
JAK1 Is a Novel Target of Tumor- and Invasion-Suppressive microRNA 494-5p in Colorectal Cancer


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Bioinformatics Analysis and Target Identification
2.3. Total RNA Isolation from Cell Lines and cDNA Synthesis
2.4. Genomic DNA Isolation and Plasmid Isolation
2.5. Quantitative Real-Time PCR Primers, miRs, and si-RNAs
2.6. Plasmid Generation and Site-Directed Mutagenesis
2.7. Transfection and Stimulation of Cultured Human CRC Cells
2.8. Dual-Luciferase Reporter Assays
2.9. Proliferation Assay
2.10. Migration Assays and Invasion Assays
2.11. Immunoblot (Western Blot) Analysis
2.12. Statistical Analysis
3. Results
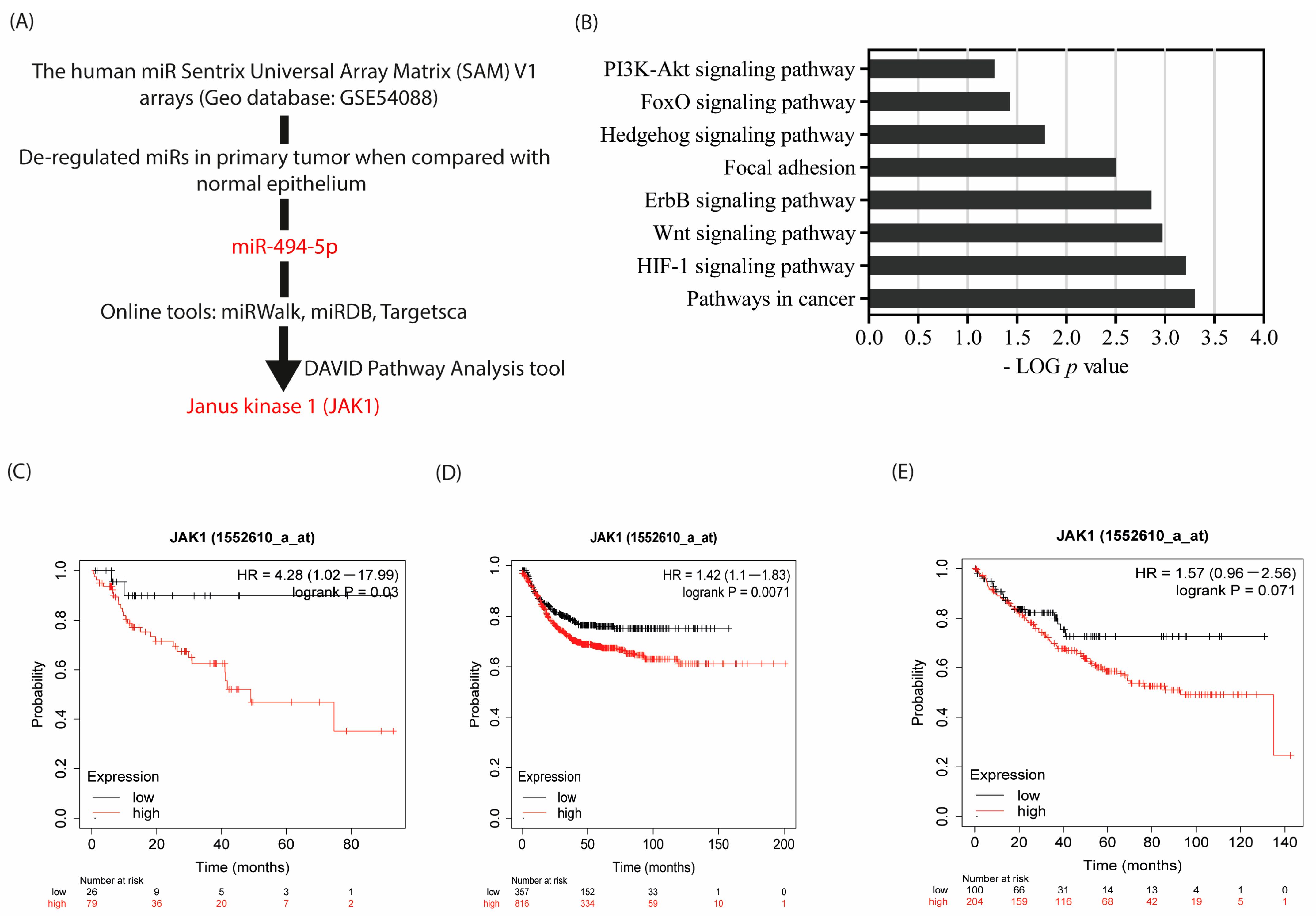
3.1. JAK 1 Is an In Silico Target of miR-494-5p, and Associated with Poor Prognosis
3.2. Endogenous Expression of JAK1 and miR-494-5p in CRC Cell Lines
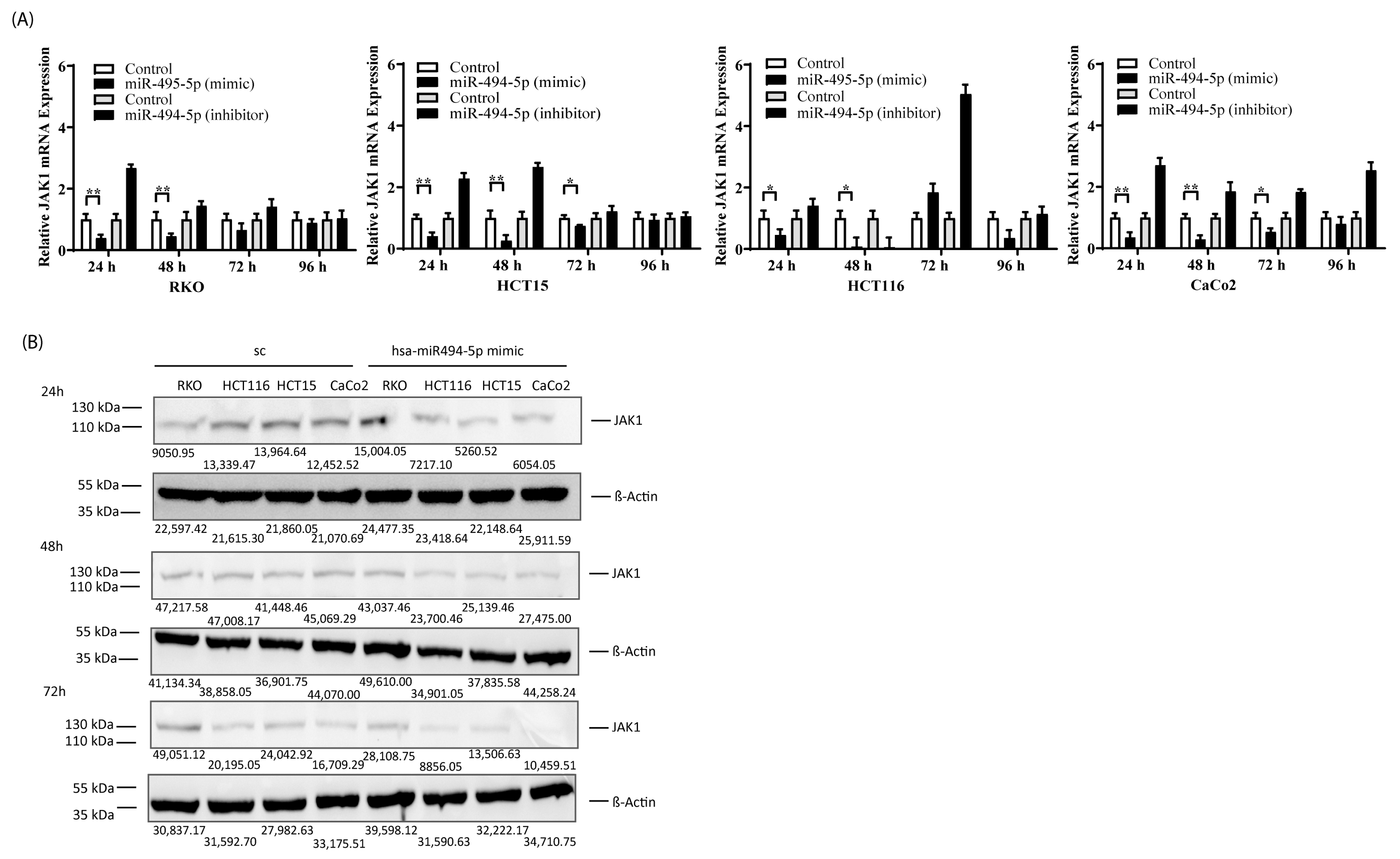
3.3. miR-494-5p Represses JAK1 Expression In Vitro
3.4. JAK1-3′UTR Is a Direct Physical Target of miR-494-5p
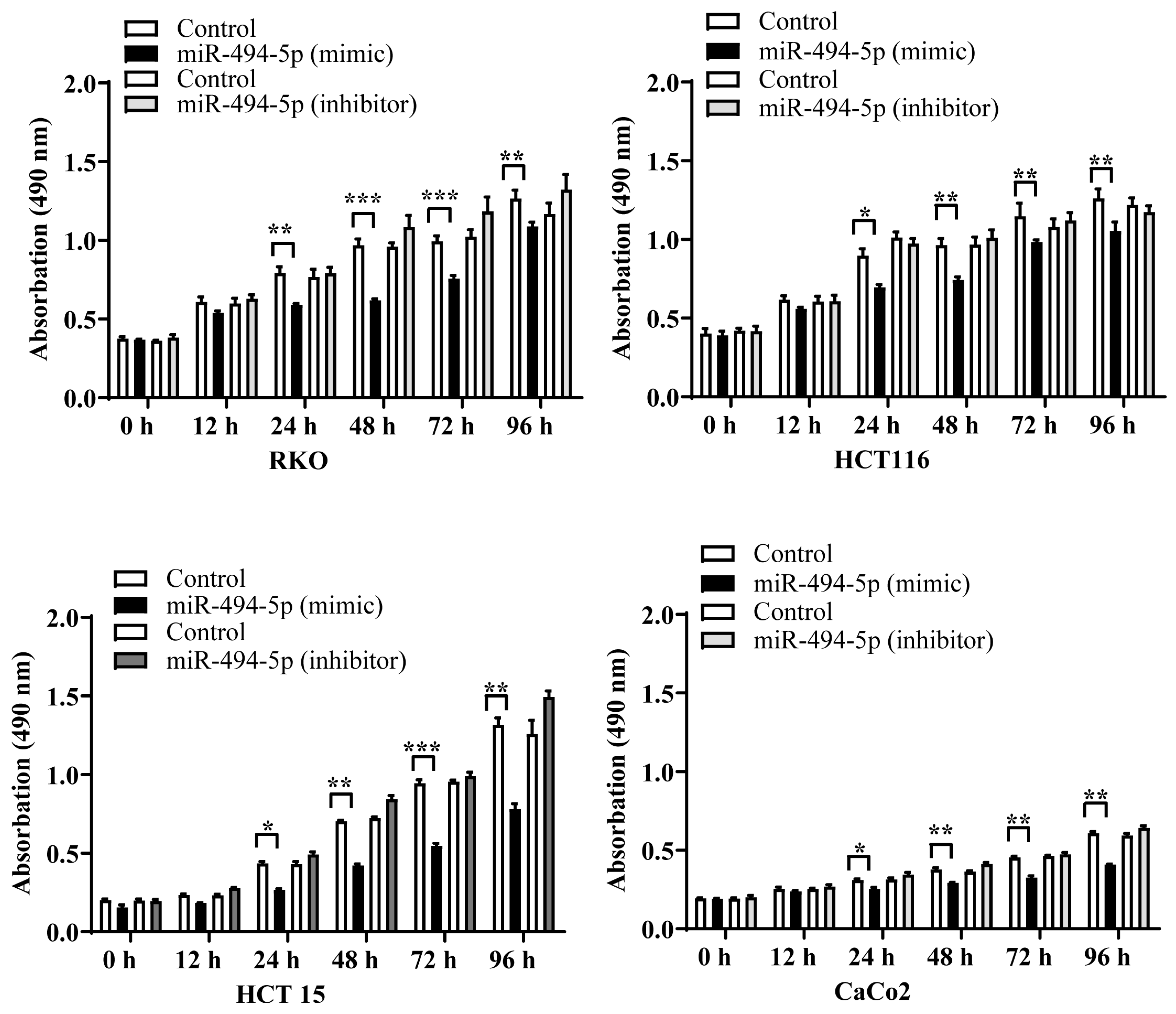
3.5. MiR-494-5p Reduces CRC Cell Proliferation
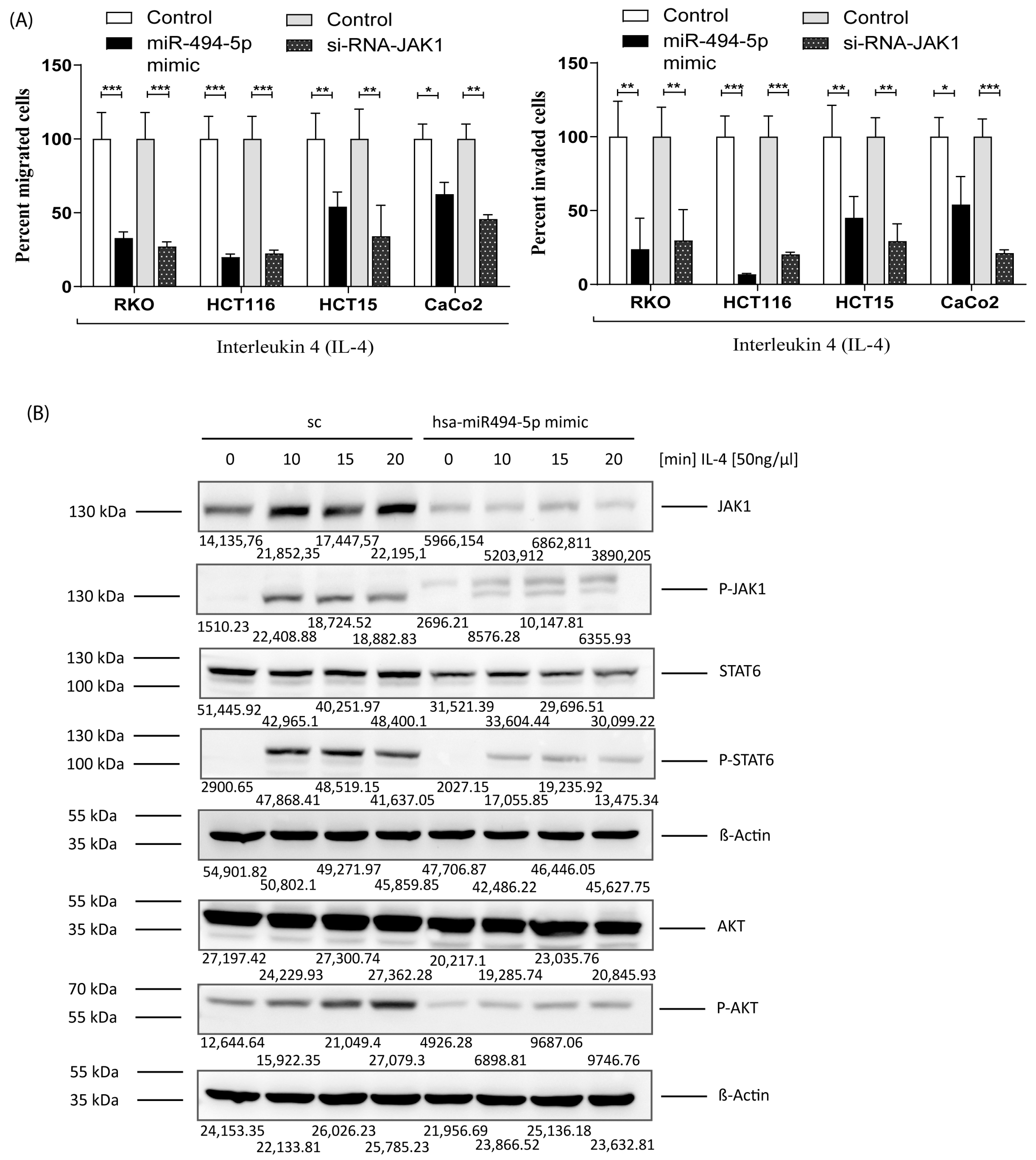
3.6. MiR-494-5p Inhibits Constitutive Migration and Invasion of CRC Cells
3.7. MiR-494-5p Inhibits IL-4 Stimulated JAK1, STAT6 and AKT Activation and IL-4 Induced Migration/Invasion
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.; Desantis, C.; Jemal, A. Colorectal cancer statistics, 2014. CA A Cancer J. Clin. 2014, 64, 104–117. [Google Scholar] [CrossRef] [PubMed]
- Takami, K.; Yana, I.; Kurahashi, H.; Nishisho, I. Multistep carcinogenesis in colorectal cancers. Southeast Asian J. Trop. Med. Public Health 1995, 26 (Suppl. S1), 190–196. [Google Scholar] [PubMed]
- Nana-Sinkam, S.P.; Croce, C.M. MicroRNA regulation of tumorigenesis, cancer progression and interpatient heterogeneity: Towards clinical use. Genome Biol. 2014, 15, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Asangani, I.A.; Rasheed, S.A.; Nikolova, D.A.; Leupold, J.H.; Colburn, N.H.; Post, S.; Allgayer, H. MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene 2008, 27, 2128–2136. [Google Scholar] [CrossRef] [PubMed]
- Ceppi, P.; Mudduluru, G.; Kumarswamy, R.; Rapa, I.; Scagliotti, G.V.; Papotti, M.; Allgayer, H. Loss of miR-200c expression induces an aggressive, invasive, and chemoresistant phenotype in non-small cell lung cancer. Mol. Cancer Res. 2010, 8, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Laudato, S.; Patil, N.; Abba, M.L.; Leupold, J.H.; Benner, A.; Gaiser, T.; Marx, A.; Allgayer, H. P53-induced miR-30e-5p inhibits colorectal cancer invasion and metastasis by targeting ITGA6 and ITGB1. Int. J. Cancer 2017, 141, 1879–1890. [Google Scholar] [CrossRef]
- Mudduluru, G.; Abba, M.; Batliner, J.; Patil, N.; Scharp, M.; Lunavat, T.R.; Leupold, J.H.; Oleksiuk, O.; Juraeva, D.; Thiele, W.; et al. A Systematic Approach to Defining the microRNA Landscape in Metastasis. Cancer Res. 2015, 75, 3010–3019. [Google Scholar] [CrossRef]
- El-Daly, S.M.; Abba, M.L.; Patil, N.; Allgayer, H. miRs-134 and -370 function as tumor suppressors in colorectal cancer by independently suppressing EGFR and PI3K signalling. Sci. Rep. 2016, 6, 24720–24731. [Google Scholar] [CrossRef]
- Patil, N.; Allgayer, H.; Leupold, J.H. MicroRNAs in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1277, 1–31. [Google Scholar] [CrossRef]
- Darwiche, N. Epigenetic mechanisms and the hallmarks of cancer: An intimate affair. Am. J. Cancer Res. 2020, 10, 1954–1978. [Google Scholar] [PubMed]
- Abba, M.L.; Patil, N.; Leupold, J.H.; Allgayer, H. MicroRNA Regulation of Epithelial to Mesenchymal Transition. J. Clin. Med. 2016, 5, 8–28. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal Transduct. Target. Ther. 2016, 1, 15004–15012. [Google Scholar] [CrossRef] [PubMed]
- Grillone, K.; Riillo, C.; Scionti, F.; Rocca, R.; Tradigo, G.; Guzzi, P.H.; Alcaro, S.; Di Martino, M.T.; Tagliaferri, P.; Tassone, P. Non-coding RNAs in cancer: Platforms and strategies for investigating the genomic “dark matter”. J. Exp. Clin. Cancer Res. 2020, 39, 117–135. [Google Scholar] [CrossRef] [PubMed]
- Jansson, M.D.; Lund, A.H. MicroRNA and cancer. Mol. Oncol. 2012, 6, 590–610. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.; Peruzzi, P.P.; Lawler, S. MicroRNAs in cancer: Biomarkers, functions and therapy. Trends Mol. Med. 2014, 20, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Ishaque, N.; Abba, M.L.; Hauser, C.; Patil, N.; Paramasivam, N.; Huebschmann, D.; Leupold, J.H.; Balasubramanian, G.P.; Kleinheinz, K.; Toprak, U.H.; et al. Whole genome sequencing puts forward hypotheses on metastasis evolution and therapy in colorectal cancer. Nat. Commun. 2018, 9, 4782–4795. [Google Scholar] [CrossRef]
- Patil, N.; Abba, M.L.; Zhou, C.; Chang, S.; Gaiser, T.; Leupold, J.H.; Allgayer, H. Changes in Methylation across Structural and MicroRNA Genes Relevant for Progression and Metastasis in Colorectal Cancer. Cancers 2021, 13, 5951. [Google Scholar] [CrossRef]
- Mudduluru, G.; Ceppi, P.; Kumarswamy, R.; Scagliotti, G.V.; Papotti, M.; Allgayer, H. Regulation of Axl receptor tyrosine kinase expression by miR-34a and miR-199a/b in solid cancer. Oncogene 2011, 30, 2888–2899. [Google Scholar] [CrossRef]
- Pfeifer, G.P. Defining Driver DNA Methylation Changes in Human Cancer. Int. J. Mol. Sci. 2018, 19, 1166. [Google Scholar] [CrossRef]
- Abba, M.L.; Patil, N.; Leupold, J.H.; Moniuszko, M.; Utikal, J.; Niklinski, J.; Allgayer, H. MicroRNAs as novel targets and tools in cancer therapy. Cancer Lett. 2017, 387, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Marongiu, L.; Allgayer, H. Viruses in colorectal cancer. Mol. Oncol. 2022, 16, 1423–1450. [Google Scholar] [CrossRef] [PubMed]
- Marino, A.L.F.; Evangelista, A.F.; Vieira, R.A.C.; Macedo, T.; Kerr, L.M.; Abrahão-Machado, L.F.; Longatto-Filho, A.; Silveira, H.C.S.; Marques, M.M.C. MicroRNA expression as risk biomarker of breast cancer metastasis: A pilot retrospective case-cohort study. BMC Cancer 2014, 14, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Zhan, M.N.; Yu, X.T.; Tang, J.; Zhou, C.X.; Wang, C.L.; Yin, Q.Q.; Gong, X.F.; He, M.; He, J.R.; Chen, G.Q.; et al. MicroRNA-494 inhibits breast cancer progression by directly targeting PAK1. Cell Death Dis. 2017, 8, e2529. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.Q.; Liang, T.J.; Fu, J.W. miR-494 inhibits invasion and proliferation of gastric cancer by targeting IGF-1R. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 3818–3824. [Google Scholar]
- Yuan, J.; Wang, K.; Xi, M. MiR-494 Inhibits Epithelial Ovarian Cancer Growth by Targeting c-Myc. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2016, 22, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Fang, Z.; Wang, H.; Jiao, R.; Zhou, J.; Fang, N. CUL4A functions as an oncogene in ovarian cancer and is directly regulated by miR-494. Biochem. Biophys. Res. Commun. 2016, 480, 675–681. [Google Scholar] [CrossRef]
- Liu, Y.; Li, X.; Zhu, S.; Zhang, J.G.; Yang, M.; Qin, Q.; Deng, S.C.; Wang, B.; Tian, K.; Liu, L.; et al. Ectopic expression of miR-494 inhibited the proliferation, invasion and chemoresistance of pancreatic cancer by regulating SIRT1 and c-Myc. Gene Ther. 2015, 22, 729–738. [Google Scholar] [CrossRef]
- Wan, B.S.; Cheng, M.; Zhang, L. Insulin-like growth factor 2 mRNA-binding protein 1 promotes cell proliferation via activation of AKT and is directly targeted by microRNA-494 in pancreatic cancer. World J. Gastroenterol. 2019, 25, 6063–6076. [Google Scholar] [CrossRef]
- Yang, Y.; Tao, X.; Li, C.B.; Wang, C.M. MicroRNA-494 acts as a tumor suppressor in pancreatic cancer, inhibiting epithelial-mesenchymal transition, migration and invasion by binding to SDC1. Int. J. Oncol. 2018, 53, 1204–1214. [Google Scholar] [CrossRef]
- Liu, L.; Jiang, Y.; Zhang, H.; Greenlee, A.R.; Han, Z. Overexpressed miR-494 down-regulates PTEN gene expression in cells transformed by anti-benzo(a)pyrene-trans-7,8-dihydrodiol-9,10-epoxide. Life Sci. 2010, 86, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Cheung, K.S.C.; Liu, Z.L.; Leung, F.; Lu, W.W. Matrix metallopeptidase 9 targeted by hsa-miR-494 promotes silybin-inhibited osteosarcoma. Mol. Carcinog. 2018, 57, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhu, Y.; Hu, L.; Yan, F.; Chen, J. miR-494 induces EndMT and promotes the development of HCC (Hepatocellular Carcinoma) by targeting SIRT3/TGF-β/SMAD signaling pathway. Sci. Rep. 2019, 9, 7213–7225. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Peng, J.H. Increased Expression of miR-494 in Serum of Patients with Prostate Cancer and its Potential Diagnostic Value. Clin. Lab. 2019, 65, 1507–1512. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT signaling pathway: From bench to clinic. Signal Transduct. Target. Ther. 2021, 6, 402. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.J.; Snowden, J.A.; Zeidler, M.P.; Danson, S.J. The role of JAK/STAT signalling in the pathogenesis, prognosis and treatment of solid tumours. Br. J. Cancer 2015, 113, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Aittomäki, S.; Pesu, M. Therapeutic targeting of the Jak/STAT pathway. Basic. Clin. Pharmacol. Toxicol. 2014, 114, 18–23. [Google Scholar] [CrossRef]
- Murray, P.J. The JAK-STAT signaling pathway: Input and output integration. J. Immunol. 2007, 178, 2623–2629. [Google Scholar] [CrossRef]
- Rawlings, J.S.; Rosler, K.M.; Harrison, D.A. The JAK/STAT signaling pathway. J. Cell Sci. 2004, 117, 1281–1283. [Google Scholar] [CrossRef]
- Jiang, H.; Harris, M.B.; Rothman, P. IL-4/IL-13 signaling beyond JAK/STAT. J. Allergy Clin. Immunol. 2000, 105, 1063–1070. [Google Scholar] [CrossRef]
- Qureshy, Z.; Johnson, D.E.; Grandis, J.R. Targeting the JAK/STAT pathway in solid tumors. J. Cancer Metastasis Treat. 2020, 6, 27–45. [Google Scholar] [CrossRef] [PubMed]
- Lánczky, A.; Győrffy, B. Web-Based Survival Analysis Tool Tailored for Medical Research (KMplot): Development and Implementation. J. Med. Internet Res. 2021, 23, e27633. [Google Scholar] [CrossRef] [PubMed]
- Murata, T.; Noguchi, P.D.; Puri, R.K. IL-13 induces phosphorylation and activation of JAK2 Janus kinase in human colon carcinoma cell lines: Similarities between IL-4 and IL-13 signaling. J. Immunol. 1996, 156, 2972–2978. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I.; Batzer, A.G.; Blaikie, P.; Obermeier, A.; Ullrich, A.; Schlessinger, J.; Margolis, B. Shc binding to nerve growth factor receptor is mediated by the phosphotyrosine interaction domain. J. Biol. Chem. 1995, 270, 15125–15129. [Google Scholar] [CrossRef] [PubMed]
- Wick, K.R.; Berton, M.T. IL-4 induces serine phosphorylation of the STAT6 transactivation domain in B lymphocytes. Mol. Immunol. 2000, 37, 641–652. [Google Scholar] [CrossRef] [PubMed]
- Yin, T.; Tsang, M.L.; Yang, Y.C. JAK1 kinase forms complexes with interleukin-4 receptor and 4PS/insulin receptor substrate-1-like protein and is activated by interleukin-4 and interleukin-9 in T lymphocytes. J. Biol. Chem. 1994, 269, 26614–26617. [Google Scholar] [CrossRef] [PubMed]
- Pesu, M.; Takaluoma, K.; Aittomäki, S.; Lagerstedt, A.; Saksela, K.; Kovanen, P.E.; Silvennoinen, O. Interleukin-4-induced transcriptional activation by Stat6 involves multiple serine/threonine kinase pathways and serine phosphorylation of Stat6. Blood 2000, 95, 494–502. [Google Scholar] [CrossRef]
- Chai, J.; Dong, W.; Xie, C.; Wang, L.; Han, D.-L.; Wang, S.; Guo, H.-L.; Zhang, Z.-L. MicroRNA-494 sensitizes colon cancer cells to fluorouracil through regulation of DPYD. IUBMB Life 2015, 67, 191–201. [Google Scholar] [CrossRef]
- Delgado-Ramirez, Y.; Colly, V.; Gonzalez, G.V.; Leon-Cabrera, S. Signal transducer and activator of transcription 6 as a target in colon cancer therapy. Oncol. Lett. 2020, 20, 455–464. [Google Scholar] [CrossRef]
- Schindler, C.; Darnell, J.E., Jr. Transcriptional responses to polypeptide ligands: The JAK-STAT pathway. Annu. Rev. Biochem. 1995, 64, 621–651. [Google Scholar] [CrossRef]
- Cheng, J.Q.; Godwin, A.K.; Bellacosa, A.; Taguchi, T.; Franke, T.F.; Hamilton, T.C.; Tsichlis, P.N.; Testa, J.R. AKT2, a putative oncogene encoding a member of a subfamily of protein-serine/threonine kinases, is amplified in human ovarian carcinomas. Proc. Natl. Acad. Sci. USA 1992, 89, 9267–9271. [Google Scholar] [CrossRef] [PubMed]
- Altomare, D.A.; Tanno, S.; De Rienzo, A.; Klein-Szanto, A.J.; Tanno, S.; Skele, K.L.; Hoffman, J.P.; Testa, J.R. Frequent activation of AKT2 kinase in human pancreatic carcinomas. J. Cell. Biochem. 2002, 87, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Dobashi, Y.; Kimura, M.; Matsubara, H.; Endo, S.; Inazawa, J.; Ooi, A. Molecular alterations in AKT and its protein activation in human lung carcinomas. Hum. Pathol. 2012, 43, 2229–2240. [Google Scholar] [CrossRef] [PubMed]
- Bellacosa, A.; de Feo, D.; Godwin, A.K.; Bell, D.W.; Cheng, J.Q.; Altomare, D.A.; Wan, M.; Dubeau, L.; Scambia, G.; Masciullo, V.; et al. Molecular alterations of the AKT2 oncogene in ovarian and breast carcinomas. Int. J. Cancer 1995, 64, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Stahl, J.M.; Sharma, A.; Cheung, M.; Zimmerman, M.; Cheng, J.Q.; Bosenberg, M.W.; Kester, M.; Sandirasegarane, L.; Robertson, G.P. Deregulated Akt3 activity promotes development of malignant melanoma. Cancer Res. 2004, 64, 7002–7010. [Google Scholar] [CrossRef]
- Staal, S.P. Molecular cloning of the akt oncogene and its human homologues AKT1 and AKT2: Amplification of AKT1 in a primary human gastric adenocarcinoma. Proc. Natl. Acad. Sci. USA 1987, 84, 5034–5037. [Google Scholar] [CrossRef]
- Roy, H.K.; Olusola, B.F.; Clemens, D.L.; Karolski, W.J.; Ratashak, A.; Lynch, H.T.; Smyrk, T.C. AKT proto-oncogene overexpression is an early event during sporadic colon carcinogenesis. Carcinogenesis 2002, 23, 201–205. [Google Scholar] [CrossRef]
- Xu, X.; Sakon, M.; Nagano, H.; Hiraoka, N.; Yamamoto, H.; Hayashi, N.; Dono, K.; Nakamori, S.; Umeshita, K.; Ito, Y.; et al. Akt2 expression correlates with prognosis of human hepatocellular carcinoma. Oncol. Rep. 2004, 11, 25–32. [Google Scholar] [CrossRef]
- Chomarat, P.; Banchereau, J. An update on interleukin-4 and its receptor. Eur. Cytokine Netw. 1997, 8, 333–344. [Google Scholar]
- Yang, N.; Zhu, S.; Lv, X.; Qiao, Y.; Liu, Y.J.; Chen, J. MicroRNAs: Pleiotropic Regulators in the Tumor Microenvironment. Front. Immunol. 2018, 9, 2491. [Google Scholar] [CrossRef]
- Zitvogel, L.; Tesniere, A.; Kroemer, G. Cancer despite immunosurveillance: Immunoselection and immunosubversion. Nat. Rev. Immunol. 2006, 6, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Allavena, P.; Sica, A.; Solinas, G.; Porta, C.; Mantovani, A. The inflammatory micro-environment in tumor progression: The role of tumor-associated macrophages. Crit. Rev. Oncol. Hematol. 2008, 66, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Muller, A.J.; Scherle, P.A. Targeting the mechanisms of tumoral immune tolerance with small-molecule inhibitors. Nat. Rev. Cancer 2006, 6, 613–625. [Google Scholar] [CrossRef] [PubMed]
- Locati, M.; Mantovani, A.; Sica, A. Macrophage activation and polarization as an adaptive component of innate immunity. Adv. Immunol. 2013, 120, 163–184. [Google Scholar] [CrossRef] [PubMed]
- Shirota, H.; Klinman, D.M.; Ito, S.E.; Ito, H.; Kubo, M.; Ishioka, C. IL4 from T Follicular Helper Cells Downregulates Antitumor Immunity. Cancer Immunol. Res. 2017, 5, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Pedroza-Gonzalez, A.; Xu, K.; Wu, T.C.; Aspord, C.; Tindle, S.; Marches, F.; Gallegos, M.; Burton, E.C.; Savino, D.; Hori, T.; et al. Thymic stromal lymphopoietin fosters human breast tumor growth by promoting type 2 inflammation. J. Exp. Med. 2011, 208, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Nevala, W.K.; Vachon, C.M.; Leontovich, A.A.; Scott, C.G.; Thompson, M.A.; Markovic, S.N.; Melanoma Study Group of the Mayo Clinic Cancer, C. Evidence of systemic Th2-driven chronic inflammation in patients with metastatic melanoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 1931–1939. [Google Scholar] [CrossRef]
- Gao, J.; Wu, Y.; Su, Z.; Amoah Barnie, P.; Jiao, Z.; Bie, Q.; Lu, L.; Wang, S.; Xu, H. Infiltration of alternatively activated macrophages in cancer tissue is associated with MDSC and Th2 polarization in patients with esophageal cancer. PLoS ONE 2014, 9, e104453. [Google Scholar] [CrossRef]
- Li, J.; Wang, Z.; Mao, K.; Guo, X. Clinical significance of serum T helper 1/T helper 2 cytokine shift in patients with non-small cell lung cancer. Oncol. Lett. 2014, 8, 1682–1686. [Google Scholar] [CrossRef]
- Todaro, M.; Alea, M.P.; Di Stefano, A.B.; Cammareri, P.; Vermeulen, L.; Iovino, F.; Tripodo, C.; Russo, A.; Gulotta, G.; Medema, J.P.; et al. Colon cancer stem cells dictate tumor growth and resist cell death by production of interleukin-4. Cell Stem Cell 2007, 1, 389–402. [Google Scholar] [CrossRef]
- Li, Z.; Jiang, J.; Wang, Z.; Zhang, J.; Xiao, M.; Wang, C.; Lu, Y.; Qin, Z. Endogenous interleukin-4 promotes tumor development by increasing tumor cell resistance to apoptosis. Cancer Res. 2008, 68, 8687–8694. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Lai, J.; Dai, D.; Chen, R.; Li, X.; Liao, N. JAK1 as a prognostic marker and its correlation with immune infiltrates in breast cancer. Aging 2019, 11, 11124–11135. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Huang, Y.; Zhang, L.; Liang, D.N.; Li, L. Activation of Janus kinase 1 confers poor prognosis in patients with non-small cell lung cancer. Oncol. Lett. 2017, 14, 3959–3966. [Google Scholar] [CrossRef] [PubMed]
- Wen, W.; Liang, W.; Wu, J.; Kowolik, C.M.; Buettner, R.; Scuto, A.; Hsieh, M.Y.; Hong, H.; Brown, C.E.; Forman, S.J.; et al. Targeting JAK1/STAT3 signaling suppresses tumor progression and metastasis in a peritoneal model of human ovarian cancer. Mol. Cancer Ther. 2014, 13, 3037–3048. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, Z.; Zhang, S.; Yao, Q.; Chen, W.; Liu, F. Ruxolitinib induces apoptosis of human colorectal cancer cells by downregulating the JAK1/2-STAT1-Mcl-1 axis. Oncol. Lett. 2021, 21, 352–366. [Google Scholar] [CrossRef] [PubMed]
- Vodenkova, S.; Buchler, T.; Cervena, K.; Veskrnova, V.; Vodicka, P.; Vymetalkova, V. 5-fluorouracil and other fluoropyrimidines in colorectal cancer: Past, present and future. Pharmacol. Ther. 2020, 206, 107447. [Google Scholar] [CrossRef]
- Zou, Q.; Lei, X.; Xu, A.; Li, Z.; He, Q.; Huang, X.; Xu, G.; Tian, F.; Ding, Y.; Zhu, W. Chemokines in progression, chemoresistance, diagnosis, and prognosis of colorectal cancer. Front. Immunol. 2022, 13, 724139. [Google Scholar] [CrossRef]
- Elrebehy, M.A.; Al-Saeed, S.; Gamal, S.; El-Sayed, A.; Ahmed, A.A.; Waheed, O.; Ismail, A.; El-Mahdy, H.A.; Sallam, A.M.; Doghish, A.S. miRNAs as cornerstones in colorectal cancer pathogenesis and resistance to therapy: A spotlight on signaling pathways interplay—A review. Int. J. Biol. Macromol. 2022, 214, 583–600. [Google Scholar] [CrossRef]
- Peng, Q.P.; Du, D.B.; Ming, Q.; Hu, F.; Wu, Z.B.; Qiu, S. MicroRNA 494 increases chemosensitivity to doxorubicin in gastric cancer cells by targeting phosphodiesterases 4D. Cell. Mol. Biol. 2018, 64, 62–66. [Google Scholar] [CrossRef]
- Chang, N.; Ge, N.; Zhao, Y.; Yang, L.; Qin, W.; Cui, Y. Hsa_circ_0007142 contributes to cisplatin resistance in esophageal squamous cell carcinoma via miR-494-3p/LASP1 axis. J. Clin. Lab. Anal. 2022, 36, e24304. [Google Scholar] [CrossRef]
- Atari-Hajipirloo, S.; Nikanfar, S.; Heydari, A.; Kheradmand, F. Imatinib and its combination with 2,5-dimethyl-celecoxibinduces apoptosis of human HT-29 colorectal cancer cells. Res. Pharm. Sci. 2017, 12, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.M.; Chang, Y.L.; Hsieh, C.C.; Huang, S.M. The Synergistic Cytotoxic Effects of GW5074 and Sorafenib by Impacting Mitochondrial Functions in Human Colorectal Cancer Cell Lines. Front. Oncol. 2022, 12, 925653. [Google Scholar] [CrossRef] [PubMed]
- Sartore-Bianchi, A.; Trusolino, L.; Martino, C.; Bencardino, K.; Lonardi, S.; Bergamo, F.; Zagonel, V.; Leone, F.; Depetris, I.; Martinelli, E.; et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): A proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016, 17, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Buzard, B.; Douglass, L.; Gustafson, B.; Buckley, J.; Roth, M.; Kujtan, L.; Bansal, D. Response to osimertinib in a colorectal cancer patient with an EGFR T790M mutation: A case report. World J. Gastrointest. Oncol. 2023, 15, 1829–1834. [Google Scholar] [CrossRef]
- Ergun, S.; Akgun, O.; Hekim, N.T.; Aslan, S.; Ari, F.; Gunes, S.; Abur, U. The Interrelationship Between FYN and miR-128/193a-5p/494 in Imatinib Resistance in Prostate Cancer. Anti-Cancer Agents Med. Chem. 2023, 23, 360–365. [Google Scholar] [CrossRef]
- Salati, S.; Salvestrini, V.; Carretta, C.; Genovese, E.; Rontauroli, S.; Zini, R.; Rossi, C.; Ruberti, S.; Bianchi, E.; Barbieri, G.; et al. Deregulated expression of miR-29a-3p, miR-494-3p and miR-660-5p affects sensitivity to tyrosine kinase inhibitors in CML leukemic stem cells. Oncotarget 2017, 8, 49451–49469. [Google Scholar] [CrossRef] [PubMed]
- Bergamini, C.; Leoni, I.; Rizzardi, N.; Melli, M.; Galvani, G.; Coada, C.A.; Giovannini, C.; Monti, E.; Liparulo, I.; Valenti, F.; et al. MiR-494 induces metabolic changes through G6pc targeting and modulates sorafenib response in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2023, 42, 145. [Google Scholar] [CrossRef] [PubMed]
- Pollutri, D.; Patrizi, C.; Marinelli, S.; Giovannini, C.; Trombetta, E.; Giannone, F.A.; Baldassarre, M.; Quarta, S.; Vandewynckel, Y.P.; Vandierendonck, A.; et al. The epigenetically regulated miR-494 associates with stem-cell phenotype and induces sorafenib resistance in hepatocellular carcinoma. Cell Death Dis. 2018, 9, 4. [Google Scholar] [CrossRef]
- Gao, Y.; Yin, Z.; Qi, Y.; Peng, H.; Ma, W.; Wang, R.; Li, W. Golgi phosphoprotein 3 promotes angiogenesis and sorafenib resistance in hepatocellular carcinoma via upregulating exosomal miR-494-3p. Cancer Cell Int. 2022, 22, 35. [Google Scholar] [CrossRef]
- Yu, Y.; Yu, X.; Liu, H.; Song, Q.; Yang, Y. miR-494 inhibits cancer-initiating cell phenotypes and reverses resistance to lapatinib by downregulating FGFR2 in HER2-positive gastric cancer. Int. J. Mol. Med. 2018, 42, 998–1007. [Google Scholar] [CrossRef]
- Hermawan, A.; Putri, H. Integrative bioinformatics analysis reveals miR-494 and its target genes as predictive biomarkers of trastuzumab-resistant breast cancer. J. Egypt. Natl. Cancer Inst. 2020, 32, 16. [Google Scholar] [CrossRef] [PubMed]
- Alanazi, I.; Hoffmann, P.; Adelson, D.L. MicroRNAs are part of the regulatory network that controls EGF induced apoptosis, including elements of the JAK/STAT pathway, in A431 cells. PLoS ONE 2015, 10, e0120337. [Google Scholar] [CrossRef] [PubMed]
- Olaru, A.V.; Ghiaur, G.; Yamanaka, S.; Luvsanjav, D.; An, F.; Popescu, I.; Alexandrescu, S.; Allen, S.; Pawlik, T.M.; Torbenson, M.; et al. MicroRNA down-regulated in human cholangiocarcinoma control cell cycle through multiple targets involved in the G1/S checkpoint. Hepatology 2011, 54, 2089–2098. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.K.; Park, M.; Kim, Y.-K.; Tae, Y.K.; Yang, H.-K.; Lee, J.M.; Kim, H. MicroRNA-494 Downregulates KIT and Inhibits Gastrointestinal Stromal Tumor Cell Proliferation. Clin. Cancer Res. 2011, 17, 7584–7594. [Google Scholar] [CrossRef] [PubMed]
- Ghorbanhosseini, S.S.; Nourbakhsh, M.; Zangooei, M.; Abdolvahabi, Z.; Bolandghamtpour, Z.; Hesari, Z.; Yousefi, Z.; Panahi, G.; Meshkani, R. MicroRNA-494 induces breast cancer cell apoptosis and reduces cell viability by inhibition of nicotinamide phosphoribosyltransferase expression and activity. EXCLI J. 2019, 18, 838–851. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lai, L.; Chen, Q.; Song, Y.; Xu, S.; Ma, F.; Wang, X.; Wang, J.; Yu, H.; Cao, X.; et al. MicroRNA-494 is required for the accumulation and functions of tumor-expanded myeloid-derived suppressor cells via targeting of PTEN. J. Immunol. 2012, 188, 5500–5510. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.; Wang, D.; Liu, Y.; Tian, D.; Wang, Y.; Zhang, R.; Yin, L.; Deng, Z. miR-494 inhibits cell proliferation and metastasis via targeting of CDK6 in osteosarcoma. Mol. Med. Rep. 2017, 16, 8627–8634. [Google Scholar] [CrossRef] [PubMed]
- Heiss, M.M.; Allgayer, H.; Gruetzner, K.U.; Funke, I.; Babic, R.; Jauch, K.W.; Schildberg, F.W. Individual development and uPA-receptor expression of disseminated tumour cells in bone marrow: A reference to early systemic disease in solid cancer. Nat. Med. 1995, 1, 1035–1039. [Google Scholar] [CrossRef]
- Allgayer, H.; Heiss, M.M.; Riesenberg, R.; Grutzner, K.U.; Tarabichi, A.; Babic, R.; Schildberg, F.W. Urokinase plasminogen activator receptor (uPA-R): One potential characteristic of metastatic phenotypes in minimal residual tumor disease. Cancer Res. 1997, 57, 1394–1399. [Google Scholar]
- Leupold, J.H.; Yang, H.S.; Colburn, N.H.; Asangani, I.; Post, S.; Allgayer, H. Tumor suppressor Pdcd4 inhibits invasion/intravasation and regulates urokinase receptor (u-PAR) gene expression via Sp-transcription factors. Oncogene 2007, 26, 4550–4562. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, Y.; Zhao, M.; Lin, H.; Wang, W.; Li, D.; Cui, W.; Zhou, C.; Zhong, J.; Huang, C. MiR-494 acts as a tumor promoter by targeting CASP2 in non-small cell lung cancer. Sci. Rep. 2019, 9, 3008–3017. [Google Scholar] [CrossRef] [PubMed]
- Tomasello, L.; Distefano, R.; Nigita, G.; Croce, C.M. The MicroRNA Family Gets Wider: The IsomiRs Classification and Role. Front. Cell Dev. Biol. 2021, 9, 668648. [Google Scholar] [CrossRef] [PubMed]
- Telonis, A.G.; Magee, R.; Loher, P.; Chervoneva, I.; Londin, E.; Rigoutsos, I. Knowledge about the presence or absence of miRNA isoforms (isomiRs) can successfully discriminate amongst 32 TCGA cancer types. Nucleic Acids Res. 2017, 45, 2973–2985. [Google Scholar] [CrossRef] [PubMed]
- Salem, O.; Erdem, N.; Jung, J.; Munstermann, E.; Worner, A.; Wilhelm, H.; Wiemann, S.; Korner, C. The highly expressed 5’isomiR of hsa-miR-140-3p contributes to the tumor-suppressive effects of miR-140 by reducing breast cancer proliferation and migration. BMC Genom. 2016, 17, 566. [Google Scholar] [CrossRef] [PubMed]
- van der Kwast, R.; Woudenberg, T.; Quax, P.H.A.; Nossent, A.Y. MicroRNA-411 and Its 5′-IsomiR Have Distinct Targets and Functions and Are Differentially Regulated in the Vasculature under Ischemia. Mol. Ther. 2020, 28, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Freedman, J.E.; Tanriverdi, K. Defining miRNA targets: Balancing simplicity with complexity. Circulation 2013, 127, 2075–2077. [Google Scholar] [CrossRef]
- Wen, N.; Zhang, J.; Zhang, Q. MiR-494 Inhibits the Proliferation, Migration and Invasion of Cervical Cancer Cells by Regulating LETMD1. Cell. Mol. Biol. 2022, 67, 81–87. [Google Scholar] [CrossRef]
- Yang, Y.; Yang, T.; Zhao, Z.; Zhang, H.; Yuan, P.; Wang, G.; Zhao, Z.; An, J.; Lyu, Z.; Xing, J.; et al. Down-regulation of BMAL1 by MiR-494-3p Promotes Hepatocellular Carcinoma Growth and Metastasis by Increasing GPAM-mediated Lipid Biosynthesis. Int. J. Biol. Sci. 2022, 18, 6129–6144. [Google Scholar] [CrossRef]
- Zhao, X.; Zhou, Y.; Chen, Y.U.; Yu, F. miR-494 inhibits ovarian cancer cell proliferation and promotes apoptosis by targeting FGFR2. Oncol. Lett. 2016, 11, 4245–4251. [Google Scholar] [CrossRef]
- Liborio-Kimura, T.N.; Jung, H.M.; Chan, E.K. miR-494 represses HOXA10 expression and inhibits cell proliferation in oral cancer. Oral. Oncol. 2015, 51, 151–157. [Google Scholar] [CrossRef]
- Cheng, L.; Kong, B.; Zhao, Y.; Jiang, J. miR-494 inhibits cervical cancer cell proliferation through upregulation of SOCS6 expression. Oncol. Lett. 2018, 15, 3075–3080. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patil, N.; Abdelrahim, O.G.; Leupold, J.H.; Allgayer, H. JAK1 Is a Novel Target of Tumor- and Invasion-Suppressive microRNA 494-5p in Colorectal Cancer. Cancers 2024, 16, 24. https://doi.org/10.3390/cancers16010024
Patil N, Abdelrahim OG, Leupold JH, Allgayer H. JAK1 Is a Novel Target of Tumor- and Invasion-Suppressive microRNA 494-5p in Colorectal Cancer. Cancers. 2024; 16(1):24. https://doi.org/10.3390/cancers16010024
Chicago/Turabian StylePatil, Nitin, Omar G. Abdelrahim, Jörg H. Leupold, and Heike Allgayer. 2024. "JAK1 Is a Novel Target of Tumor- and Invasion-Suppressive microRNA 494-5p in Colorectal Cancer" Cancers 16, no. 1: 24. https://doi.org/10.3390/cancers16010024
APA StylePatil, N., Abdelrahim, O. G., Leupold, J. H., & Allgayer, H. (2024). JAK1 Is a Novel Target of Tumor- and Invasion-Suppressive microRNA 494-5p in Colorectal Cancer. Cancers, 16(1), 24. https://doi.org/10.3390/cancers16010024