
Design and Biological Evaluation of Small-Molecule PET-Tracers for Imaging of Programmed Death Ligand 1

 , , ,
, , ,  and
and 
Abstract
Simple Summary
Abstract
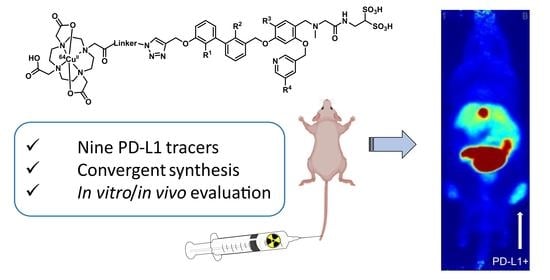
1. Introduction
2. Results
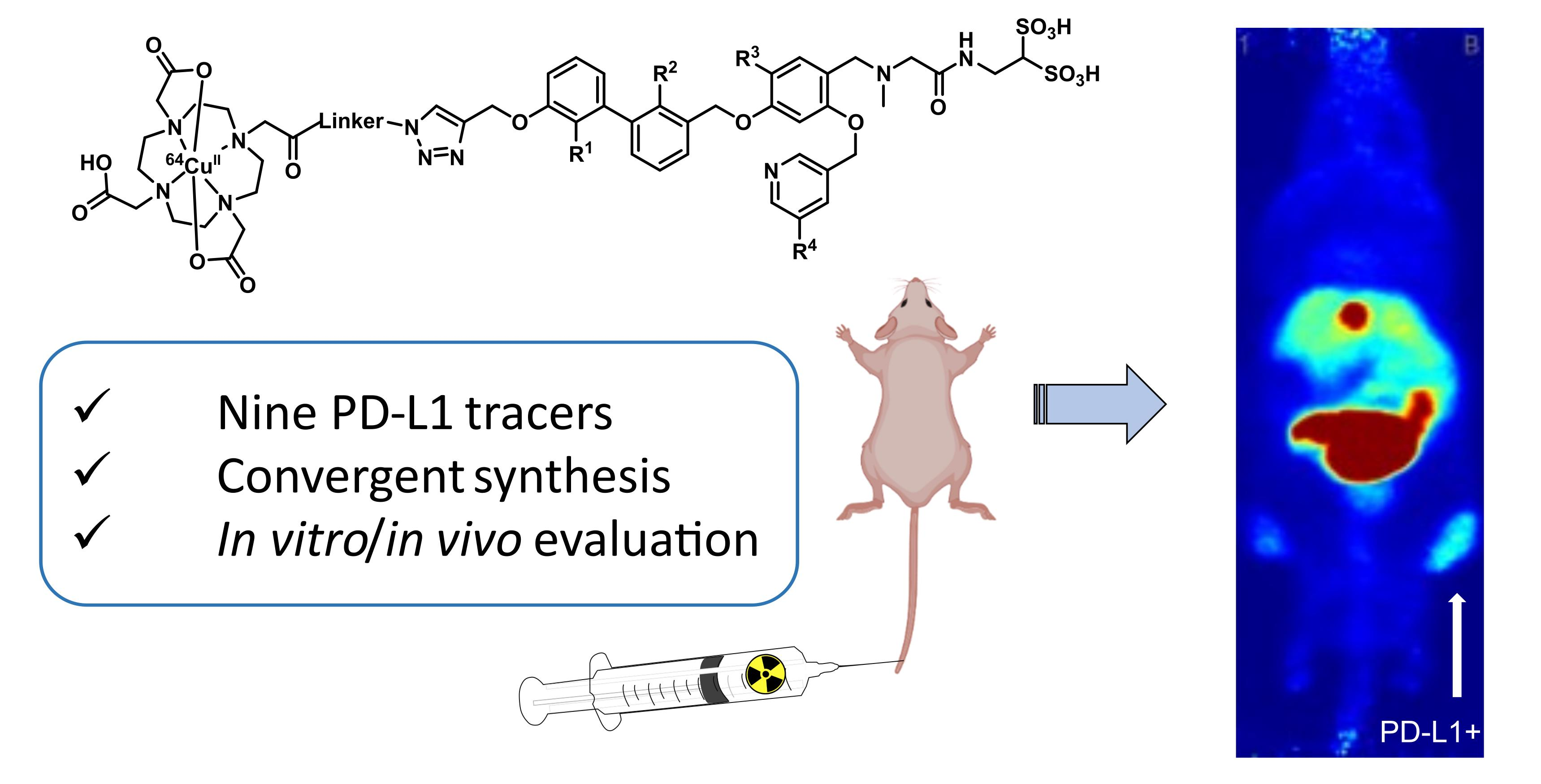
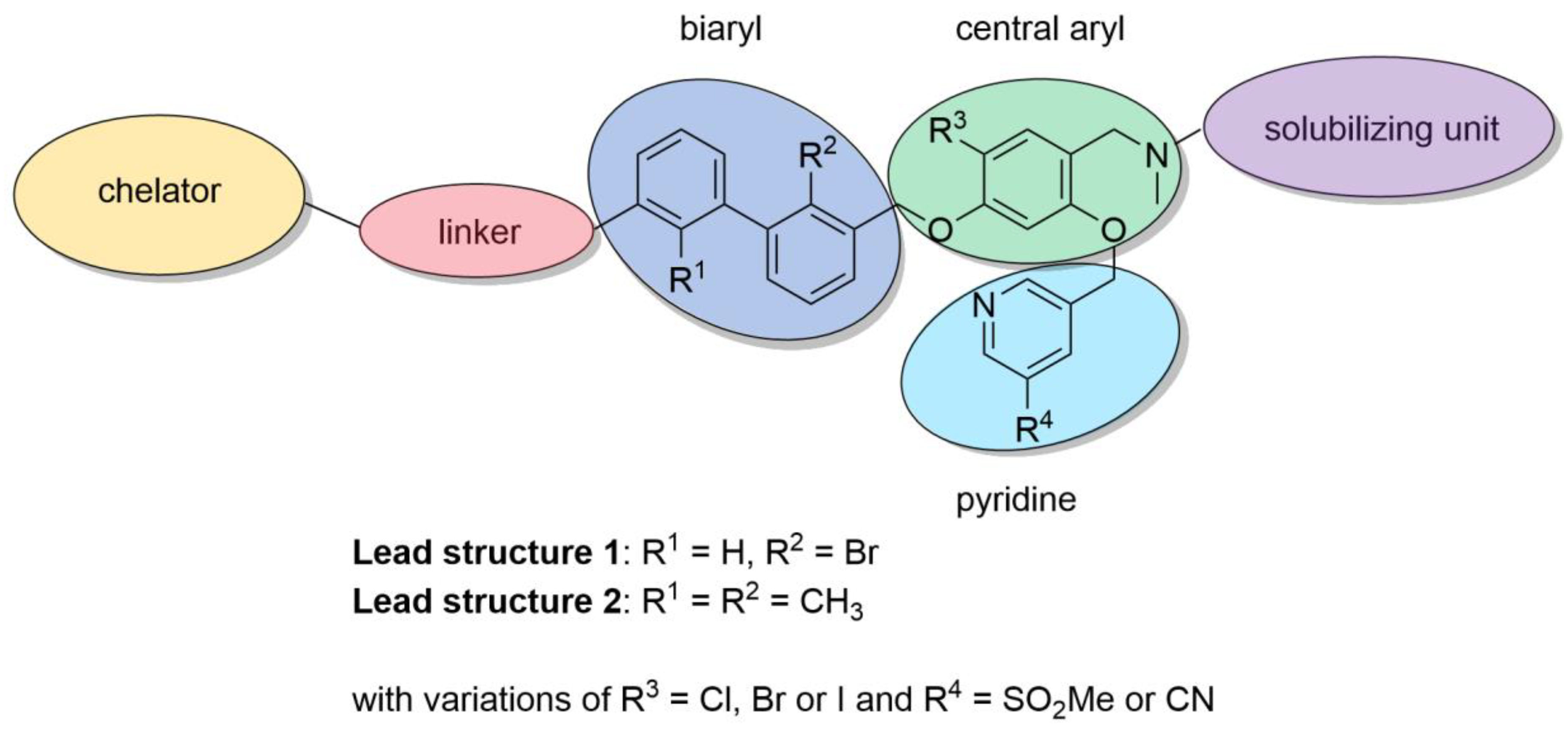
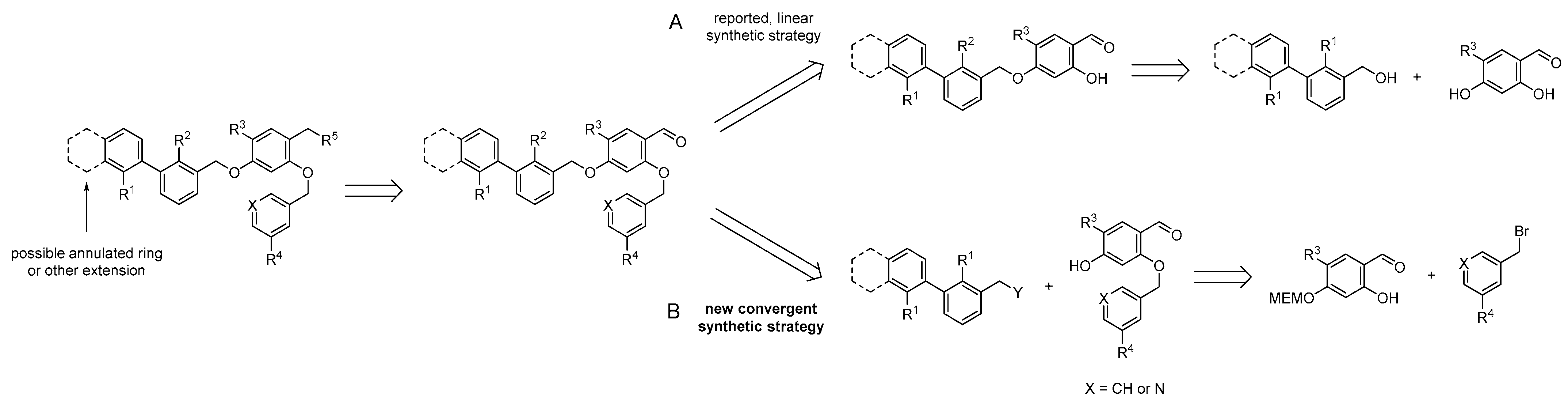
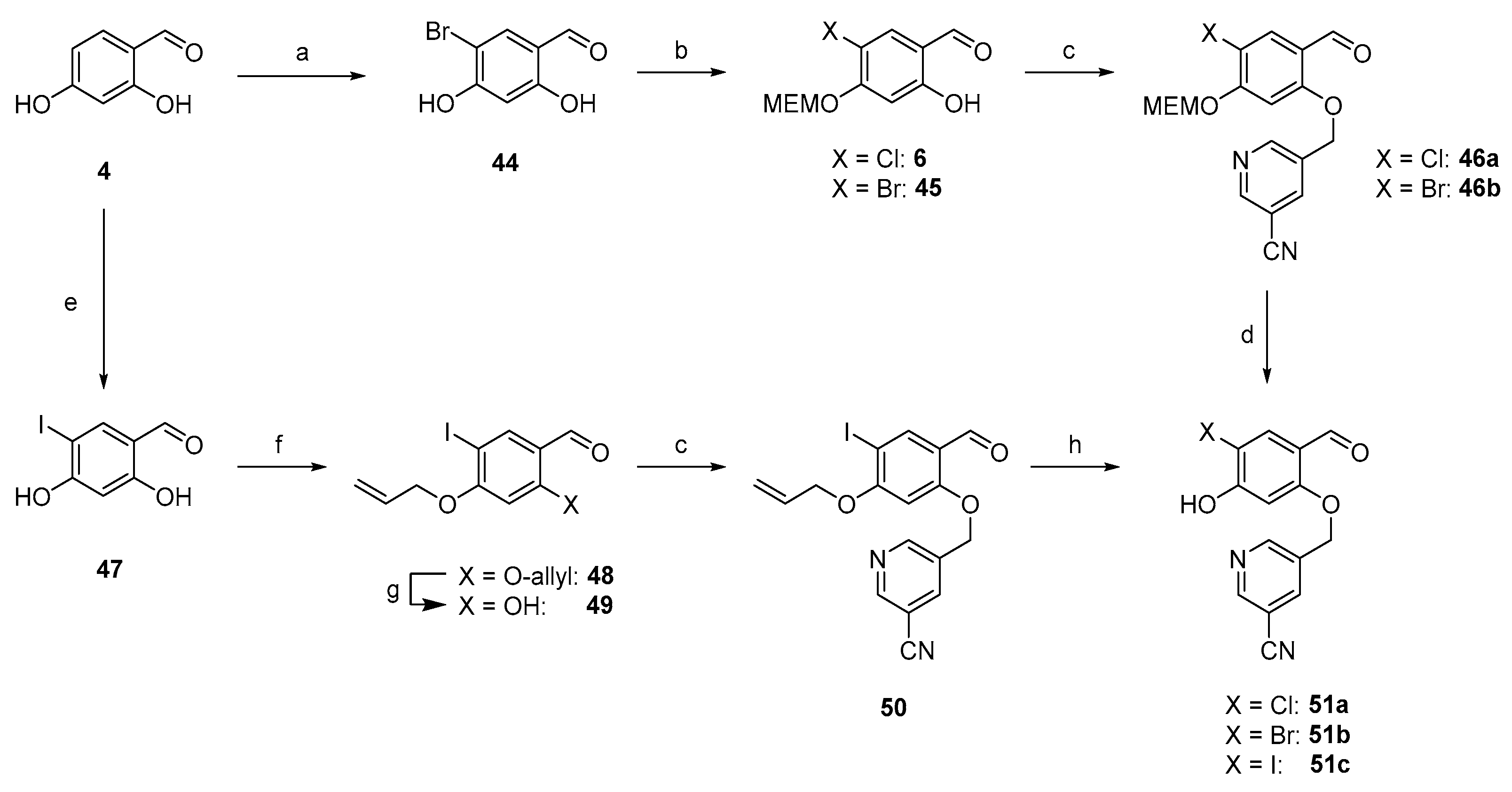
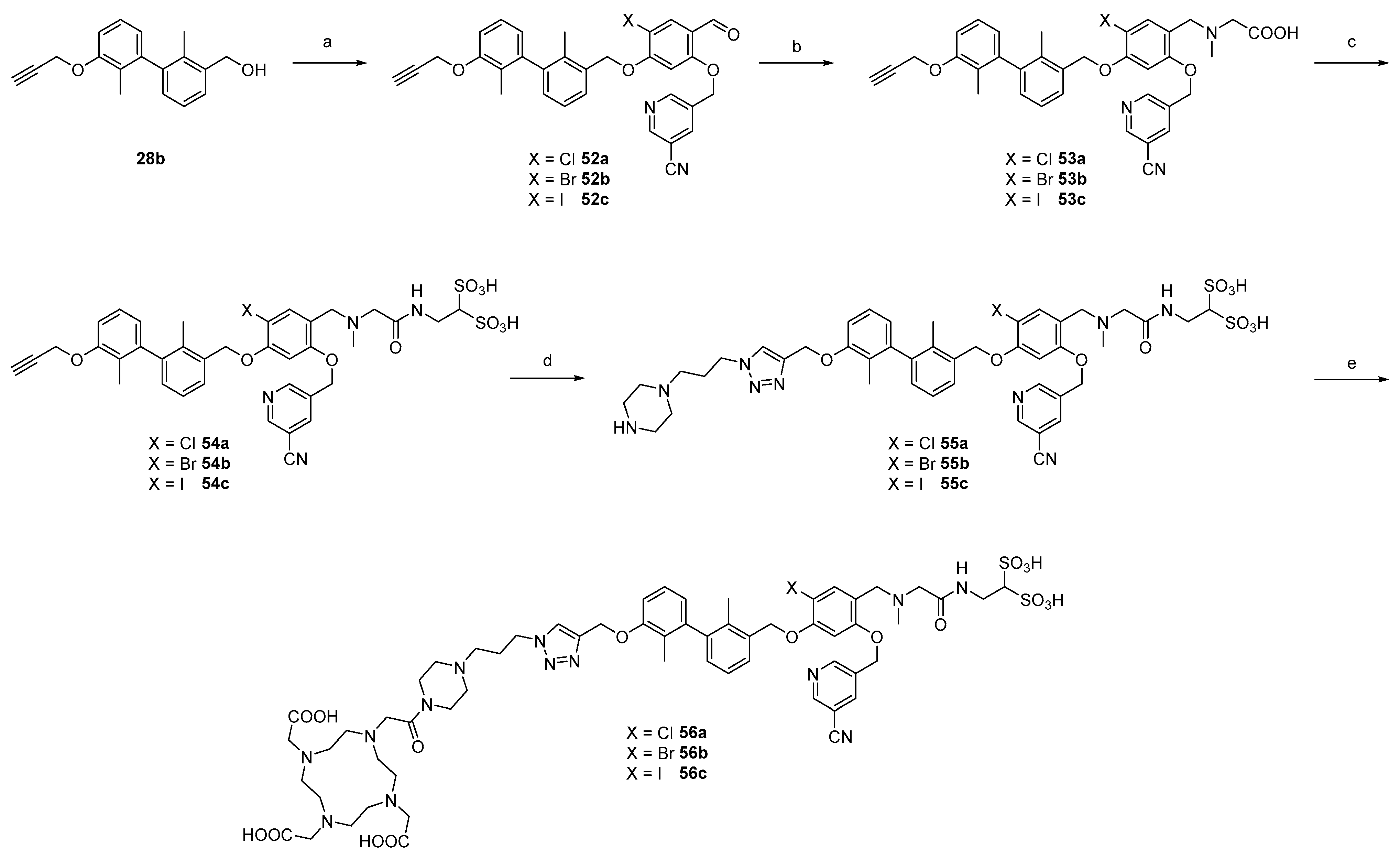
2.1. Organic Synthesis
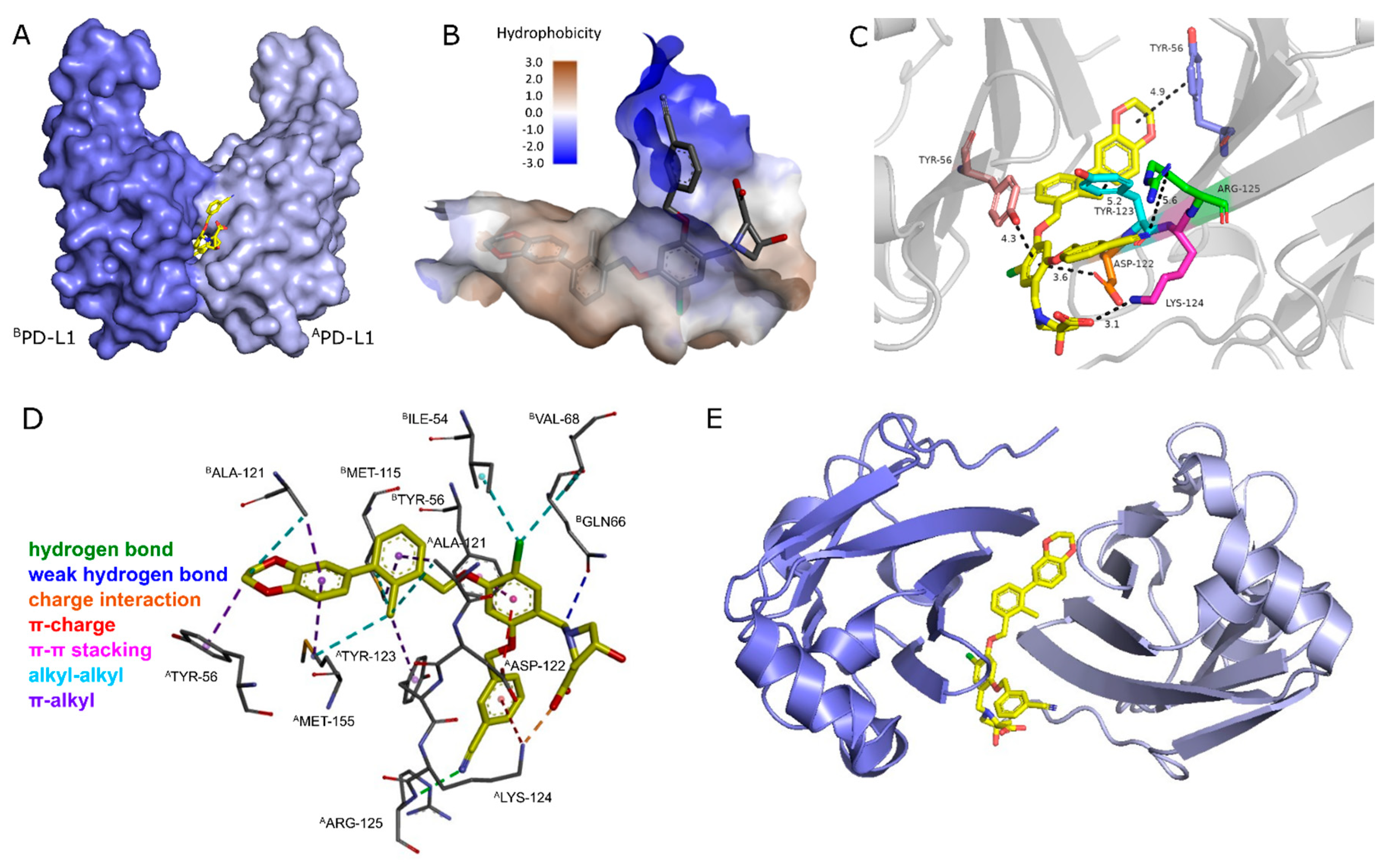
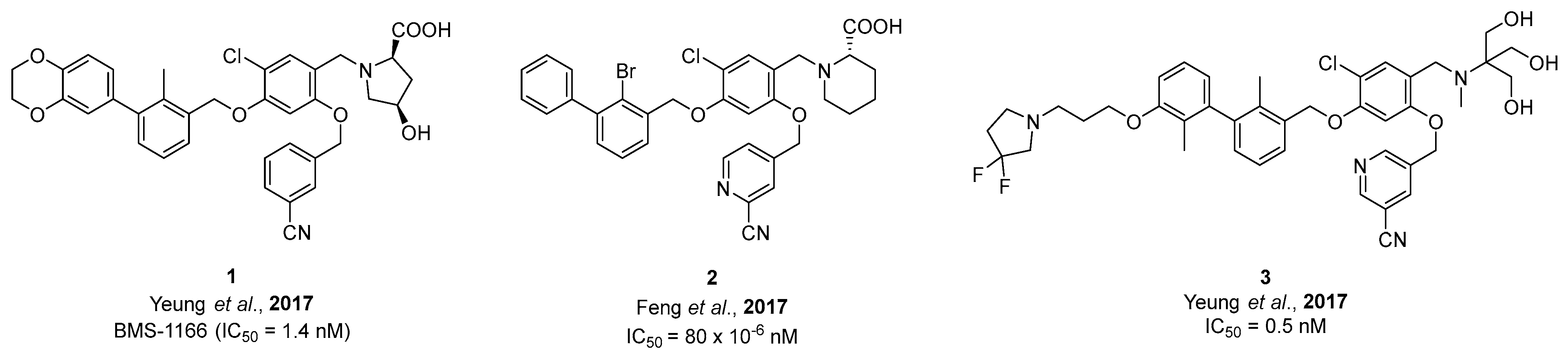
2.1.1. Synthetic Strategy Based on PD-L1 Cocrystal Structures
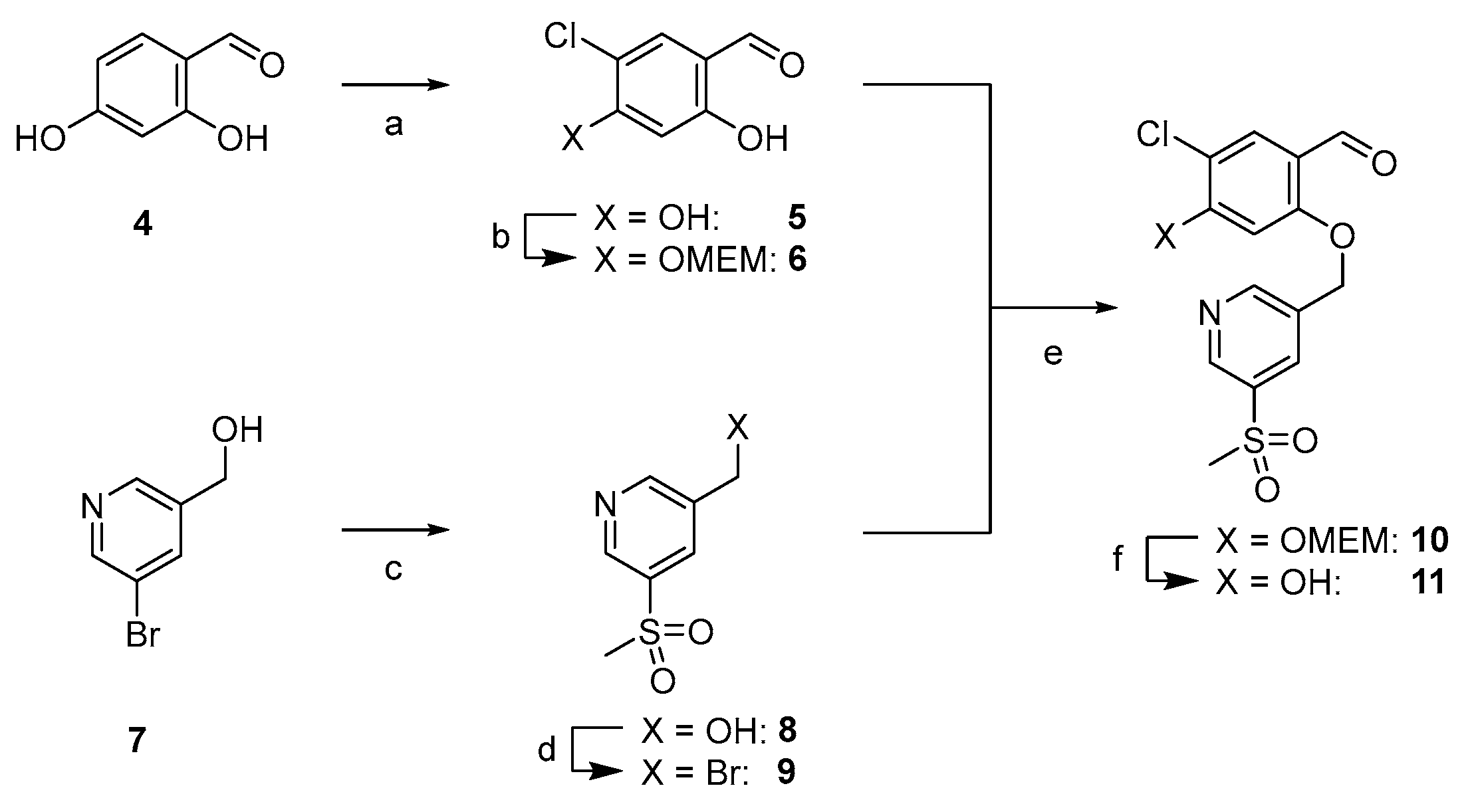
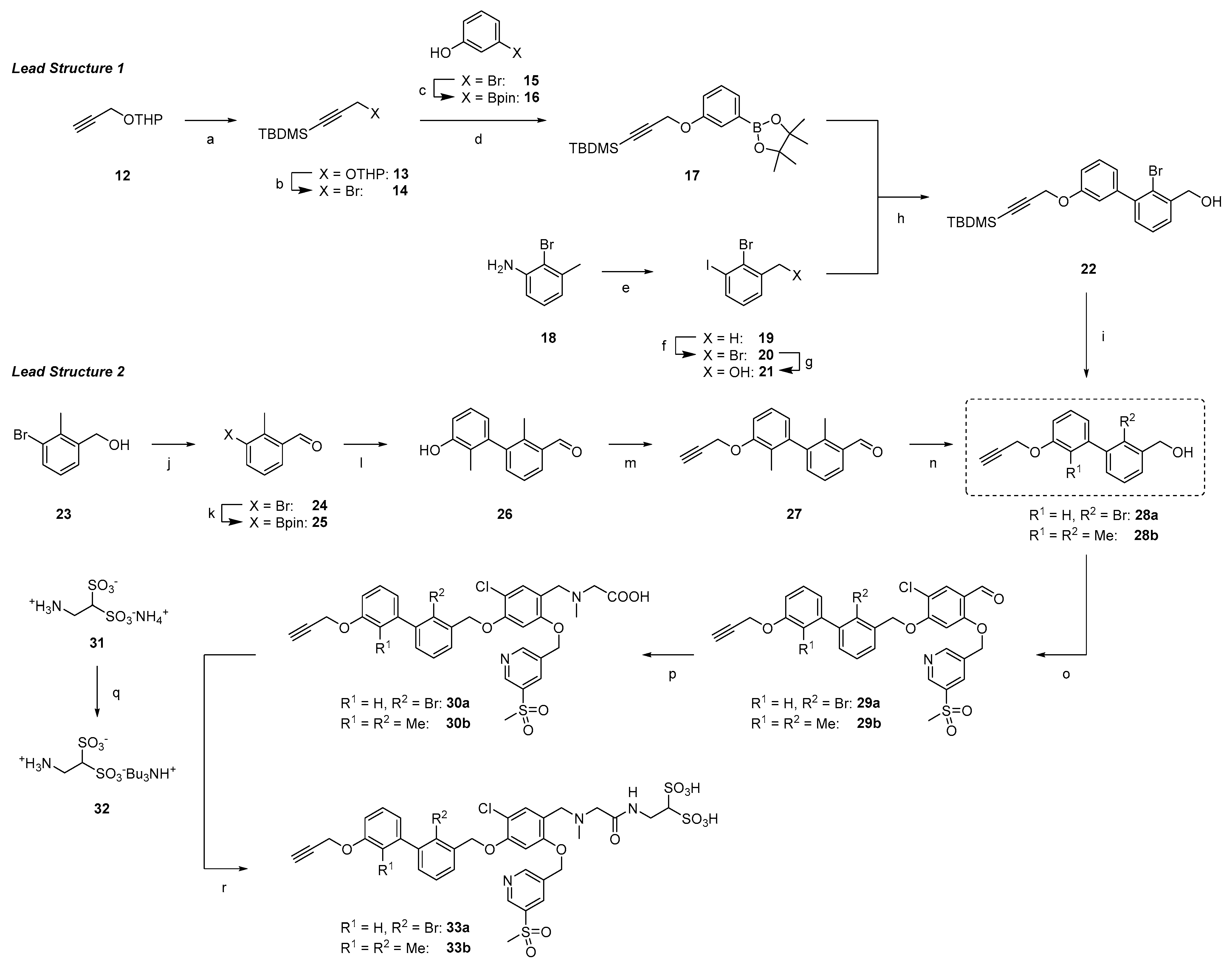
2.1.2. Synthesis of First Series of PD-L1 Ligands
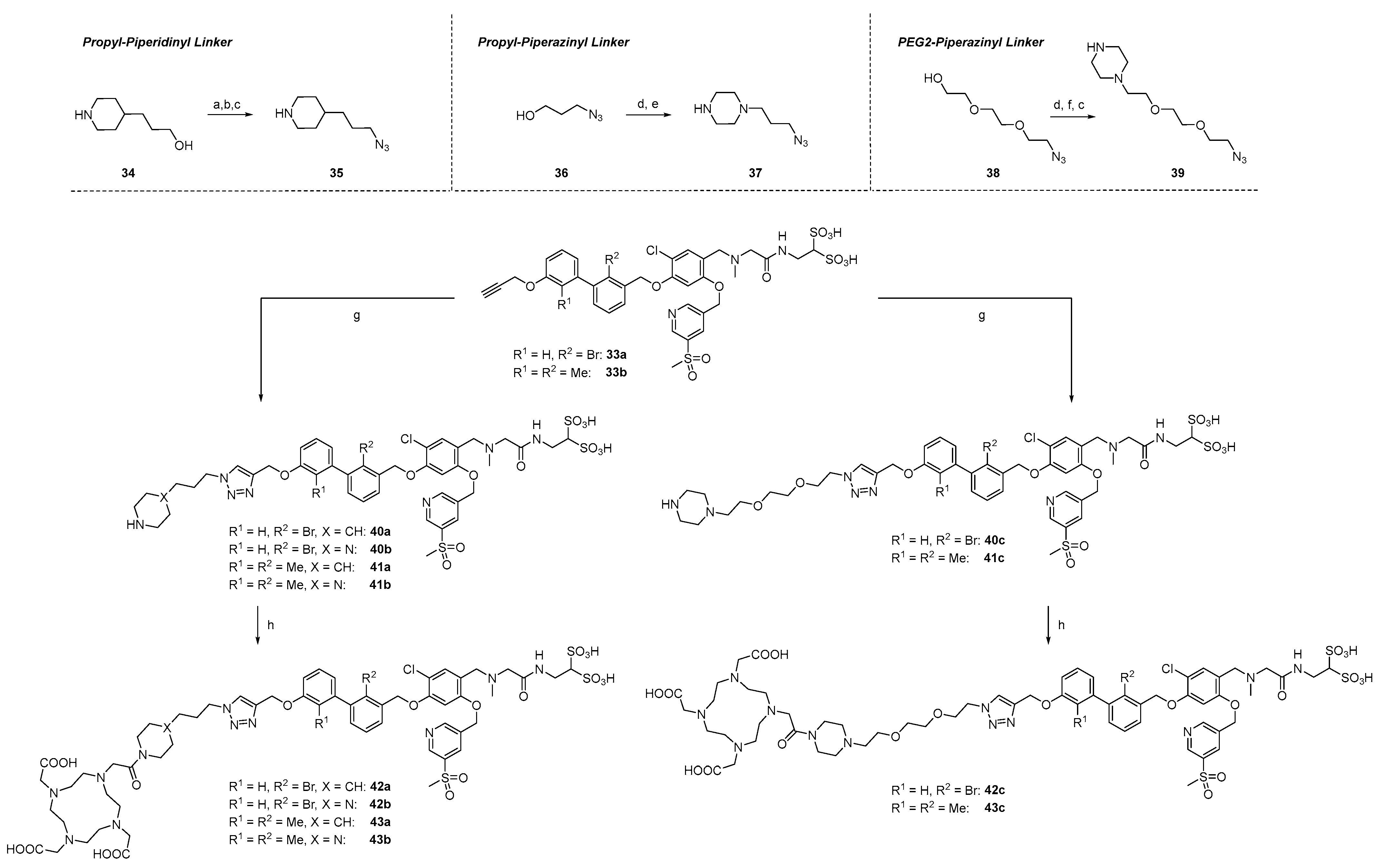
2.1.3. Exploration of Larger Halogens at the Central Aryl Moiety
2.2. Radiochemistry
2.2.1. Radiolabeling with 64Cu, 68Ga, and 177Lu
2.2.2. Determination of logD7.4 Values
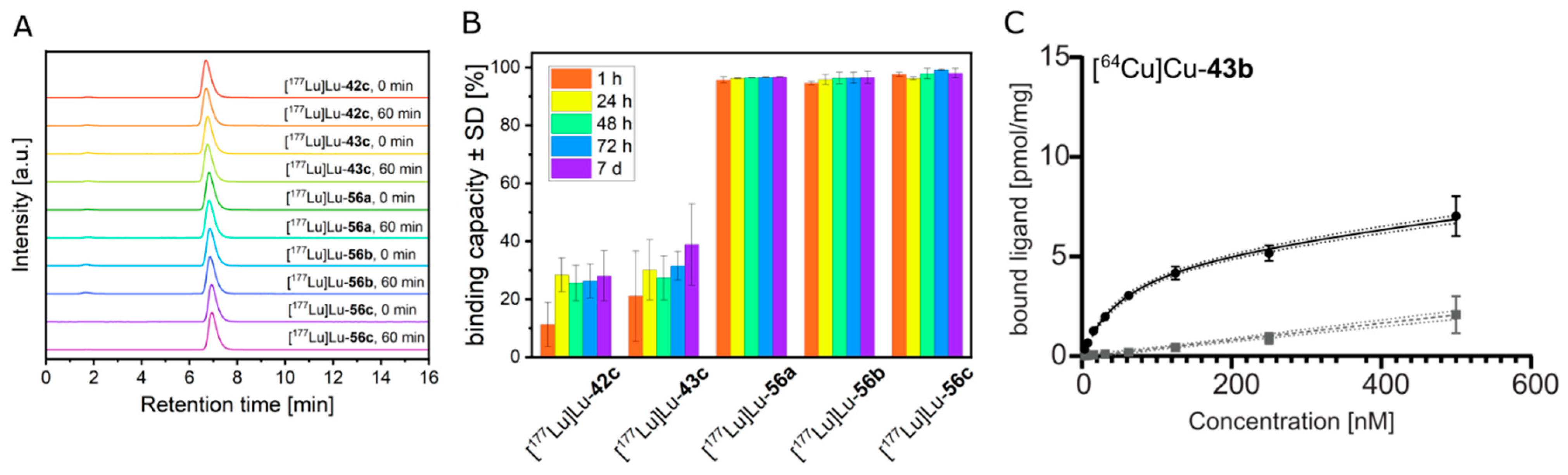
2.3. Stability Studies
2.3.1. Kinetic Stability in Buffer Solutions
2.3.2. Proteolytic Stability in Human Serum
2.3.3. Metabolic Stability
2.4. Albumin Binding Experiments
2.5. In Vitro Evaluation
2.5.1. Saturation Binding
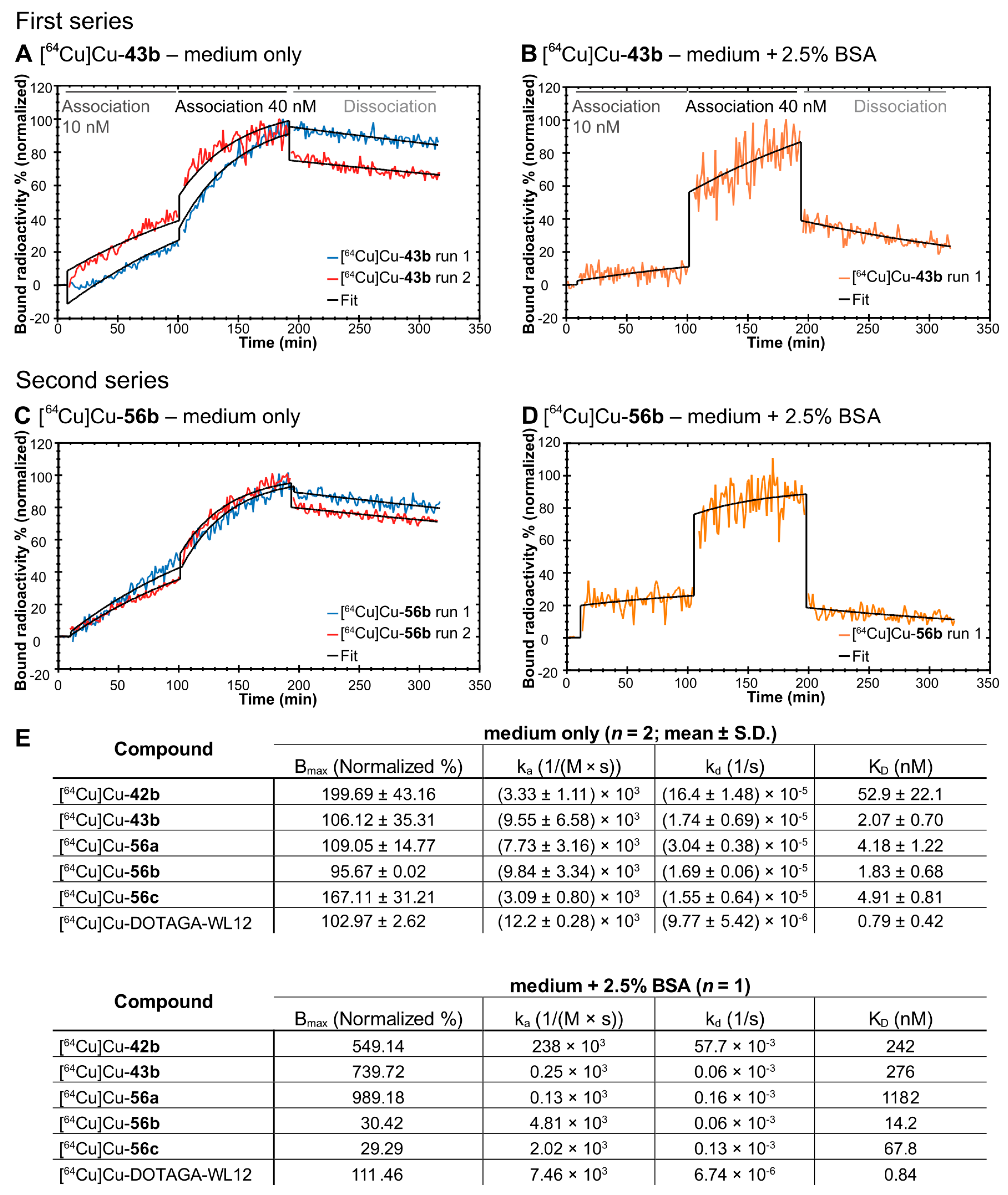
2.5.2. Real-Time Radioligand Binding
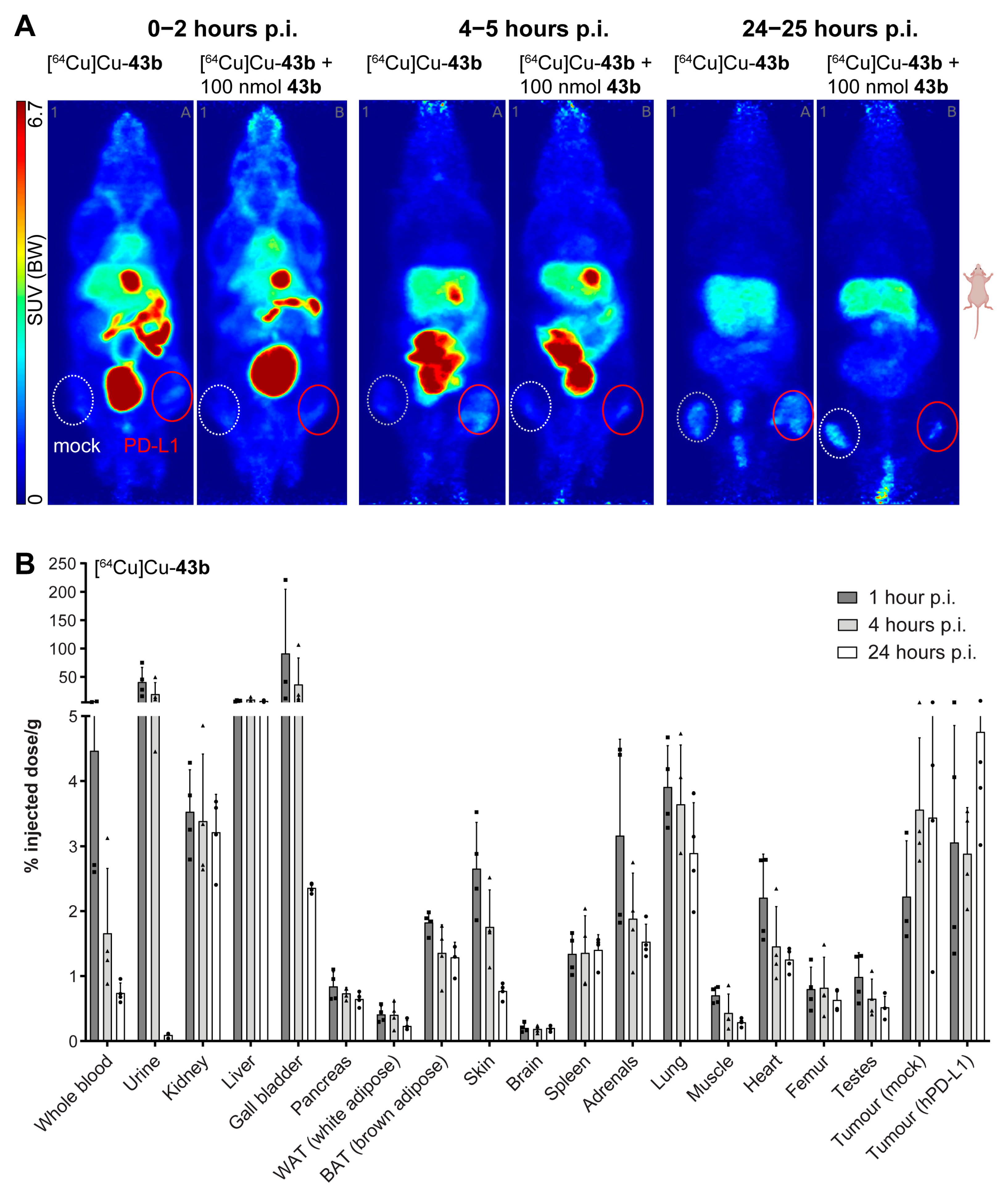
2.6. PET Imaging and Biodistribution
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Organic Synthesis
5.1.1. General Remarks
5.1.2. Synthetic Procedures
5.2. Visualization of PD-L1 Cocrystals
5.3. Radiochemistry
5.3.1. Gallium-68
5.3.2. Copper-64
5.3.3. Lutetium-177
5.3.4. Radiolabeling Procedure
5.4. Log D7.4 Determination
5.5. Stability Studies in Different Buffer Systems
5.5.1. Stability Studies in Human Serum
By Radio-TLC
By Precipitation and Radio-HPLC
Radiotracer Stability in the Presence of Liver Microsomes
5.6. Serum Binding Studies
5.7. Cell Lines and Cell Culture
5.8. Saturation Binding Studies
5.9. Real-Time Radioligand Binding Studies
5.10. PD-L1 Immunostaining
5.11. Animals, Biodistribution and PET Imaging
5.12. Data and Statistical Analysis
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Anderson, N.M.; Simon, M.C. The tumor microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef]
- Hakozaki, T.; Hosomi, Y.; Kitadai, R.; Kitagawa, S.; Okuma, Y. Efficacy of Immune Checkpoint Inhibitor Monotherapy for Patients With Massive Non-small-Cell Lung Cancer. J. Cancer Res. Clin. Oncol. 2020, 146, 2957–2966. [Google Scholar] [CrossRef]
- Guardascione, M.; Toffoli, G. Immune Checkpoint Inhibitors as Monotherapy or Within a Combinatorial Strategy in Advanced Hepatocellular Carcinoma. Int. J. Mol. Sci. 2020, 21, 6302. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Su, W.; Lu, T.; Wang, Y.; Dong, Y.; Qin, Y.; Liu, D.; Sun, L.; Jiao, W. First-Line Immune-Checkpoint Inhibitors in Non-small Cell Lung Cancer: Current Landscape and Future Progress. Front. Pharmacol. 2020, 11, 578091. [Google Scholar] [CrossRef] [PubMed]
- Krutzek, F.; Kopka, K.; Stadlbauer, S. Development of Radiotracers for Imaging of the PD-1/PD-L1 Axis. Pharmaceuticals 2022, 15, 747. [Google Scholar] [CrossRef] [PubMed]
- Lesniak, W.G.; Chatterjee, S.; Gabrielson, M.; Lisok, A.; Wharram, B.; Pomper, M.G.; Nimmagadda, S. PD-L1 Detection in Tumors Using [64Cu]Atezolizumab with PET. Bioconjugate Chem. 2016, 27, 2103–2110. [Google Scholar] [CrossRef]
- Xu, M.; Han, Y.; Liu, G.; Xu, Y.; Duan, D.; Liu, H.; Du, F.; Luo, P.; Liu, Z. Preclinical Study of a Fully Human Anti-PD-L1 Antibody as a Theranostic Agent for Cancer Immunotherapy. Mol. Pharm. 2018, 15, 4426–4433. [Google Scholar] [CrossRef]
- Jagoda, E.M.; Vasalatiy, O.; Basuli, F.; Opina, A.C.L.; Williams, M.R.; Wong, K.; Lane, K.C.; Adler, S.; Ton, A.T.; Szajek, L.P.; et al. Immuno-PET Imaging of the Programmed Cell Death-1 Ligand (PD-L1) Using a Zirconium-89 Labeled Therapeutic Antibody, Avelumab. Mol. Imaging 2019, 18, 1–14. [Google Scholar] [CrossRef]
- Li, D.; Zou, S.; Cheng, S.; Song, S.; Wang, P.; Zhu, X. Monitoring the Response of PD-L1 Expression to Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors in Nonsmall-Cell Lung Cancer Xenografts by Immuno-PET Imaging. Mol. Pharm. 2019, 16, 3469–3476. [Google Scholar] [CrossRef]
- Kikuchi, M.; Clump, D.A.; Srivastava, R.M.; Sun, L.; Zeng, D.; Diaz-Perez, J.A.; Anderson, C.J.; Edwards, W.B.; Ferris, R.L. Preclinical ImmunoPET/CT Imaging Using Zr-89-Labeled Anti-PD-L1 Monoclonal Antibody for Assessing Radiation-Induced PD-L1 Upregulation in head and neck cancer and melanoma. OncoImmunology 2017, 6, e1329071. [Google Scholar] [CrossRef]
- Josefsson, A.; Nedrow, J.R.; Park, S.; Banerjee, S.R.; Rittenbach, A.; Jammes, F.; Tsui, B.; Sgouros, G. Imaging, Biodistribution, and Dosimetry of Radionuclide-Labeled PD-L1 Antibody in an Immunocompetent Mouse Model of Breast Cancer. Cancer Res. 2016, 76, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Ingram, J.R.; Dougan, M.; Rashidian, M.; Knoll, M.; Keliher, E.J.; Garrett, S.; Garforth, S.; Blomberg, O.S.; Espinosa, C.; Bhan, A.; et al. PD-L1 is an Activation-Independent Marker of Brown Adipocytes. Nat. Commun. 2017, 8, 647. [Google Scholar] [CrossRef] [PubMed]
- Lv, G.; Sun, X.; Qiu, L.; Sun, Y.; Li, K.; Liu, Q.; Zhao, Q.; Qin, S.; Lin, J. PET Imaging of Tumor PD-L1 Expression with a Highly Specific Nonblocking Single-Domain Antibody. J. Nucl. Med. 2020, 61, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Bridoux, J.; Broos, K.; Lecocq, Q.; Debie, P.; Martin, C.; Ballet, S.; Raes, G.; Neyt, S.; Vanhove, C.; Breckpot, K.; et al. Anti-Human PD-L1 Nanobody for Immuno-PET Imaging: Validation of a Conjugation Strategy for Clinical Translation. Biomolecules 2020, 10, 1388. [Google Scholar] [CrossRef] [PubMed]
- Broos, K.; Keyaerts, M.; Lecocq, Q.; Renmans, D.; Nguyen, T.; Escors, D.; Liston, A.; Raes, G.; Breckpot, K.; Devoogdt, N. Non-Invasive Assessment of Murine PD-L1 Levels in Syngeneic Tumor Models by Nuclear Imaging with Nanobody Rracers. Oncotarget 2017, 8, 41932–41946. [Google Scholar] [CrossRef]
- Rubins, D.J.; Meng, X.; McQuade, P.; Klimas, M.; Getty, K.; Lin, S.-A.; Connolly, B.M.; O’Malley, S.S.; Haley, H.; Purcell, M.; et al. In Vivo Evaluation and Dosimetry Estimate for a High Affinity Affibody PET Tracer Targeting PD-L1. Mol. Imaging Biol. 2020, 23, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Stutvoet, T.S.; Van Der Veen, E.L.; Kol, A.; Antunes, I.F.; De Vries, E.F.J.; Hospers, G.A.P.; De Vries, E.G.E.; De Jong, S.; Lub-De Hooge, M.N. Molecular Imaging of PD-L1 Expression and Dynamics with the Adnectin-Based PET Tracer 18F-BMS-986192. J. Nucl. Med. 2020, 12, 1839–1844. [Google Scholar] [CrossRef] [PubMed]
- Huisman, M.C.; Niemeijer, A.-L.N.; Windhorst, A.D.; Schuit, R.C.; Leung, D.; Hayes, W.; Poot, A.; Bahce, I.; Radonic, T.; Oprea-Lager, D.E.; et al. Quantification of PD-L1 Expression with 18F-BMS-986192 PET/CT in Patients with Advanced-Stage Non–Small Cell Lung Cancer. J. Nucl. Med. 2020, 61, 1455–1460. [Google Scholar] [CrossRef]
- Vento, J.; Mulgaonkar, A.; Woolford, L.; Nham, K.; Christie, A.; Bagrodia, A.; De Leon, A.D.; Hannan, R.; Bowman, I.; McKay, R.M.; et al. PD-L1 Detection Using 89Zr-Atezolizumab Immuno-PET in Renal Cell Carcinoma Tumorgrafts from a Patient with Favorable Nivolumab Response. J. Immunother. Cancer 2019, 7, 144. [Google Scholar] [CrossRef]
- Xing, Y.; Chand, G.; Liu, C.; Cook, G.J.R.; O’Doherty, J.; Zhao, L.; Wong, N.C.L.; Meszaros, L.K.; Ting, H.H.; Zhao, J. Early Phase I Study of a 99mTc-Labeled Anti–Programmed Death Ligand-1 (PD-L1) Single-Domain Antibody in SPECT/CT Assessment of PD-L1 Expression in Non–Small Cell Lung Cancer. J. Nucl. Med. 2019, 60, 1213–1220. [Google Scholar] [CrossRef]
- Huck, B.R.; Kötzner, L.; Urbahns, K. Small Molecules Drive Big Improvements in Immuno-Oncology Therapies. Angew. Chem. Int. Ed. 2018, 57, 4412–4428. [Google Scholar] [CrossRef]
- Pollack, M.; Betof, A.; Dearden, H.; Rapazzo, K.; Valentine, I.; Brohl, A.; Ancell, K.; Long, G.; Menzies, A.; Eroglu, Z.; et al. Safety of Resuming Anti-PD-1 in Patients With Immune-Related Adverse Events (irAEs) During Combined Anti-CTLA-4 and Anti-PD1 in Metastatic Melanoma. Ann. Oncol. 2018, 29, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Farid, S.S. Process Economics of Industrial Monoclonal Antibody Manufacture. J. Chromatogr. B Biomed. Appl. 2007, 848, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.L.; Smothers, J.; Srinivasan, R.; Hoos, A. Big Opportunities for Small Molecules in Immuno-Oncology. Nat. Rev. Drug Discov. 2015, 14, 603–622. [Google Scholar] [CrossRef]
- De Silva, R.A.; Kumar, D.; Lisok, A.; Chatterjee, S.; Wharram, B.; Rao, K.V.; Mease, R.C.; Dannals, R.F.; Pomper, M.G.; Nimmagadda, S. Peptide-Based 68Ga-PET Radiotracer for Imaging PD-L1 Expression in Cancer. Mol. Pharm. 2018, 15, 3946–3952. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Van Valkenburgh, J.; Hong, X.; Conti, P.S.; Zhang, X.; Chen, K. Small Molecules as Theranostic Agents in Cancer Immunology. Theranostics 2019, 9, 7849–7871. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Lesniak, W.G.; Miller, M.S.; Lisok, A.; Sikorska, E.; Wharram, B.; Kumar, D.; Gabrielson, M.; Pomper, M.G.; Gabelli, S.B.; et al. Rapid PD-L1 Detection in Tumors with PET Using a Highly Specific Peptide. Biochem. Biophys. Res. Commun. 2017, 483, 258–263. [Google Scholar] [CrossRef]
- Liu, H.; Hu, M.; Deng, J.; Zhao, Y.; Peng, D.; Feng, Y.; Wang, L.; Chen, Y.; Qiu, L. A Novel Small Cyclic Peptide-Based 68Ga-Radiotracer for Positron Emission Tomography Imaging of PD-L1 Expression in Tumors. Mol. Pharm. 2021, 19, 138–147. [Google Scholar] [CrossRef]
- Hu, K.; Kuan, H.; Hanyu, M.; Masayuki, H.; Xie, L.; Zhang, Y.; Nagatsu, K.; Kotaro, N.; Suzuki, H.; Hisashi, S.; et al. Developing Native Peptide-Based Radiotracers for PD-L1 Pet Imaging and Improving Imaging Contrast by Pegylation. Chem. Commun. 2019, 55, 4162–4165. [Google Scholar] [CrossRef]
- Zhou, X.; Jiang, J.; Yang, X.; Liu, T.; Ding, J.; Nimmagadda, S.; Pomper, M.G.; Zhu, H.; Zhao, J.; Yang, Z. First-In-Human Evaluation of a PD-L1-Binding Peptide Radiotracer in Non-small Cell Lung Cancer Patients With PET. J. Nucl. Med. 2022, 63, 536–542. [Google Scholar] [CrossRef]
- Chen, T.; Li, Q.; Liu, Z.; Chen, Y.; Feng, F.; Sun, H. Peptide-Based and Small Synthetic Molecule Inhibitors on PD-1/PD-L1 Pathway: A New Choice for Immunotherapy? Eur. J. Med. Chem. 2019, 161, 378–398. [Google Scholar] [CrossRef] [PubMed]
- Guzik, K.; Tomala, M.; Muszak, D.; Konieczny, M.; Hec, A.; Blaszkiewicz, U.; Pustula, M.; Butera, R.; Domling, A.; Holak, T.A. Development of the Inhibitors that Target the PD-1/PD-L1 Interaction-A Brief Look at Progress on Small Molecules, Peptides and Macrocycles. Molecules 2019, 24, 2071. [Google Scholar] [CrossRef]
- Surmiak, E.; Magiera-Mularz, K.; Musielak, B.; Muszak, D.; Kocik-Krol, J.; Kitel, R.; Plewka, J.; Holak, T.A.; Skalniak, L. PD-L1 Inhibitors: Different Classes, Activities, and Mechanisms of Action. Int. J. Mol. Sci. 2021, 22, 11797. [Google Scholar] [CrossRef]
- Miao, Y.; Lv, G.; Chen, Y.; Qiu, L.; Xie, M.; Lin, J. One-Step Radiosynthesis and Initial Evaluation of a Small Molecule PET Tracer for PD-L1 Imaging. Bioorg. Med. Chem. Lett. 2020, 30, 127572. [Google Scholar] [CrossRef] [PubMed]
- Lv, G.; Miao, Y.; Chen, Y.; Lu, C.; Wang, X.; Xie, M.; Qiu, L.; Lin, J. Promising Potential of a 18F-Labelled Small-Molecular Radiotracer to Evaluate PD-L1 Expression in Tumors by PET Imaging. Bioorg. Chem. 2021, 115, 105294. [Google Scholar] [CrossRef]
- Brust, P.; van den Hoff, J.; Steinbach, J. Development of 18 F-Labeled Radiotracers for Neuroreceptor Imaging With Positron Emission Tomography. Neurosci. Bull. 2014, 30, 777–811. [Google Scholar] [CrossRef] [PubMed]
- Jennings, M.; Marcu, L.G.; Bezak, E. PET-Specific Parameters and Radiotracers in Theoretical Tumour Modelling. Comput. Math. Methods Med. 2015, 2015, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Fowler, J.S.; Volkow, N.D.; Wang, G.J.; Ding, Y.S.; Dewey, S.L. PET and Drug Research and Development. J. Nucl. Med. 1999, 40, 1154–1163. [Google Scholar] [PubMed]
- Imlimthan, S.; Moon, E.S.; Rathke, H.; Afshar-Oromieh, A.; Rösch, F.; Rominger, A.; Gourni, E. New Frontiers in Cancer Imaging and Therapy Based on Radiolabeled Fibroblast Activation Protein Inhibitors: A Rational Review and Current Progress. Pharmaceuticals 2021, 14, 1023. [Google Scholar] [CrossRef]
- Altmann, A.; Haberkorn, U.A.; Siveke, J. The Latest Developments in Imaging of Fibroblast Activation Protein. J. Nucl. Med. 2021, 62, 160–167. [Google Scholar] [CrossRef]
- Lindner, T.; Altmann, A.; Giesel, F.; Kratochwil, C.; Kleist, C.; Krämer, S.; Mier, W.; Cardinale, J.; Kauczor, H.-U.; Jäger, D.; et al. 18F-Labeled Tracers Targeting Fibroblast Activation Protein. EJNMMI Radiopharm. Chem. 2021, 6, 26. [Google Scholar]
- Lindner, T.; Loktev, A.; Giesel, F.; Kratochwil, C.; Altmann, A.; Haberkorn, U. Targeting of Activated Fibroblasts for Imaging and Therapy. EJNMMI Radiopharm. Chem. 2019, 4, 16. [Google Scholar] [CrossRef]
- Muszak, D.; Surmiak, E.; Plewka, J.; Magiera-Mularz, K.; Kocik, J.; Musielak, B.; Sala, D.; Kitel, R.; Stec, M.; Siedlar, M. Terphenyl-based Small-Molecule Inhibitors of Programmed Cell Death-1/Programmed Death-Ligand 1 Protein− Protein Interaction. J. Med. Chem. 2021, 64, 11614–11636. [Google Scholar] [CrossRef]
- Guzik, K.; Zak, K.M.; Grudnik, P.; Magiera, K.; Musielak, B.; Torner, R.; Skalniak, L.; Domling, A.; Dubin, G.; Holak, T.A. Small-Molecule Inhibitors of the Programmed Cell Death-1/Programmed Death-Ligand 1 (PD-1/PD-L1) Interaction via Transiently Induced Protein States and Dimerization of PD-L1. J. Med. Chem. 2017, 60, 5857–5867. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, K.; Chen, H.; Feng, Z. Design, Synthesis, Evaluation, and SAR of 4-Phenylindoline Derivatives, a Novel Class of Small-Molecule Inhibitors of the Programmed Cell Death-1/Programmed Cell Death-Ligand 1 (PD-1/PD-L1) Interaction. Eur. J. Med. Chem. 2021, 211, 113001. [Google Scholar] [CrossRef]
- Kitel, R.; Rodríguez, I.; Del Corte, X.; Atmaj, J.; Zarnik, M.; Surmiak, E.; Muszak, D.; Magiera-Mularz, K.; Popowicz, G.M.; Holak, T.A. Exploring the Surface of the Ectodomain of the PD-L1 Immune Checkpoint with Small-Molecule Fragments. ACS Chem. Biol. 2022, 17, 2655–2663. [Google Scholar] [CrossRef] [PubMed]
- Konieczny, M.; Musielak, B.; Kocik, J.; Skalniak, L.; Sala, D.; Czub, M.; Magiera-Mularz, K.; Rodriguez, I.; Myrcha, M.; Stec, M.; et al. Di-Bromo-Based Small-Molecule Inhibitors of the PD-1/PD-L1 Immune Checkpoint. J. Med. Chem. 2020, 63, 11271–11285. [Google Scholar] [CrossRef]
- Yeung, K.-S.; Connolly, T.P.; Frennesson, D.B.; Grant-Young, K.A.; Hewawasam, P.; Langley, D.R.; Meng, Z.; Mull, E.; Parcella, K.E.; Saulnier, M.G.; et al. Compounds Useful as Immunomodulators. Patent WO 2017/066227 A1, 20 April 2017. [Google Scholar]
- Basu, S.; Yang, J.; Xu, B.; Magiera-Mularz, K.; Skalniak, L.; Musielak, B.; Kholodovych, V.; Holak, T.A.; Hu, L. Design, Synthesis, Evaluation, and Structural Studies of C2-Symmetric Small Molecule Inhibitors of Programmed Cell Death-1/Programmed Death-Ligand 1 Protein–Protein Interaction. J. Med. Chem. 2019, 62, 7250–7263. [Google Scholar] [CrossRef]
- Zhu, P.; Zhang, J.; Yang, Y.; Wang, L.; Zhou, J.; Zhang, H. Design, Synthesis and Biological Evaluation of Isoxazole-Containing Biphenyl Derivatives as Small-Molecule Inhibitors Targeting the Programmed Cell Death-1/Programmed Cell Death-Ligand 1 Immune Checkpoint. Mol. Divers. 2022, 26, 245–264. [Google Scholar] [CrossRef]
- Feng, Z.; Chen, X.; Yang, Y.; Lai, F. Bromobenzyl ether derivatives as PD-1/PD-L1 inhibitors, preparation method therefor, and pharmaceutical composition and uses thereof. Patent WO2017/202275 A1, 30 November 2017. [Google Scholar]
- Musso, L.; Cincinelli, R.; Giannini, G.; Manetti, F.; Dallavalle, S. Synthesis of 5,6-dihydro-4H-benzo[d]isoxazol-7-one and 5,6-dihydro-4H-isoxazolo[5,4-c]pyridin-7-one Derivatives as Potential Hsp90 Inhibitors. Chem. Biol. Drug. Des. 2015, 86, 1030–1035. [Google Scholar] [CrossRef]
- Wünsch, S.; Breit, B. Probing o-Diphenylphosphanyl Benzoate (o-DPPB)-Directed C=C Bond Formation: Total Synthesis of Dictyostatin. Chem. Eur. J. 2015, 21, 2358–2363. [Google Scholar] [CrossRef] [PubMed]
- König, B.; Pitsch, W.; Dix, I.; Jones, P. Synthesis and Structure of Bicyclic Enediynes IA Twofold Carbenoid Ring Closure. New J. Chem. 2001, 25, 912–916. [Google Scholar] [CrossRef]
- Xiong, X.; Yeung, Y.-Y. Ammonium Salt-Catalyzed Highly Practical Ortho-Selective Monohalogenation and Phenylselenation of Phenols: Scope and Applications. ACS Catal. 2018, 8, 4033–4043. [Google Scholar] [CrossRef]
- Chougala, B.M.; Samundeeswari, S.; Holiyachi, M.; Shastri, L.A. Mild, Efficient and Catalyst-Free Hydroxylation of Alkyl Halides in Water: Significant Enhancement of Water Nucleophilicity in Dipolar Solvents. ChemistrySelect 2017, 2, 1290–1296. [Google Scholar] [CrossRef]
- Fleming, P.E.; Shi, Z.; Chen, S.; Schmidt, J.F.; Reader, J.C.; Hone, N.D.; Civarri, J.P. 2-(Amino-Substituted)-4-Aryl Pyrimidines and Related Compounds Useful for Treating Inflammatory Diseases. Patent WO 2005/066139 A2, 21 July 2005. [Google Scholar]
- Romieu, A.; Tavernier-Lohr, D.; Pellet-Rostaing, S.; Lemaire, M.; Renard, P.-Y. Water solubilization of xanthene dyes by post-synthetic sulfonation in organic media. Tetrahedron Lett. 2010, 51, 3304–3308. [Google Scholar] [CrossRef]
- Zhang, R.-H.; Guo, H.-Y.; Deng, H.; Li, J.; Quan, Z.-S. Piperazine skeleton in the structural modification of natural products: A review. J. Enzym. Inhib. Med. Chem. 2021, 36, 1165–1197. [Google Scholar] [CrossRef]
- Lindner, T.; Loktev, A.; Altmann, A.; Giesel, F.; Kratochwil, C.; Debus, J.; Jäger, D.; Mier, W.; Haberkorn, U. Development of quinoline-based theranostic ligands for the targeting of fibroblast activation protein. J. Nucl. Med. 2018, 59, 1415–1422. [Google Scholar] [CrossRef]
- Kitamura, M.; Koga, T.; Yano, M.; Okauchi, T. Direct Synthesis of Organic Azides from Alchols Using 2-Azido-1,3-dimethyl-imiadzolinium Hexafluorophosphate. Synlett 2012, 23, 1335–1338. [Google Scholar] [CrossRef]
- Pretze, M.; Mamat, C. Automated Preparation of [18F]AFP and [18F]BFP: Two Novel Bifunctional 18F-Labeling Building Blocks for Huisgen-Click. J. Fluor. Chem. 2013, 150, 25–35. [Google Scholar] [CrossRef]
- Mier, W.; Hoffend, J.; Krämer, S.; Schuhmacher, J.; Hull, W.E.; Eisenhut, M.; Haberkorn, U. Conjugation of DOTA Using Isolated Phenolic Active Esters: The Labeling and Biodistribution of Albumin as Blood Pool Marker. Bioconjugate Chem. 2005, 16, 237–240. [Google Scholar] [CrossRef]
- Liu, Z.; Yoon, G.; Cheon, S.H. An Enantioselective Total Synthesis of (S)-(−)-Licochalcone E: Determination of the Absolute Configuration. Tetrahedron 2010, 66, 3165–3172. [Google Scholar] [CrossRef]
- Enders, D.; Fronert, J.; Bisschops, T.; Boeck, F. Asymmetric Total Synthesis of Smyrindiol Employing an Organocatalytic Aldol Key Step. Beilstein J. Org. Chem. 2012, 8, 1112–1117. [Google Scholar] [CrossRef] [PubMed]
- Vincent, J.B.; Love, S. The Binding and Transport of Alternative Metals by Transferrin. Biochim. Biophys. Acta 2012, 1820, 362–378. [Google Scholar] [CrossRef]
- Benjamín-Rivera, J.A.; Cardona-Rivera, A.E.; Vázquez-Maldonado, Á.L.; Dones-Lassalle, C.Y.; Pabón-Colon, H.L.; Rodríguez-Rivera, H.M.; Rodríguez, I.; González-Espiet, J.C.; Pazol, J.; Pérez-Ríos, J.D. Exploring serum transferrin regulation of nonferric metal therapeutic function and toxicity. Inorganics 2020, 8, 48. [Google Scholar] [CrossRef]
- Jiang, J.; Li, D.; Liu, T.; Xia, L.; Guo, X.; Meng, X.; Liu, F.; Wang, F.; Yang, Z.; Zhu, H. Noninvasive evaluation of PD-L1 expression using Copper-64 labeled peptide WL12 by micro-PET imaging in Chinese hamster ovary cell tumor model. Bioorganic Med. Chem. Lett. 2021, 40, 127901. [Google Scholar] [CrossRef] [PubMed]
- Björke, H.; Andersson, K. Automated, high-resolution cellular retention and uptake studies in vitro. Appl. Radiat. Isot. 2006, 64, 901–905. [Google Scholar] [CrossRef]
- Li, H.; van der Merwe, P.A.; Sivakumar, S. Biomarkers of Response to PD-1 Pathway Blockade. Br. J. Cancer 2022, 126, 1663–1675. [Google Scholar] [CrossRef]
- Yamashita, K.; Iwatsuki, M.; Harada, K.; Koga, Y.; Kiyozumi, Y.; Eto, K.; Hiyoshi, Y.; Ishimoto, T.; Iwagami, S.; Baba, Y.; et al. Can PD-L1 Expression Evaluated by Biopsy Sample Accurately Reflect Its Expression in the Whole Tumour in Gastric Cancer? Br. J. Cancer 2019, 121, 278–280. [Google Scholar] [CrossRef]
- Torlakovic, E.; Lim, H.J.; Adam, J.; Barnes, P.; Bigras, G.; Chan, A.W.H.; Cheung, C.C.; Chung, J.-H.; Couture, C.; Fiset, P.O.; et al. “Interchangeability” of PD-L1 Immunohistochemistry Assays: A Meta-Analysis of Diagnostic Accuracy. Mod. Pathol. 2020, 33, 4–17. [Google Scholar] [CrossRef]
- O’Malley, D.P.; Yang, Y.; Boisot, S.; Sudarsanam, S.; Wang, J.-F.; Chizhevsky, V.; Zhao, G.; Arain, S.; Weiss, L.M. Immunohistochemical Detection of PD-L1 Among Diverse Human Neoplasms in a Reference Laboratory: Observations Based Upon 62,896 Cases. Mod. Pathol. 2019, 32, 929–942. [Google Scholar] [CrossRef]
- Parra, E.R.; Villalobos, P.; Mino, B.; Rodriguez-Canales, J. Comparison of Different Antibody Clones for Immunohistochemistry Detection of Programmed Cell Death Ligand 1 (PD-L1) on Non-Small Cell Lung Carcinoma. Appl. Immunohistochem. Mol. Morphol. 2018, 26, 83–93. [Google Scholar] [CrossRef]
- Haslam, A.; Gill, J.; Prasad, V. Estimation of the Percentage of US Patients With Cancer Who Are Eligible for Immune Checkpoint Inhibitor Drugs. JAMA Netw. Open 2020, 3, e200423. [Google Scholar] [CrossRef] [PubMed]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.; Weber, J.S.; et al. Five-Year Survival Outcomes for Patients With Advanced Melanoma Treated With Pembrolizumab in Keynote-001. Ann. Oncol. 2019, 30, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Verhoeff, S.R.; van den Heuvel, M.M.; van Herpen, C.M.L.; Piet, B.; Aarntzen, E.; Heskamp, S. Programmed Cell Death-1/Ligand-1 PET Imaging: A Novel Tool to Optimize Immunotherapy? PET Clin. 2020, 15, 35–43. [Google Scholar] [CrossRef]
- Liu, C.; Zhou, F.; Yan, Z.; Shen, L.; Zhang, X.; He, F.; Wang, H.; Lu, X.; Yu, K.; Zhao, Y. Discovery of a Novel, Potent and Selective Small-Molecule Inhibitor of PD-1/PD-L1 Interaction With Robust in Vivo Anti-Tumour Efficacy. Br. J. Pharmacol. 2021, 178, 2651–2670. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Jin, Y.; Wang, B.; Liu, B. Molecular Mechanism of Small-Molecule Inhibitors in Blocking the PD-1/PD-L1 Pathway Through PD-L1 Dimerization. Int J Mol Sci 2021, 22, 4766. [Google Scholar] [CrossRef]
- Jouini, N.; Cardinale, J.; Mindt, T.L. Evaluation of a Radiolabeled Macrocyclic Peptide as Potential PET Imaging Probe for PD-L1. ChemMedChem 2022, 17, e202200091. [Google Scholar] [CrossRef] [PubMed]
- Lesniak, W.G.; Mease, R.C.; Chatterjee, S.; Kumar, D.; Lisok, A.; Wharram, B.; Kalagadda, V.R.; Emens, L.A.; Pomper, M.G.; Nimmagadda, S. Development of [18F]FPy-WL12 as a PD-L1 Specific PET Imaging Peptide. Mol. Imaging 2019, 18, 1–9. [Google Scholar] [CrossRef]
- Brandt, M.; Cardinale, J.; Giammei, C.; Guarrochena, X.; Happl, B.; Jouini, N.; Mindt, T.L. Mini-Review: Targeted Radiopharmaceuticals Incorporating Reversible, Low Molecular Weight Albumin Binders. Nucl. Med. Biol. 2019, 70, 46–52. [Google Scholar] [CrossRef]
- Tsopelas, C.; Sutton, R. Why certain dyes are useful for localizing the sentinel lymph node. J. Nucl. Med. 2002, 43, 1377–1382. [Google Scholar]
- Maes, V.; Engelborghs, Y.; Hoebeke, J.; Maras, Y.; Vercruysse, A. Fluorimetric analysis of the binding of warfarin to human serum albumin. Equilibrium and kinetic study. Mol. Pharmacol. 1982, 21, 100–107. [Google Scholar] [PubMed]
- Zheng, X.; Li, Z.; Podariu, M.I.; Hage, D.S. Determination of rate constants and equilibrium constants for solution-phase drug–protein interactions by ultrafast affinity extraction. Anal. Chem. 2014, 86, 6454–6460. [Google Scholar] [CrossRef] [PubMed]
- Peters, T., Jr. All About Albumin: Biochemistry, Genetics, and Medical Applications; Academic Press: Cambridge, MA, USA, 1995. [Google Scholar]
- Brandt, F.; Ullrich, M.; Laube, M.; Kopka, K.; Bachmann, M.; Löser, R.; Pietzsch, J.; Pietzsch, H.-J.; Hoff, J.V.D.; Wodtke, R. “Clickable” Albumin Binders for Modulating the Tumor Uptake of Targeted Radiopharmaceuticals. J. Med. Chem. 2021, 65, 710–733. [Google Scholar] [CrossRef] [PubMed]
- Boswell, C.A.; Sun, X.; Niu, W.; Weisman, G.R.; Wong, E.H.; Rheingold, A.L.; Anderson, C.J. Comparative in Vivo Stability of Copper-64-Labeled Cross-Bridged and Conventional Tetraazamacrocyclic Complexes. J. Med. Chem. 2004, 47, 1465–1474. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Han, Y.; Hu, K.; Huang, S.; Wang, M.; Zhou, K.; Fu, L.; Chen, H.; Tang, G. Synthesis and Biological Evaluation of Al[18F]-NOTA-IPB-PDL1P as a Molecular Probe for PET Imaging of PD-L1 Positive Tumors. Bioorg. Chem. 2022, 122, 105682. [Google Scholar] [CrossRef]
- Riccio, A.; Coletti, A.; Dolciami, D.; Mammoli, A.; Cerra, B.; Moretti, S.; Gioiello, A.; Ferlin, S.; Puxeddu, E.; Macchiarulo, A. The Stone Guest: How Does pH Affect Binding Properties of PD-1/PD-L1 Inhibitors? ChemMedChem 2021, 16, 568–577. [Google Scholar] [CrossRef]
- Thieme, S.; Walther, M.; Pietzsch, H.-J.; Henniger, J.; Preusche, S.; Mäding, P.; Steinbach, J. Module-Assisted Preparation of 64Cu With High Specific Activity. Appl. Radiat. Isot. 2012, 70, 602–608. [Google Scholar] [CrossRef]
- Wessel, D.; Flügge, U.I. A Method for the Quantitative Recovery of Protein in Dilute Solution in the Presence of Detergents and Lipids. Anal. Biochem. 1984, 138, 141–143. [Google Scholar] [CrossRef]
- Ludwig, F.-A.; Fischer, S.; Houska, R.; Hoepping, A.; Deuther-Conrad, W.; Schepmann, D.; Patt, M.; Meyer, P.M.; Hesse, S.; Becker, G.-A.; et al. In vitro and in vivo Human Metabolism of (S)-[18F] Fluspidine–A Radioligand for Imaging σ1 Receptors With Positron Emission Tomography (PET). Front. Pharmacol. 2019, 10, 534. [Google Scholar] [CrossRef]
- Önell, A.; Andersson, K. Kinetic determinations of molecular interactions using Biacore—Minimum data requirements for efficient experimental design. J. Mol. Recognit. 2005, 18, 307–317. [Google Scholar] [CrossRef]
- Rombouts, F.; Peschiulli, A. Preparation of Macrocyclic Indole Compounds as MCL-1 Inhibitor. Patent WO/2020/254471, 18 June 2020. [Google Scholar]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | KD (nM) a | Bmax (pmol/mg) a | log D7.4 b |
|---|---|---|---|
| [64Cu]Cu-42a | 585 ± 53.0 | 11.7 ± 0.77 | −2.73 ± 0.04 |
| [64Cu]Cu-42b | 487 ± 58.6 | 8.91 ± 0.63 | −3.03 ± 0.05 |
| [64Cu]Cu-42c | 351 ± 8.31 | 5.85 ± 1.72 | −3.47 ± 0.10 |
| [64Cu]Cu-43a | 123 ± 17.3 | 13.5 ± 1.38 | −2.75 ± 0.04 |
| [64Cu]Cu-43b | 59.9 ± 6.05 | 5.43 ± 0.63 | −3.14 ± 0.02 |
| [64Cu]Cu-43c | 71.3 ± 13.2 | 7.04 ± 0.63 | −3.50 ± 0.01 |
| [64Cu]Cu-56a | 128 ± 17.2 | 20.9 ± 2.82 | −2.99 ± 0.02 |
| [64Cu]Cu-56b | 135 ± 3.37 | 8.28 ± 0.18 | −3.48 ± 0.03 |
| [64Cu]Cu-56c | 300 ± 26.8 | 49.6 ± 1.78 | −2.99 ± 0.02 |
| [64Cu]Cu-DOTAGA-WL12 [27] | 50.2 ± 7.2 | 56.7 ± 3.87 | −2.57 ± 0.02 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krutzek, F.; Donat, C.K.; Ullrich, M.; Zarschler, K.; Ludik, M.-C.; Feldmann, A.; Loureiro, L.R.; Kopka, K.; Stadlbauer, S. Design and Biological Evaluation of Small-Molecule PET-Tracers for Imaging of Programmed Death Ligand 1. Cancers 2023, 15, 2638. https://doi.org/10.3390/cancers15092638
Krutzek F, Donat CK, Ullrich M, Zarschler K, Ludik M-C, Feldmann A, Loureiro LR, Kopka K, Stadlbauer S. Design and Biological Evaluation of Small-Molecule PET-Tracers for Imaging of Programmed Death Ligand 1. Cancers. 2023; 15(9):2638. https://doi.org/10.3390/cancers15092638
Chicago/Turabian StyleKrutzek, Fabian, Cornelius K. Donat, Martin Ullrich, Kristof Zarschler, Marie-Charlotte Ludik, Anja Feldmann, Liliana R. Loureiro, Klaus Kopka, and Sven Stadlbauer. 2023. "Design and Biological Evaluation of Small-Molecule PET-Tracers for Imaging of Programmed Death Ligand 1" Cancers 15, no. 9: 2638. https://doi.org/10.3390/cancers15092638
APA StyleKrutzek, F., Donat, C. K., Ullrich, M., Zarschler, K., Ludik, M.-C., Feldmann, A., Loureiro, L. R., Kopka, K., & Stadlbauer, S. (2023). Design and Biological Evaluation of Small-Molecule PET-Tracers for Imaging of Programmed Death Ligand 1. Cancers, 15(9), 2638. https://doi.org/10.3390/cancers15092638