
A New Hypothesis Describing the Pathogenesis of Oral Mucosal Injury Associated with the Mammalian Target of Rapamycin (mTOR) Inhibitors



Abstract
:Simple Summary
Abstract
1. Introduction
- Do mTORi cause direct cell death and, if yes, by what mechanism?
- If mIAS is initiated by mTORi-mediated direct cell death, why is the mucosal lesion presentation different from conventional cytotoxic therapy?
- Is there a relationship between the level of mTORi immunosuppression and mIAS?
- What drives the anatomic site predilection for mIAS and why, given the systemic nature and pharmacodynamics of treatment, and are oral lesions not widespread?
- Is there a relationship between the dermatologic toxicities of mTORi and mIAS?
- Why is the oral mucosal predisposed to mTORi-mediated injury compared to another stratified squamous epithelium, i.e., the vaginal mucosa?
- Is mIAS primarily mediated by the mTORC1 or mTORC2 pathways? Or does it require both?
- How closely does mIAS resemble aphthous pathobiologically?
- Is autophagy vs. apoptosis a component of mIAS pathogenesis?
- Is the efficacy of corticosteroids as an intervention for mIAS independent of their immunosuppressive activity?
2. Background
3. mTOR Inhibitor Stomatitis Background
3.1. Clinical Presentation, Incidence and Risk
3.2. mIAS Trajectory and Pharmacokinetics
3.3. mIAS Pathogenesis
3.4. Initiation of mIAS
3.5. Ulceration and Resolution and Rationale for Steroids
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed]
- Seto, B. Rapamycin and mTOR: A serendipitous discovery and implications in breast cancer. Clin. Transl. Med. 2012, 1, 29. [Google Scholar] [CrossRef] [PubMed]
- Hobby, G.; Clark, R.; Woywodt, A. A treasure from a barren island: The discovery of rapamycin. Clin. Kidney J. 2022, 15, 1971–1972. [Google Scholar] [CrossRef] [PubMed]
- Samanta, D. Letter to the Editor. Indian J. Cancer 2017, 53, 697–698. [Google Scholar] [CrossRef] [PubMed]
- Douros, J.; Suffness, M. New antitumor substances of natural origin. Cancer Treat. Rev. 1981, 8, 63–87. [Google Scholar] [CrossRef] [PubMed]
- Jhanwar-Uniyal, M.; Amin, A.G.; Cooper, J.B.; Das, K.; Schmidt, M.H.; Murali, R. Discrete signaling mechanisms of mTORC1 and mTORC2: Connected yet apart in cellular and molecular aspects. Adv. Biol. Reg. 2017, 64, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Kurdi, A.; De Meyer, R.Y.; Martinet, W. Potential therapeutic effects of mTOR inhibition in atherosclerosis. Br. J. Clin. Pharm. 2016, 82, 1267–1279. [Google Scholar] [CrossRef]
- Schreiber, K.H.; Arriola Apelo, S.I.; Yu, D.; Brinkman, J.A.; Velarde, M.C.; Syed, F.A.; Liao, C.Y.; Baar, E.L.; Carbajal, K.A.; Sherman, D.S.; et al. A novel rapamycin analog is highly selective for mTORC1 in vivo. Nat. Commun. 2019, 10, 3194. [Google Scholar] [CrossRef]
- Van Gelder, T.; Ter Meulen, C.G.; Hene, R.; Weimer, W.; Hoitsma, A. Oral ulcers in kidney transplant recipients treated with sirolimus and mycophenolate mofetil. Transplant 2003, 75, 786–791. [Google Scholar] [CrossRef]
- Sonis, S.; Treister, N.; Chawla, S.; Demetri, G.; Haluska, F. Preliminary characterization of oral lesions associated with inhibitors of Mammalian Target of Rapamycin in cancer patients. Cancer 2010, 116, 210–215. [Google Scholar] [CrossRef]
- Martins, F.; de Oliveirs, M.A.; Wang, Q.; Sonis, S.; Gallottini, M.; George, S.; Treister, N. A review of oral toxicity associated with mTOR inhibitor therapy in cancer patients. Oral Oncol. 2013, 49, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Shameem, R.; Lacouture, M.; Wu, S. Incidence and risk of high-grade stomatitis with mTOR inhibitors in cancer patients. Cancer Investig. 2015, 33, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Villa, A.; Aboalela, A.; Luskin, K.A.; Cutler, C.S.; Sonis, S.T.; Woo, S.B.; Peterson, D.E.; Treister, N.S. Mammalian Target of Rapamycin Inhibitor-associated stomatitis in hematopoietic stem cell transplantation patients receiving sirolimus prophylaxis for graft-versus-host disease. Biol. Blood Marrow Transplant. 2015, 21, 503–508. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, M.A.; Martins E Martins, F.; Wang, Q.; Sonis, S.; Demetri, G.; George, S.; Butrynski, J.; Treister, N.S. Clinical presentation and management of mTOR inhibitor-associated stomatitis. Oral Oncol. 2011, 47, 998–1003. [Google Scholar] [CrossRef]
- Baur, B.; Oroszian, M.; Carrel, T.; Mohacs, P. Efficacy and safety of sirolimus and everolimus in heart transplant patients: A retrospective analysis. Transplant. Proc. 2011, 43, 1853–1861. [Google Scholar] [CrossRef]
- Hwangbo, Y.; Lee, E.Y. Acute hyperglycemia associated with anti-cancer medication. Endocrinol. Metab. 2017, 32, 23–29. [Google Scholar] [CrossRef]
- Rugo, H.S.; Hortobagy, G.N.; Yao, J.; Pavel, M.; Ravaud, A.; Franz, D.; Ringeisen, F.; Gallo, J.; Rouyrre, N.; Anak, O.; et al. Meta-analysis of stomatitis in clinical studies of everolimus: Incidence and relationship with efficacy. Ann. Oncol. 2016, 27, 519–525. [Google Scholar] [CrossRef]
- Ferte, C.; Paci, A.; Zizi, M.; Gonzales, D.B.; Goubar, A.; Gomez-Roca, C.; Massard, C.; Sahmoud, T.; Andre, F.; Soria, J. Natural history, management and pharmacokinetics of everolimus-induced-oral ulcers. Insights into compliance issues. Eur. J. Cancer 2011, 47, 2249–2255. [Google Scholar] [CrossRef]
- Sonis, S.; Andreotta, P.W.; Lyng, G. On the pathogenesis of mTOR inhibitor-associated stomatitis (mIAS)—Studies using an organotypic model of the oral mucosa. Oral Dis. 2017, 23, 247–252. [Google Scholar] [CrossRef]
- Peterson, D.E.; O’Shaughnessy, J.A.; Rugo, H.S.; Elad, S.; Schubert, M.M.; Viet, C.T.; Campbell-Baird, C.; Hronek, J.; Seery, V.; Divers, J.; et al. Oral mucosal injury caused by mammalian target of rapamycin inhibitors: Emerging perspectives on pathobiology and impact on clinical practice. Cancer Med. 2016, 5, 1897–1907. [Google Scholar] [CrossRef]
- Al-Samadi, A.; Drozd, A.; Salem, A.; Hietanen, J.; Häyrinen-Immonen, R.; Konttinen, Y.T. Epithelial Cell Apoptosis in Recurrent Aphthous Ulcers. J. Dent. Res. 2015, 94, 928–935. [Google Scholar] [CrossRef] [PubMed]
- Sonis, S.T. New thoughts on the initiation of mucositis. Oral Dis. 2010, 16, 597–600. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Banakanakere, M.R.; Hosur, K.B.; Galicia, J.C.; Martin, M.; Kinane, D.F. Mammalian target of rapamycin (mTOR) regulates TLR3 induced cytokines in human oral keratinocytes. Mol. Immunol. 2010, 48, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, U.; Groscurth, P. Morphological features of cell death. News Physiol. Sci. 2004, 19, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Kaloni, D.; Diepstraten, S.T.; Strasser, A.; Kelly, G.L. BCL-2 protein family: Attractive targets for cancer therapy. Apoptosis 2023, 28, 20–38. [Google Scholar] [CrossRef] [PubMed]
- Ciołczyk-Wierzbicka, D.; Zarzycka, M.; Gil, D.; Laidler, P. mTOR inhibitor Everolimus-induced apoptosis in melanoma cells. J. Cell Commun. Signal. 2019, 13, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Bossenmeyer-Pourié, C.; Kannan, R.; Ribieras, S.; Wendling, C.; Stoll, I.; Thim, L.; Tomasetto, C.; Rio, M.C. The trefoil factor 1 participates in gastrointestinal cell differentiation by delaying G1-S phase transition and reducing apoptosis. J. Cell Biol. 2002, 157, 761–770. [Google Scholar] [CrossRef]
- Merry, R.; Belfield, L.; McArdle, P.; McLennan, A.; Crean, S.; Foey, A. Oral health and pathology: A macrophage account. Br. J. Oral Maxillofac. Surg. 2012, 50, 2–7. [Google Scholar] [CrossRef]
- Martinet, W.; Verheye, S.; De Meyer, I.; Timmermans, J.P.; Schrijvers, D.M.; Van Brussel, I.; Bult, H.; De Meyer, G.R. Everolimus triggers cytokine release by macrophages: Rationale for stents eluting everolimus and a glucocorticoid. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1228–1235. [Google Scholar] [CrossRef]
- Denton, D.; Xu, T.; Kumar, S. Autophagy as a pro-death pathway. Immunol. Cell Biol. 2015, 93, 35–42. [Google Scholar] [CrossRef]
- Denton, D.; Kumar, S. Autophagy-dependent cell death. Cell Death Differ. 2019, 26, 605–616. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Kurdi, A.; De Doncker, M.; Leloup, A.; Neels, H.; Timmermans, J.P.; Lemmens, K.; Apers, S.; De Meyer, G.R.Y.; Martinet, W. Continuous administration of the mTORC1 inhibitor everolimus induces tolerance and decreases autophagy in mice. Br. J. Pharmacol. 2016, 173, 3359–3371. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Li, X.; Zhen, L.; Chen, W.; Mu, L.; Zhang, Y.; Song, A. Everolimus inhibits breast cancer cell growth through PI3K/AKT/mTOR signaling pathway. Mol. Med. Rep. 2018, 17, 7163–7169. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.Y.; Cheng, X.B.; Li, Y.; Jin, L.D.; Yin, H.P. Effect of everolimus on the expression of Ki-67 and caspase-3 in patients with neuroendocrine tumors. Genet. Mol. Res. 2017, 16, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Meiller, T.F.; Varlotta, S.; Weikel, D. Recognition and Management of Oral Mucosal Injury Caused by Mammalian Target of Rapamycin Inhibitors: A Case Series. Case Rep. Oncol. 2015, 8, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, A.E.; Chapman, K.E. The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol. Cell. Endocrinol. 2011, 335, 2–13. [Google Scholar] [CrossRef]
- Kim, J.; Park, M.Y.; Kim, H.K.; Park, Y.; Whang, K.-Y. Cortisone and dexamethasone inhibit myogenesis by modulating the AKT/mTOR signaling pathway in C2C12. Biosci. Biotechnol. Biochem. 2016, 80, 2093–2099. [Google Scholar] [CrossRef]
- Ruddy, K.J.; Zahrieh, D.; He, J.; Waechter, B.; Holleran, J.L.; Lewis, L.D.; Chow, S.; Beumer, J.; Weiss, M.; Trikalinos, N.; et al. Dexamethasone to prevent everolimus-induced stomatitis (Alliance MIST Trial: A221701). Semin. Oncol. 2023, 50, 7–10. [Google Scholar] [CrossRef]
- Afinitor Market Market Size Is Expanding at a CAGR of 6.5% Forecasted for Period from 2023–2030 and Provide Market Analysis of Report Associated with It. Available online: https://www.digitaljournal.com/pr/news/prime-pr-wire/afinitor-market-market-size-is-expanding-at-a-cagr-of-6-5-forecasted-for-period-from-2023-2030-and-provide-market-analysis-of-report-associated-with-it (accessed on 18 September 2023).
- Shen, R.; Richter, H.E.; Clements, R.H.; Novak, L.; Huff, K.; Bimczok, D.; Sankaran-Walters, S.; Dandekar, S.; Clapham, P.R.; Smythies, L.E.; et al. Macrophages in vaginal but not intestinal mucosa are monocyte-like and permissive to human immunodeficiency virus type 1 infection. J. Virol. 2009, 83, 3258–3267. [Google Scholar] [CrossRef]
- Nudel, I.; Elnekave, M.; Furmanov, K.; Arizon, M.; Björn; Clausen, E.; Wilensky, A.; Hovav, A.-H. Dendritic Cells in Distinct Oral Mucosal Tissues Engage Different Mechanisms to Prime CD8+ T Cells. J. Immunol. 2011, 186, 891–900. [Google Scholar] [CrossRef] [PubMed]
- Iijima, N.; Thompson, J.M.; Iwasaki, A. Dendritic cells and macrophages in the genitourinary tract. Mucosal Immunol. 2008, 1, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Ji, D.J.; Aboalela, A.; Villa, A. Everolimus-associated stomatitis in a patient who had renal transplant. BMJ Case Rep. 2016, 2016, bcr2016217513. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.J.; Lee, J.; Shin, N.R.; Kim, M.S.; Hyun, D.W.; Yun, J.U.; Kim, P.S.; Whon, T.W.; Bae, J.-W. Chronic repression of mTOR Complex 2 induces changes in the gut microbiota of diet-induced obese mice. Sci. Rep. 2016, 6, 30887. [Google Scholar] [CrossRef]
- Hertz, D.L.; Lustberg, M.B.; Sonis, S. Evolution of predictive risk factor analysis for chemotherapy-related toxicity. Support. Care Cancer 2023, 31, 601. [Google Scholar] [CrossRef]
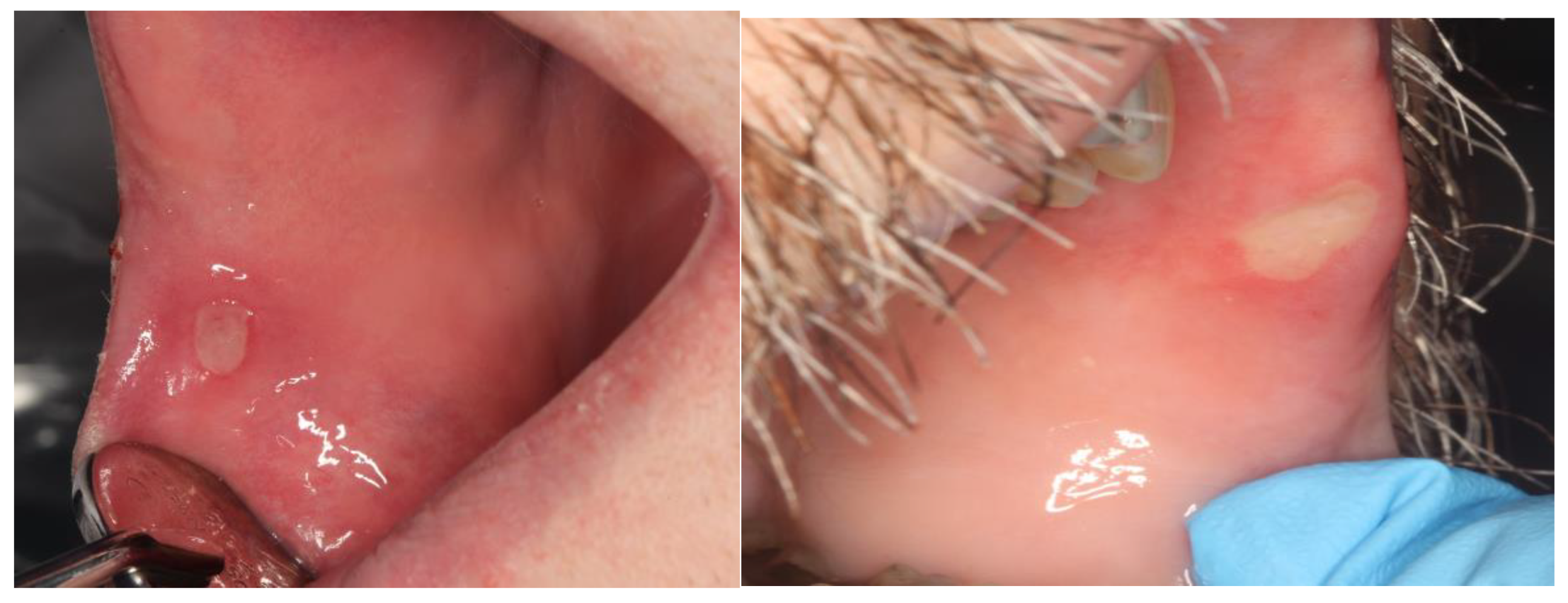
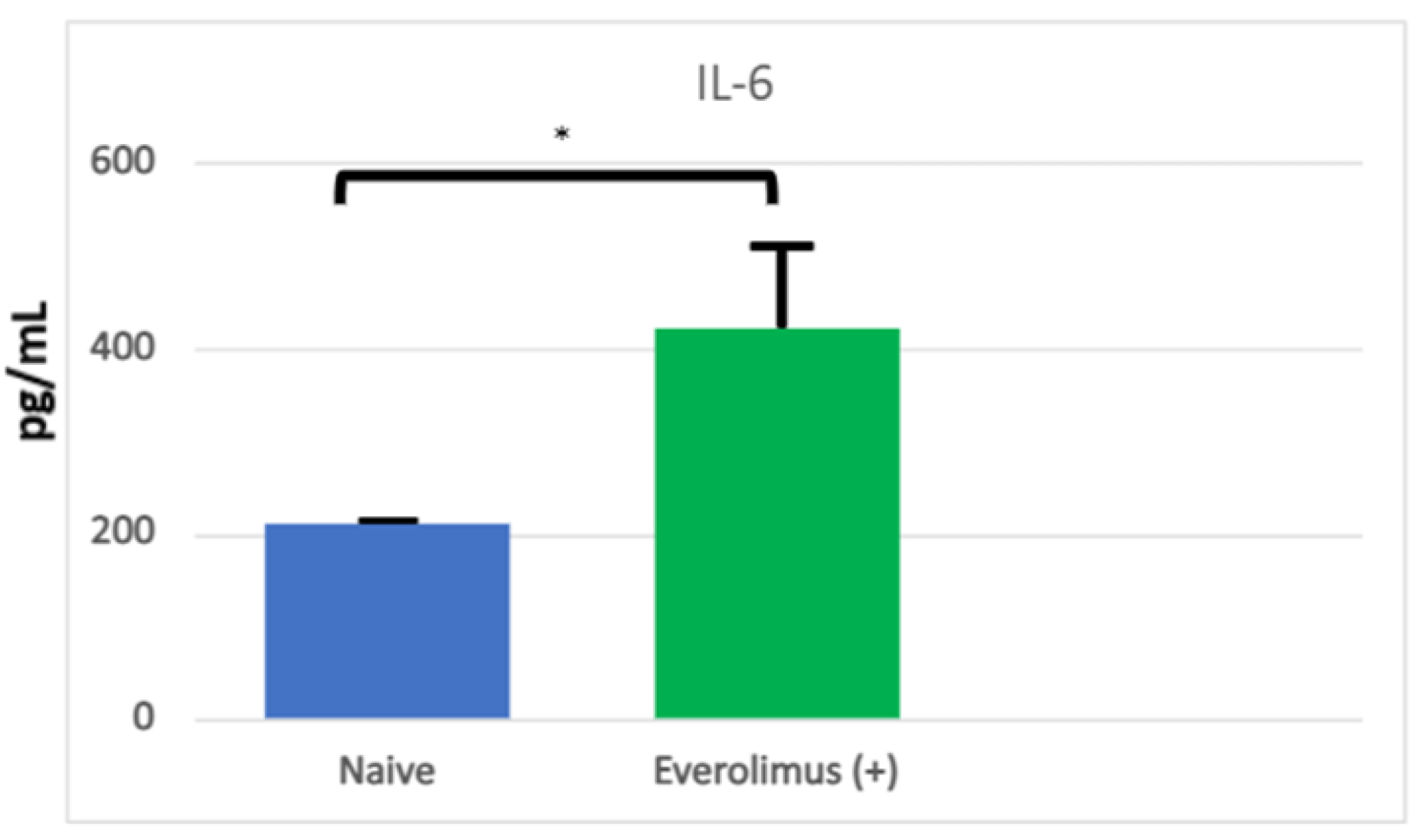
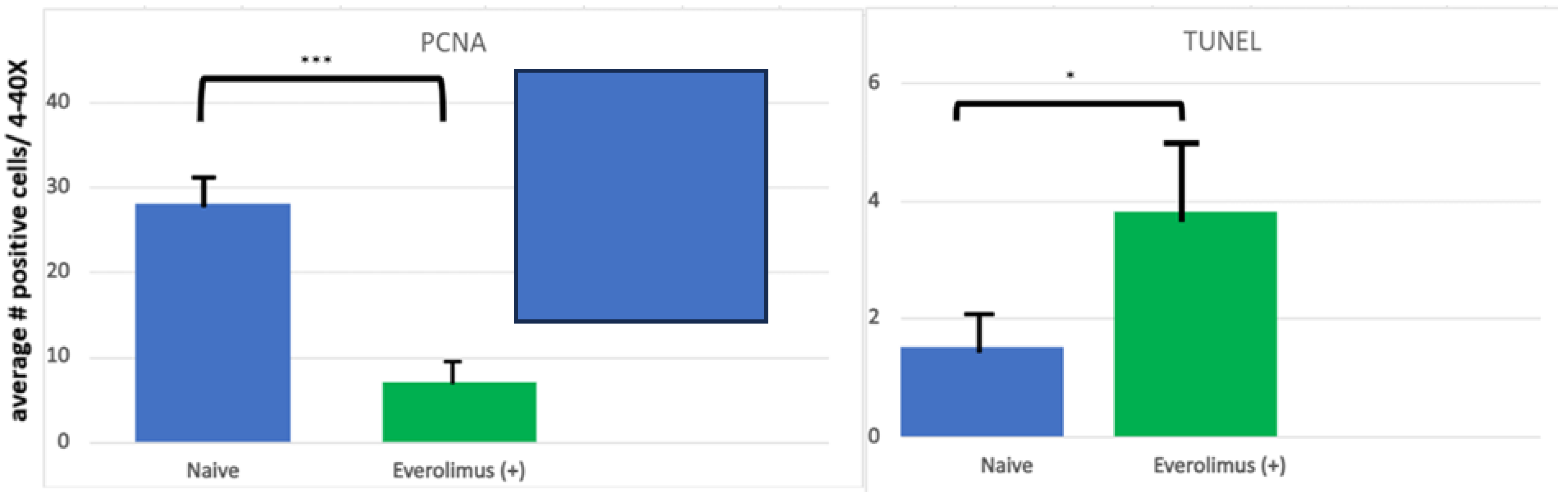

{kind=link}
{kind=link}
{kind=link}
| Overall | % Of Adverse Events | Overall | % Of Dose Reductions | Overall | % Of Discontinuations | |||
|---|---|---|---|---|---|---|---|---|
| All Grades | Grades 3, 4 | All Grades | Grades 3, 4 | |||||
| All mTOR Inhibitors | 52.9% (1493/2822) | 5.4% (153/2822) | 73.4% (1493/2033) | 20.7% (153/739) | 19.2% (176/917) | 27.3% (48/176) | 11.5% (99/862) | 13.1% (13/99) |
| Temsirolimus | 60.8% (819/1347) | 5.2% (70/1347) | 88.5% (819/925) | 16.0% (70/437) | 28.1% (140/499) | 30% (42/140) | 10.2% (40/393) | 0 (0/40) |
| Everolimus | 44.3% (568/1281) | 5.2% (67/1281) | 58.4% (568/972) | 27.2% (67/246) | 8.2% (28/340) | 7.1% (2/28) | 12.4% (38/307) | 18.4% (7/38) |
| Ridaforolimus | 54.6% (106/194) | 8.2% (16/194) | 77.9% (106/136) | 28.6% (16/56) | 10.3% (8/78) | 50% (4/8) | 13.0% (21/162) | 28.6% (6/21) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sonis, S.T.; Villa, A. A New Hypothesis Describing the Pathogenesis of Oral Mucosal Injury Associated with the Mammalian Target of Rapamycin (mTOR) Inhibitors. Cancers 2024, 16, 68. https://doi.org/10.3390/cancers16010068
Sonis ST, Villa A. A New Hypothesis Describing the Pathogenesis of Oral Mucosal Injury Associated with the Mammalian Target of Rapamycin (mTOR) Inhibitors. Cancers. 2024; 16(1):68. https://doi.org/10.3390/cancers16010068
Chicago/Turabian StyleSonis, Stephen T., and Alessandro Villa. 2024. "A New Hypothesis Describing the Pathogenesis of Oral Mucosal Injury Associated with the Mammalian Target of Rapamycin (mTOR) Inhibitors" Cancers 16, no. 1: 68. https://doi.org/10.3390/cancers16010068
APA StyleSonis, S. T., & Villa, A. (2024). A New Hypothesis Describing the Pathogenesis of Oral Mucosal Injury Associated with the Mammalian Target of Rapamycin (mTOR) Inhibitors. Cancers, 16(1), 68. https://doi.org/10.3390/cancers16010068