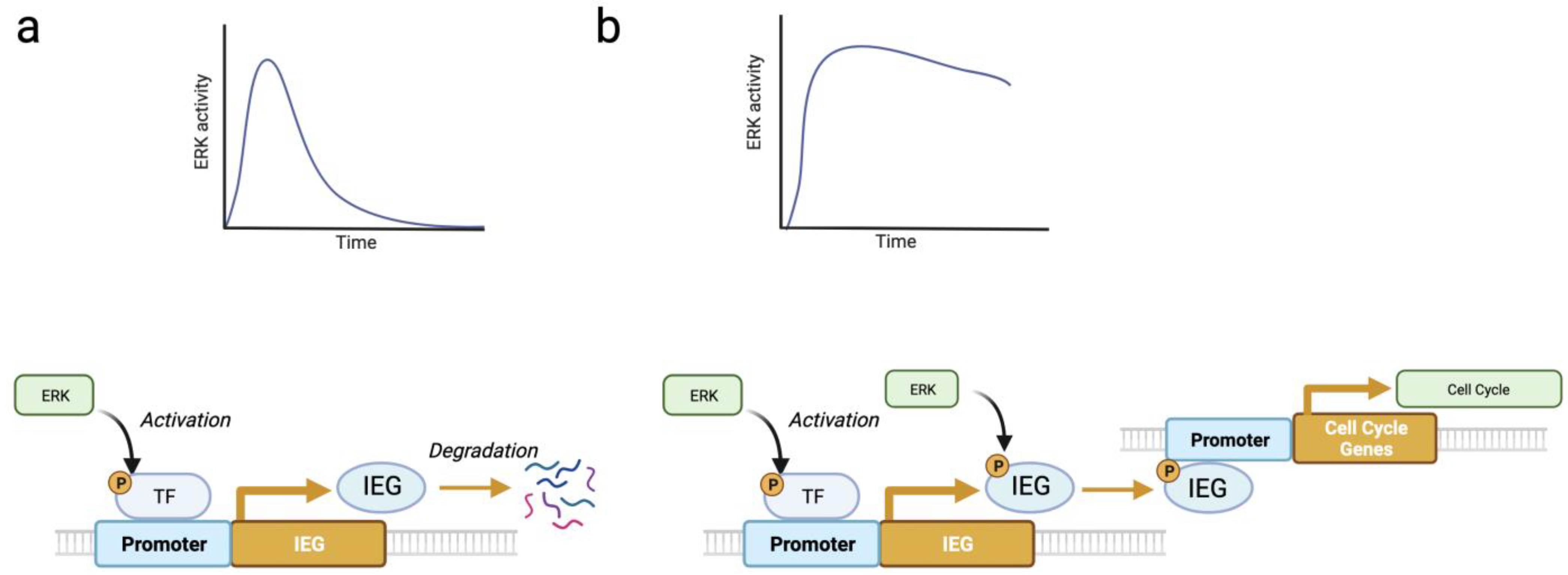{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
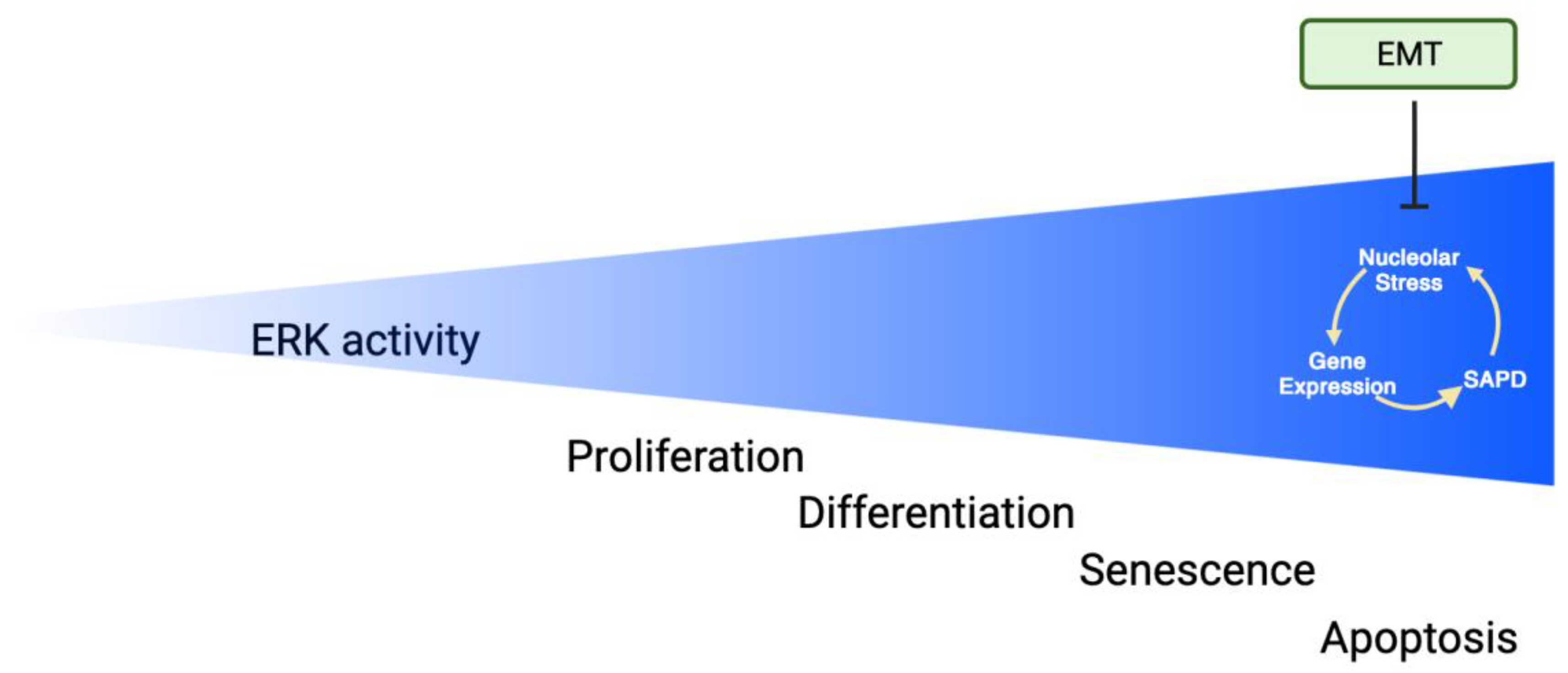{kind=link}
1. Introduction
2. Proliferation and Survival
2.1. ERK Localization in Cell Proliferation and Survival
2.2. ERK Pulses in Cell Proliferation and Survival
3. Differentiation
4. Pluripotency
5. Senescence
6. EMT
7. Apoptosis
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Deschenes-Simard, X.; Kottakis, F.; Meloche, S.; Ferbeyre, G. ERKs in cancer: Friends or foes? Cancer Res. 2014, 74, 412–419. [Google Scholar] [CrossRef] [PubMed]
- von Kriegsheim, A.; Baiocchi, D.; Birtwistle, M.; Sumpton, D.; Bienvenut, W.; Morrice, N.; Yamada, K.; Lamond, A.; Kalna, G.; Orton, R.; et al. Cell fate decisions are specified by the dynamic ERK interactome. Nat. Cell Biol. 2009, 11, 1458–1464. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.; Wang, P.; Cao, P.; Zhu, J.K.; Tao, W.A. Identification of extracellular signal-regulated kinase 1 (ERK1) direct substrates using stable isotope labeled kinase assay-linked phosphoproteomics. Mol. Cell Proteom. 2014, 13, 3199–3210. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.; Chung, M.; Dobrzynski, M.; Fey, D.; Blum, Y.; Lee, S.S.; Peter, M.; Kholodenko, B.N.; Jeon, N.L.; Pertz, O. Frequency modulation of ERK activation dynamics rewires cell fate. Mol. Syst. Biol. 2015, 11, 838. [Google Scholar] [CrossRef] [PubMed]
- Inder, K.; Hancock, J.F. System output of the MAPK module is spatially regulated. Commun. Integr. Biol. 2008, 1, 178–179. [Google Scholar] [CrossRef][Green Version]
- Herrero, A.; Casar, B.; Colon-Bolea, P.; Agudo-Ibanez, L.; Crespo, P. Defined spatiotemporal features of RAS-ERK signals dictate cell fate in MCF-7 mammary epithelial cells. Mol. Biol. Cell 2016, 27, 1958–1968. [Google Scholar] [CrossRef] [PubMed]
- Kholodenko, B.N.; Hancock, J.F.; Kolch, W. Signalling ballet in space and time. Nat. Rev. Mol. Cell Biol. 2010, 11, 414–426. [Google Scholar] [CrossRef]
- Lavoie, H.; Gagnon, J.; Therrien, M. ERK signalling: A master regulator of cell behaviour, life and fate. Nat. Rev. Mol. Cell Biol. 2020, 21, 607–632. [Google Scholar] [CrossRef]
- Boulton, T.G.; Nye, S.H.; Robbins, D.J.; Ip, N.Y.; Radziejewska, E.; Morgenbesser, S.D.; DePinho, R.A.; Panayotatos, N.; Cobb, M.H.; Yancopoulos, G.D. ERKs: A family of protein-serine/threonine kinases that are activated and tyrosine phosphorylated in response to insulin and NGF. Cell 1991, 65, 663–675. [Google Scholar] [CrossRef]
- Marshall, C.J. Specificity of receptor tyrosine kinase signaling: Transient versus sustained extracellular signal-regulated kinase activation. Cell 1995, 80, 179–185. [Google Scholar] [CrossRef]
- Hernandez, M.A.; Patel, B.; Hey, F.; Giblett, S.; Davis, H.; Pritchard, C. Regulation of BRAF protein stability by a negative feedback loop involving the MEK-ERK pathway but not the FBXW7 tumour suppressor. Cell Signal. 2016, 28, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Santos, S.D.; Verveer, P.J.; Bastiaens, P.I. Growth factor-induced MAPK network topology shapes Erk response determining PC-12 cell fate. Nat. Cell Biol. 2007, 9, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Kaur, N.; Lum, M.A.; Lewis, R.E.; Black, A.R.; Black, J.D. A novel antiproliferative PKCalpha-Ras-ERK signaling axis in intestinal epithelial cells. J. Biol. Chem. 2022, 298, 102121. [Google Scholar] [CrossRef] [PubMed]
- Lawan, A.; Al-Harthi, S.; Cadalbert, L.; McCluskey, A.G.; Shweash, M.; Grassia, G.; Grant, A.; Boyd, M.; Currie, S.; Plevin, R. Deletion of the dual specific phosphatase-4 (DUSP-4) gene reveals an essential non-redundant role for MAP kinase phosphatase-2 (MKP-2) in proliferation and cell survival. J. Biol. Chem. 2011, 286, 12933–12943. [Google Scholar] [CrossRef]
- Unni, A.M.; Harbourne, B.; Oh, M.H.; Wild, S.; Ferrarone, J.R.; Lockwood, W.W.; Varmus, H. Hyperactivation of ERK by multiple mechanisms is toxic to RTK-RAS mutation-driven lung adenocarcinoma cells. ELife 2018, 7, e33718. [Google Scholar] [CrossRef] [PubMed]
- Shojaee, S.; Caeser, R.; Buchner, M.; Park, E.; Swaminathan, S.; Hurtz, C.; Geng, H.; Chan, L.N.; Klemm, L.; Hofmann, W.K.; et al. Erk Negative Feedback Control Enables Pre-B Cell Transformation and Represents a Therapeutic Target in Acute Lymphoblastic Leukemia. Cancer Cell 2015, 28, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, S.H.; Harrison, T.A.; Phipps, A.I.; Steinfelder, R.; Trinh, Q.M.; Qu, C.; Banbury, B.L.; Georgeson, P.; Grasso, C.S.; Giannakis, M.; et al. Landscape of somatic single nucleotide variants and indels in colorectal cancer and impact on survival. Nat. Commun. 2020, 11, 3644. [Google Scholar] [CrossRef]
- Holck, S.; Bonde, J.; Pedersen, H.; Petersen, A.A.; Chaube, A.; Nielsen, H.J.; Larsson, L.I. Localization of active, dually phosphorylated extracellular signal-regulated kinase 1 and 2 in colorectal cancer with or without activating BRAF and KRAS mutations. Hum. Pathol. 2016, 54, 37–46. [Google Scholar] [CrossRef]
- Murphy, L.O.; Smith, S.; Chen, R.H.; Fingar, D.C.; Blenis, J. Molecular interpretation of ERK signal duration by immediate early gene products. Nat. Cell Biol. 2002, 4, 556–564. [Google Scholar] [CrossRef]
- Nagashima, T.; Inoue, N.; Yumoto, N.; Saeki, Y.; Magi, S.; Volinsky, N.; Sorkin, A.; Kholodenko, B.N.; Okada-Hatakeyama, M. Feedforward regulation of mRNA stability by prolonged extracellular signal-regulated kinase activity. FEBS J. 2015, 282, 613–629. [Google Scholar] [CrossRef]
- Mylona, A.; Theillet, F.X.; Foster, C.; Cheng, T.M.; Miralles, F.; Bates, P.A.; Selenko, P.; Treisman, R. Opposing effects of Elk-1 multisite phosphorylation shape its response to ERK activation. Science 2016, 354, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.Z.; Ravindran, P.T.; Lim, W.A.; Toettcher, J.E. Tracing Information Flow from Erk to Target Gene Induction Reveals Mechanisms of Dynamic and Combinatorial Control. Mol. Cell 2017, 67, 757–769.e755. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, C.M.; Johnson, L.A.; Piccioni, F.; Townes, A.; Frederick, D.T.; Donahue, M.K.; Narayan, R.; Flaherty, K.T.; Wargo, J.A.; Root, D.E.; et al. A melanocyte lineage program confers resistance to MAP kinase pathway inhibition. Nature 2013, 504, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Sears, R.; Nuckolls, F.; Haura, E.; Taya, Y.; Tamai, K.; Nevins, J.R. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes. Dev. 2000, 14, 2501–2514. [Google Scholar] [CrossRef] [PubMed]
- Stefanovsky, V.Y.; Moss, T. The splice variants of UBF differentially regulate RNA polymerase I transcription elongation in response to ERK phosphorylation. Nucleic Acids Res. 2008, 36, 5093–5101. [Google Scholar] [CrossRef] [PubMed]
- Goetz, E.M.; Ghandi, M.; Treacy, D.J.; Wagle, N.; Garraway, L.A. ERK mutations confer resistance to mitogen-activated protein kinase pathway inhibitors. Cancer Res. 2014, 74, 7079–7089. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef]
- Smorodinsky-Atias, K.; Goshen-Lago, T.; Goldberg-Carp, A.; Melamed, D.; Shir, A.; Mooshayef, N.; Beenstock, J.; Karamansha, Y.; Darlyuk-Saadon, I.; Livnah, O.; et al. Intrinsically active variants of Erk oncogenically transform cells and disclose unexpected autophosphorylation capability that is independent of TEY phosphorylation. Mol. Biol. Cell 2016, 27, 1026–1039. [Google Scholar] [CrossRef]
- Albeck, J.G.; Mills, G.B.; Brugge, J.S. Frequency-modulated pulses of ERK activity transmit quantitative proliferation signals. Mol. Cell 2013, 49, 249–261. [Google Scholar] [CrossRef]
- Lito, P.; Pratilas, C.A.; Joseph, E.W.; Tadi, M.; Halilovic, E.; Zubrowski, M.; Huang, A.; Wong, W.L.; Callahan, M.K.; Merghoub, T.; et al. Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell 2012, 22, 668–682. [Google Scholar] [CrossRef]
- Sale, M.J.; Balmanno, K.; Saxena, J.; Ozono, E.; Wojdyla, K.; McIntyre, R.E.; Gilley, R.; Woroniuk, A.; Howarth, K.D.; Hughes, G.; et al. MEK1/2 inhibitor withdrawal reverses acquired resistance driven by BRAF(V600E) amplification whereas KRAS(G13D) amplification promotes EMT-chemoresistance. Nat. Commun. 2019, 10, 2030. [Google Scholar] [CrossRef] [PubMed]
- Finlay, D.; Healy, V.; Furlong, F.; O′Connell, F.C.; Keon, N.K.; Martin, F. MAP kinase pathway signalling is essential for extracellular matrix determined mammary epithelial cell survival. Cell Death Differ. 2000, 7, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Dehan, E.; Bassermann, F.; Guardavaccaro, D.; Vasiliver-Shamis, G.; Cohen, M.; Lowes, K.N.; Dustin, M.; Huang, D.C.; Taunton, J.; Pagano, M. betaTrCP- and Rsk1/2-mediated degradation of BimEL inhibits apoptosis. Mol. Cell 2009, 33, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.Y.; Zong, C.S.; Xia, W.; Yamaguchi, H.; Ding, Q.; Xie, X.; Lang, J.Y.; Lai, C.C.; Chang, C.J.; Huang, W.C.; et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat. Cell Biol. 2008, 10, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Blasco, R.B.; Francoz, S.; Santamaria, D.; Canamero, M.; Dubus, P.; Charron, J.; Baccarini, M.; Barbacid, M. c-Raf, but Not B-Raf, Is Essential for Development of K-Ras Oncogene-Driven Non-Small Cell Lung Carcinoma. Cancer Cell 2011, 19, 652–663. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Roux, D.; Lenormand, P.; Dowd, S.; Keyse, S.; Pouyssegur, J. Nuclear translocation of p42/p44 mitogen-activated protein kinase is required for growth factor-induced gene expression and cell cycle entry. EMBO J. 1999, 18, 664–674. [Google Scholar] [CrossRef] [PubMed]
- Formstecher, E.; Ramos, J.W.; Fauquet, M.; Calderwood, D.A.; Hsieh, J.C.; Canton, B.; Nguyen, X.T.; Barnier, J.V.; Camonis, J.; Ginsberg, M.H.; et al. PEA-15 mediates cytoplasmic sequestration of ERK MAP kinase. Dev. Cell 2001, 1, 239–250. [Google Scholar] [CrossRef]
- Gaumont-Leclerc, M.F.; Mukhopadhyay, U.K.; Goumard, S.; Ferbeyre, G. PEA-15 is inhibited by adenovirus E1A and plays a role in ERK nuclear export and Ras-induced senescence. J. Biol. Chem. 2004, 279, 46802–46809. [Google Scholar] [CrossRef]
- Plotnikov, A.; Flores, K.; Maik-Rachline, G.; Zehorai, E.; Kapri-Pardes, E.; Berti, D.A.; Hanoch, T.; Besser, M.J.; Seger, R. The nuclear translocation of ERK1/2 as an anticancer target. Nat. Commun. 2015, 6, 6685. [Google Scholar] [CrossRef]
- Arafeh, R.; Flores, K.; Keren-Paz, A.; Maik-Rachline, G.; Gutkind, N.; Rosenberg, S.; Seger, R.; Samuels, Y. Combined inhibition of MEK and nuclear ERK translocation has synergistic antitumor activity in melanoma cells. Sci. Rep. 2017, 7, 16345. [Google Scholar] [CrossRef]
- Kwon, Y.; Mehta, S.; Clark, M.; Walters, G.; Zhong, Y.; Lee, H.N.; Sunahara, R.K.; Zhang, J. Non-canonical beta-adrenergic activation of ERK at endosomes. Nature 2022, 611, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Muta, Y.; Fujita, Y.; Sumiyama, K.; Sakurai, A.; Taketo, M.M.; Chiba, T.; Seno, H.; Aoki, K.; Matsuda, M.; Imajo, M. Composite regulation of ERK activity dynamics underlying tumour-specific traits in the intestine. Nat. Commun. 2018, 9, 2174. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Sohn, C.H.; Dalal, C.K.; Cai, L.; Elowitz, M.B. Combinatorial gene regulation by modulation of relative pulse timing. Nature 2015, 527, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Potoyan, D.A.; Wolynes, P.G. Molecular stripping, targets and decoys as modulators of oscillations in the NF-kappaB/IkappaBalpha/DNA genetic network. J. R. Soc. Interface R. Soc. 2016, 13, 20160606. [Google Scholar] [CrossRef] [PubMed]
- Ender, P.; Gagliardi, P.A.; Dobrzynski, M.; Frismantiene, A.; Dessauges, C.; Hohener, T.; Jacques, M.A.; Cohen, A.R.; Pertz, O. Spatiotemporal control of ERK pulse frequency coordinates fate decisions during mammary acinar morphogenesis. Dev. Cell 2022, 57, 2153–2167.e2156. [Google Scholar] [CrossRef] [PubMed]
- Pokrass, M.J.; Ryan, K.A.; Xin, T.; Pielstick, B.; Timp, W.; Greco, V.; Regot, S. Cell-Cycle-Dependent ERK Signaling Dynamics Direct Fate Specification in the Mammalian Preimplantation Embryo. Dev. Cell 2020, 55, 328–340.e325. [Google Scholar] [CrossRef] [PubMed]
- Byun, M.R.; Kim, A.R.; Hwang, J.H.; Kim, K.M.; Hwang, E.S.; Hong, J.H. FGF2 stimulates osteogenic differentiation through ERK induced TAZ expression. Bone 2014, 58, 72–80. [Google Scholar] [CrossRef]
- Fremin, C.; Saba-El-Leil, M.K.; Levesque, K.; Ang, S.L.; Meloche, S. Functional Redundancy of ERK1 and ERK2 MAP Kinases during Development. Cell Rep. 2015, 12, 913–921. [Google Scholar] [CrossRef]
- Newbern, J.M.; Li, X.; Shoemaker, S.E.; Zhou, J.; Zhong, J.; Wu, Y.; Bonder, D.; Hollenback, S.; Coppola, G.; Geschwind, D.H.; et al. Specific functions for ERK/MAPK signaling during PNS development. Neuron 2011, 69, 91–105. [Google Scholar] [CrossRef]
- Parada, C.; Han, D.; Grimaldi, A.; Sarrion, P.; Park, S.S.; Pelikan, R.; Sanchez-Lara, P.A.; Chai, Y. Disruption of the ERK/MAPK pathway in neural crest cells as a potential cause of Pierre Robin sequence. Development 2015, 142, 3734–3745. [Google Scholar] [CrossRef]
- Offermann, B.; Knauer, S.; Singh, A.; Fernandez-Cachon, M.L.; Klose, M.; Kowar, S.; Busch, H.; Boerries, M. Boolean Modeling Reveals the Necessity of Transcriptional Regulation for Bistability in PC12 Cell Differentiation. Front. Genet. 2016, 7, 44. [Google Scholar] [CrossRef] [PubMed]
- Johnson, H.E.; Toettcher, J.E. Signaling Dynamics Control Cell Fate in the Early Drosophila Embryo. Dev. Cell 2019, 48, 361–370.e363. [Google Scholar] [CrossRef] [PubMed]
- Doncic, A.; Skotheim, J.M. Feedforward regulation ensures stability and rapid reversibility of a cellular state. Mol. Cell 2013, 50, 856–868. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gillies, T.E.; Pargett, M.; Minguet, M.; Davies, A.E.; Albeck, J.G. Linear Integration of ERK Activity Predominates over Persistence Detection in Fra-1 Regulation. Cell Syst. 2017, 5, 549–563.e545. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.; Smith, A. Capturing pluripotency. Cell 2008, 132, 532–536. [Google Scholar] [CrossRef] [PubMed]
- Nichols, J.; Smith, A. Naive and primed pluripotent states. Cell Stem Cell 2009, 4, 487–492. [Google Scholar] [CrossRef] [PubMed]
- Pond, K.W.; Morris, J.M.; Alkhimenok, O.; Varghese, R.P.; Cabel, C.R.; Ellis, N.A.; Chakrabarti, J.; Zavros, Y.; Merchant, J.L.; Thorne, C.A.; et al. Live-cell imaging in human colonic monolayers reveals ERK waves limit the stem cell compartment to maintain epithelial homeostasis. ELife 2022, 11, e78837. [Google Scholar] [CrossRef]
- Mulas, C.; Kalkan, T.; von Meyenn, F.; Leitch, H.G.; Nichols, J.; Smith, A. Defined conditions for propagation and manipulation of mouse embryonic stem cells. Development 2019, 146, dev173146. [Google Scholar] [CrossRef]
- Chappell, J.; Sun, Y.; Singh, A.; Dalton, S. MYC/MAX control ERK signaling and pluripotency by regulation of dual-specificity phosphatases 2 and 7. Genes. Dev. 2013, 27, 725–733. [Google Scholar] [CrossRef]
- Mzoughi, S.; Zhang, J.; Hequet, D.; Teo, S.X.; Fang, H.; Xing, Q.R.; Bezzi, M.; Seah, M.K.Y.; Ong, S.L.M.; Shin, E.M.; et al. PRDM15 safeguards naive pluripotency by transcriptionally regulating WNT and MAPK-ERK signaling. Nat. Genet. 2017, 49, 1354–1363. [Google Scholar] [CrossRef]
- Nett, I.R.; Mulas, C.; Gatto, L.; Lilley, K.S.; Smith, A. Negative feedback via RSK modulates Erk-dependent progression from naive pluripotency. EMBO Rep. 2018, 19, e45642. [Google Scholar] [CrossRef]
- Gu, H.; Li, Q.; Huang, S.; Lu, W.; Cheng, F.; Gao, P.; Wang, C.; Miao, L.; Mei, Y.; Wu, M. Mitochondrial E3 ligase March5 maintains stemness of mouse ES cells via suppression of ERK signalling. Nat. Commun. 2015, 6, 7112. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, W.B.; Brickman, J.M. Erk signaling suppresses embryonic stem cell self-renewal to specify endoderm. Cell Rep. 2014, 9, 2056–2070. [Google Scholar] [CrossRef] [PubMed]
- Tee, W.W.; Shen, S.S.; Oksuz, O.; Narendra, V.; Reinberg, D. Erk1/2 activity promotes chromatin features and RNAPII phosphorylation at developmental promoters in mouse ESCs. Cell 2014, 156, 678–690. [Google Scholar] [CrossRef] [PubMed]
- Brons, I.G.; Smithers, L.E.; Trotter, M.W.; Rugg-Gunn, P.; Sun, B.; Chuva de Sousa Lopes, S.M.; Howlett, S.K.; Clarkson, A.; Ahrlund-Richter, L.; Pedersen, R.A.; et al. Derivation of pluripotent epiblast stem cells from mammalian embryos. Nature 2007, 448, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Tesar, P.J.; Chenoweth, J.G.; Brook, F.A.; Davies, T.J.; Evans, E.P.; Mack, D.L.; Gardner, R.L.; McKay, R.D. New cell lines from mouse epiblast share defining features with human embryonic stem cells. Nature 2007, 448, 196–199. [Google Scholar] [CrossRef]
- Goke, J.; Chan, Y.S.; Yan, J.; Vingron, M.; Ng, H.H. Genome-wide kinase-chromatin interactions reveal the regulatory network of ERK signaling in human embryonic stem cells. Mol. Cell 2013, 50, 844–855. [Google Scholar] [CrossRef]
- Lu, H.; Samanta, D.; Xiang, L.; Zhang, H.; Hu, H.; Chen, I.; Bullen, J.W.; Semenza, G.L. Chemotherapy triggers HIF-1-dependent glutathione synthesis and copper chelation that induces the breast cancer stem cell phenotype. Proc. Natl. Acad. Sci. USA 2015, 112, E4600–E4609. [Google Scholar] [CrossRef]
- Lu, H.; Tran, L.; Park, Y.; Chen, I.; Lan, J.; Xie, Y.; Semenza, G.L. Reciprocal Regulation of DUSP9 and DUSP16 Expression by HIF1 Controls ERK and p38 MAP Kinase Activity and Mediates Chemotherapy-Induced Breast Cancer Stem Cell Enrichment. Cancer Res. 2018, 78, 4191–4202. [Google Scholar] [CrossRef]
- Deschenes-Simard, X.; Gaumont-Leclerc, M.F.; Bourdeau, V.; Lessard, F.; Moiseeva, O.; Forest, V.; Igelmann, S.; Mallette, F.A.; Saba-El-Leil, M.K.; Meloche, S.; et al. Tumor suppressor activity of the ERK/MAPK pathway by promoting selective protein degradation. Genes. Dev. 2013, 27, 900–915. [Google Scholar] [CrossRef]
- Deschenes-Simard, X.; Parisotto, M.; Rowell, M.C.; Le Calve, B.; Igelmann, S.; Moineau-Vallee, K.; Saint-Germain, E.; Kalegari, P.; Bourdeau, V.; Kottakis, F.; et al. Circumventing senescence is associated with stem cell properties and metformin sensitivity. Aging Cell 2019, 18, e12889. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Martinez, E.; Morrisroe, C.; Sharrocks, A.D. The ubiquitin ligase UBE3A dampens ERK pathway signalling in HPV E6 transformed HeLa cells. PLoS ONE 2015, 10, e0119366. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lee, S.W.; Fang, L.; Igarashi, M.; Ouchi, T.; Lu, K.P.; Aaronson, S.A. Sustained activation of Ras/Raf/mitogen-activated protein kinase cascade by the tumor suppressor p53. Proc. Natl. Acad. Sci. USA 2000, 97, 8302–8305. [Google Scholar] [CrossRef] [PubMed]
- Deschenes-Simard, X.; Lessard, F.; Gaumont-Leclerc, M.F.; Bardeesy, N.; Ferbeyre, G. Cellular senescence and protein degradation: Breaking down cancer. Cell Cycle 2014, 13, 1840–1858. [Google Scholar] [CrossRef] [PubMed]
- Lessard, F.; Igelmann, S.; Trahan, C.; Huot, G.; Saint-Germain, E.; Mignacca, L.; Del Toro, N.; Lopes-Paciencia, S.; Le Calve, B.; Montero, M.; et al. Senescence-associated ribosome biogenesis defects contributes to cell cycle arrest through the Rb pathway. Nat. Cell Biol. 2018, 20, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Igelmann, S.; Lessard, F.; Uchenunu, O.; Bouchard, J.; Fernandez-Ruiz, A.; Rowell, M.C.; Lopes-Paciencia, S.; Papadopoli, D.; Fouillen, A.; Ponce, K.J.; et al. A hydride transfer complex reprograms NAD metabolism and bypasses senescence. Mol. Cell 2021, 81, 3848–3865.e3819. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Y.; Hug, C.; Reyes, J.; Tian, C.; Gerosa, L.; Frohlich, F.; Ponsioen, B.; Snippert, H.J.G.; Spencer, S.L.; Jambhekar, A.; et al. Multi-range ERK responses shape the proliferative trajectory of single cells following oncogene induction. Cell Rep. 2023, 42, 112252. [Google Scholar] [CrossRef]
- Rowell, M.C.; Deschenes-Simard, X.; Lopes-Paciencia, S.; Le Calve, B.; Kalegari, P.; Mignacca, L.; Fernandez-Ruiz, A.; Guillon, J.; Lessard, F.; Bourdeau, V.; et al. Targeting ribosome biogenesis reinforces ERK-dependent senescence in pancreatic cancer. Cell Cycle 2023, 22, 2172–2193. [Google Scholar] [CrossRef]
- Hong, S.K.; Wu, P.K.; Park, J.I. A cellular threshold for active ERK1/2 levels determines Raf/MEK/ERK-mediated growth arrest versus death responses. Cell Signal. 2018, 42, 11–20. [Google Scholar] [CrossRef]
- Lin, Y.K.; Wu, W.; Ponce, R.K.; Kim, J.W.; Okimoto, R.A. Negative MAPK-ERK regulation sustains CIC-DUX4 oncoprotein expression in undifferentiated sarcoma. Proc. Natl. Acad. Sci. USA 2020, 117, 20776–20784. [Google Scholar] [CrossRef]
- Ingram, K.; Samson, S.C.; Zewdu, R.; Zitnay, R.G.; Snyder, E.L.; Mendoza, M.C. NKX2-1 controls lung cancer progression by inducing DUSP6 to dampen ERK activity. Oncogene 2022, 41, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Young, M.J.; Li, R.; Jain, S.; Wernitznig, A.; Krill-Burger, J.M.; Lemke, C.T.; Monducci, D.; Rodriguez, D.J.; Chang, L.; et al. Paralog knockout profiling identifies DUSP4 and DUSP6 as a digenic dependence in MAPK pathway-driven cancers. Nat. Genet. 2021, 53, 1664–1672. [Google Scholar] [CrossRef]
- Kidger, A.M.; Rushworth, L.K.; Stellzig, J.; Davidson, J.; Bryant, C.J.; Bayley, C.; Caddye, E.; Rogers, T.; Keyse, S.M.; Caunt, C.J. Dual-specificity phosphatase 5 controls the localized inhibition, propagation, and transforming potential of ERK signaling. Proc. Natl. Acad. Sci. USA 2017, 114, E317–E326. [Google Scholar] [CrossRef]
- Sangrar, W.; Shi, C.; Mullins, G.; LeBrun, D.; Ingalls, B.; Greer, P.A. Amplified Ras-MAPK signal states correlate with accelerated EGFR internalization, cytostasis and delayed HER2 tumor onset in Fer-deficient model systems. Oncogene 2015, 34, 4109–4117. [Google Scholar] [CrossRef] [PubMed]
- Moniz, S.; Verissimo, F.; Matos, P.; Brazao, R.; Silva, E.; Kotelevets, L.; Chastre, E.; Gespach, C.; Jordan, P. Protein kinase WNK2 inhibits cell proliferation by negatively modulating the activation of MEK1/ERK1/2. Oncogene 2007, 26, 6071–6081. [Google Scholar] [CrossRef] [PubMed]
- Shankar, S.; Tien, J.C.; Siebenaler, R.F.; Chugh, S.; Dommeti, V.L.; Zelenka-Wang, S.; Wang, X.M.; Apel, I.J.; Waninger, J.; Eyunni, S.; et al. An essential role for Argonaute 2 in EGFR-KRAS signaling in pancreatic cancer development. Nat. Commun. 2020, 11, 2817. [Google Scholar] [CrossRef] [PubMed]
- Tien, J.C.; Chugh, S.; Goodrum, A.E.; Cheng, Y.; Mannan, R.; Zhang, Y.; Wang, L.; Dommeti, V.L.; Wang, X.; Xu, A.; et al. AGO2 promotes tumor progression in KRAS-driven mouse models of non-small cell lung cancer. Proc. Natl. Acad. Sci. USA 2021, 118, e2026104118. [Google Scholar] [CrossRef]
- Wu, P.K.; Hong, S.K.; Park, J.I. Steady-State Levels of Phosphorylated Mitogen-Activated Protein Kinase Kinase 1/2 Determined by Mortalin/HSPA9 and Protein Phosphatase 1 Alpha in KRAS and BRAF Tumor Cells. Mol. Cell Biol. 2017, 37, e00061-17. [Google Scholar] [CrossRef]
- Kasitinon, S.Y.; Eskiocak, U.; Martin, M.; Bezwada, D.; Khivansara, V.; Tasdogan, A.; Zhao, Z.; Mathews, T.; Aurora, A.B.; Morrison, S.J. TRPML1 Promotes Protein Homeostasis in Melanoma Cells by Negatively Regulating MAPK and mTORC1 Signaling. Cell Rep. 2019, 28, 2293–2305.e2299. [Google Scholar] [CrossRef]
- Iwamoto, N.; D′Alessandro, L.A.; Depner, S.; Hahn, B.; Kramer, B.A.; Lucarelli, P.; Vlasov, A.; Stepath, M.; Bohm, M.E.; Deharde, D.; et al. Context-specific flow through the MEK/ERK module produces cell- and ligand-specific patterns of ERK single and double phosphorylation. Sci. Signal 2016, 9, ra13. [Google Scholar] [CrossRef]
- Buonato, J.M.; Lazzara, M.J. ERK1/2 blockade prevents epithelial-mesenchymal transition in lung cancer cells and promotes their sensitivity to EGFR inhibition. Cancer Res. 2014, 74, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Caramel, J.; Papadogeorgakis, E.; Hill, L.; Browne, G.J.; Richard, G.; Wierinckx, A.; Saldanha, G.; Osborne, J.; Hutchinson, P.; Tse, G.; et al. A switch in the expression of embryonic EMT-inducers drives the development of malignant melanoma. Cancer Cell 2013, 24, 466–480. [Google Scholar] [CrossRef] [PubMed]
- Doehn, U.; Hauge, C.; Frank, S.R.; Jensen, C.J.; Duda, K.; Nielsen, J.V.; Cohen, M.S.; Johansen, J.V.; Winther, B.R.; Lund, L.R.; et al. RSK is a principal effector of the RAS-ERK pathway for eliciting a coordinate promotile/invasive gene program and phenotype in epithelial cells. Mol. Cell 2009, 35, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.; Dimitri, C.A.; Yoon, S.O.; Dowdle, W.; Blenis, J. ERK2 but not ERK1 induces epithelial-to-mesenchymal transformation via DEF motif-dependent signaling events. Mol. Cell 2010, 38, 114–127. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Guin, S.; Padhye, S.S.; Zhou, Y.Q.; Zhang, R.W.; Wang, M.H. Ribosomal protein S6 kinase (RSK)-2 as a central effector molecule in RON receptor tyrosine kinase mediated epithelial to mesenchymal transition induced by macrophage-stimulating protein. Mol. Cancer 2011, 10, 66. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, K.; Kubota, Y.; Nakamura, T.; Weng, J.S.; Tomida, T.; Saito, H.; Takekawa, M. MCRIP1, an ERK substrate, mediates ERK-induced gene silencing during epithelial-mesenchymal transition by regulating the co-repressor CtBP. Mol. Cell 2015, 58, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; El-Naggar, S.; Darling, D.S.; Higashi, Y.; Dean, D.C. Zeb1 links epithelial-mesenchymal transition and cellular senescence. Development 2008, 135, 579–588. [Google Scholar] [CrossRef]
- Liu, Y.; Lu, X.; Huang, L.; Wang, W.; Jiang, G.; Dean, K.C.; Clem, B.; Telang, S.; Jenson, A.B.; Cuatrecasas, M.; et al. Different thresholds of ZEB1 are required for Ras-mediated tumour initiation and metastasis. Nat. Commun. 2014, 5, 5660. [Google Scholar] [CrossRef]
- Ansieau, S.; Bastid, J.; Doreau, A.; Morel, A.P.; Bouchet, B.P.; Thomas, C.; Fauvet, F.; Puisieux, I.; Doglioni, C.; Piccinin, S.; et al. Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer Cell 2008, 14, 79–89. [Google Scholar] [CrossRef]
- Weiss, M.B.; Abel, E.V.; Mayberry, M.M.; Basile, K.J.; Berger, A.C.; Aplin, A.E. TWIST1 is an ERK1/2 effector that promotes invasion and regulates MMP-1 expression in human melanoma cells. Cancer Res. 2012, 72, 6382–6392. [Google Scholar] [CrossRef]
- Principe, D.R.; Diaz, A.M.; Torres, C.; Mangan, R.J.; DeCant, B.; McKinney, R.; Tsao, M.S.; Lowy, A.; Munshi, H.G.; Jung, B.; et al. TGFbeta engages MEK/ERK to differentially regulate benign and malignant pancreas cell function. Oncogene 2017, 36, 4336–4348. [Google Scholar] [CrossRef] [PubMed]
- Deschenes-Simard, X.; Rowell, M.C.; Ferbeyre, G. Metformin turns off the metabolic switch of pancreatic cancer. Aging 2019, 11, 10793–10795. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, M.E.; DeNicola, G.M.; Martins, C.P.; Jacobetz, M.A.; Maitra, A.; Hruban, R.H.; Tuveson, D.A. Cellular features of senescence during the evolution of human and murine ductal pancreatic cancer. Oncogene 2012, 31, 1599–1608. [Google Scholar] [CrossRef] [PubMed]
- Guerra, C.; Collado, M.; Navas, C.; Schuhmacher, A.J.; Hernandez-Porras, I.; Canamero, M.; Rodriguez-Justo, M.; Serrano, M.; Barbacid, M. Pancreatitis-induced inflammation contributes to pancreatic cancer by inhibiting oncogene-induced senescence. Cancer Cell 2011, 19, 728–739. [Google Scholar] [CrossRef] [PubMed]
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. EMT and dissemination precede pancreatic tumor formation. Cell 2012, 148, 349–361. [Google Scholar] [CrossRef]
- Blaj, C.; Schmidt, E.M.; Lamprecht, S.; Hermeking, H.; Jung, A.; Kirchner, T.; Horst, D. Oncogenic Effects of High MAPK Activity in Colorectal Cancer Mark Progenitor Cells and Persist Irrespective of RAS Mutations. Cancer Res. 2017, 77, 1763–1774. [Google Scholar] [CrossRef]
- Chiu, L.Y.; Hsin, I.L.; Yang, T.Y.; Sung, W.W.; Chi, J.Y.; Chang, J.T.; Ko, J.L.; Sheu, G.T. The ERK-ZEB1 pathway mediates epithelial-mesenchymal transition in pemetrexed resistant lung cancer cells with suppression by vinca alkaloids. Oncogene 2017, 36, 242–253. [Google Scholar] [CrossRef]
- Latil, M.; Nassar, D.; Beck, B.; Boumahdi, S.; Wang, L.; Brisebarre, A.; Dubois, C.; Nkusi, E.; Lenglez, S.; Checinska, A.; et al. Cell-Type-Specific Chromatin States Differentially Prime Squamous Cell Carcinoma Tumor-Initiating Cells for Epithelial to Mesenchymal Transition. Cell Stem Cell 2017, 20, 191–204.e195. [Google Scholar] [CrossRef]
- Sugiura, R.; Satoh, R.; Takasaki, T. ERK: A Double-Edged Sword in Cancer. ERK-Dependent Apoptosis as a Potential Therapeutic Strategy for Cancer. Cells 2021, 10, 2509. [Google Scholar] [CrossRef]
- Shin, S.; Buel, G.R.; Wolgamott, L.; Plas, D.R.; Asara, J.M.; Blenis, J.; Yoon, S.O. ERK2 Mediates Metabolic Stress Response to Regulate Cell Fate. Mol. Cell 2015, 59, 382–398. [Google Scholar] [CrossRef]
- Leung, G.P.; Feng, T.; Sigoillot, F.D.; Geyer, F.C.; Shirley, M.D.; Ruddy, D.A.; Rakiec, D.P.; Freeman, A.K.; Engelman, J.A.; Jaskelioff, M.; et al. Hyperactivation of MAPK Signaling Is Deleterious to RAS/RAF-mutant Melanoma. Mol. Cancer Res. 2019, 17, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Ecker, V.; Brandmeier, L.; Stumpf, M.; Giansanti, P.; Moreira, A.V.; Pfeuffer, L.; Fens, M.; Lu, J.; Kuster, B.; Engleitner, T.; et al. Negative feedback regulation of MAPK signaling is an important driver of chronic lymphocytic leukemia progression. Cell Rep. 2023, 42, 113017. [Google Scholar] [CrossRef] [PubMed]
- Viale, A.; Pettazzoni, P.; Lyssiotis, C.A.; Ying, H.; Sanchez, N.; Marchesini, M.; Carugo, A.; Green, T.; Seth, S.; Giuliani, V.; et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 2014, 514, 628–632. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deschênes-Simard, X.; Malleshaiah, M.; Ferbeyre, G. Extracellular Signal-Regulated Kinases: One Pathway, Multiple Fates. Cancers 2024, 16, 95. https://doi.org/10.3390/cancers16010095
Deschênes-Simard X, Malleshaiah M, Ferbeyre G. Extracellular Signal-Regulated Kinases: One Pathway, Multiple Fates. Cancers. 2024; 16(1):95. https://doi.org/10.3390/cancers16010095
Chicago/Turabian StyleDeschênes-Simard, Xavier, Mohan Malleshaiah, and Gerardo Ferbeyre. 2024. "Extracellular Signal-Regulated Kinases: One Pathway, Multiple Fates" Cancers 16, no. 1: 95. https://doi.org/10.3390/cancers16010095
APA StyleDeschênes-Simard, X., Malleshaiah, M., & Ferbeyre, G. (2024). Extracellular Signal-Regulated Kinases: One Pathway, Multiple Fates. Cancers, 16(1), 95. https://doi.org/10.3390/cancers16010095