

Spatially Guided and Single Cell Tools to Map the Microenvironment in Cutaneous T-Cell Lymphoma

{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
1.1. Mycosis Fungoides and Sézary Syndrome
1.2. Patient Risk Stratification: A Clinical Unmet Need in CTCL
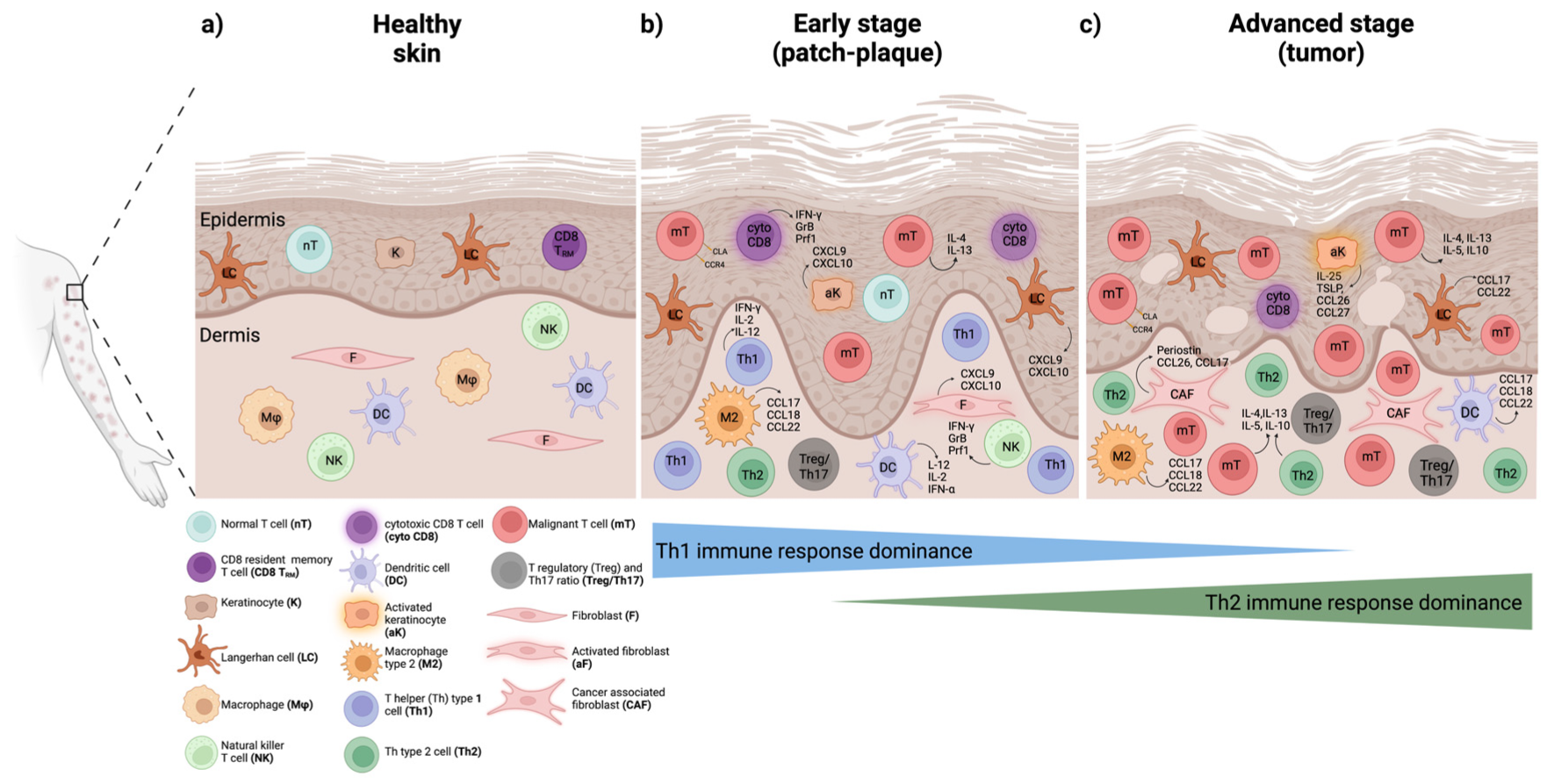
2. The Tumor Microenvironment (TME) and Immune Response in CTCL

3. Inter-Patient and Intra-Tumor Heterogeneity in MF and SS
3.1. Immunophenotypic Heterogeneity
3.2. Genomic Heterogeneity and Subclonal Evolution
3.3. Transcriptional Heterogeneity
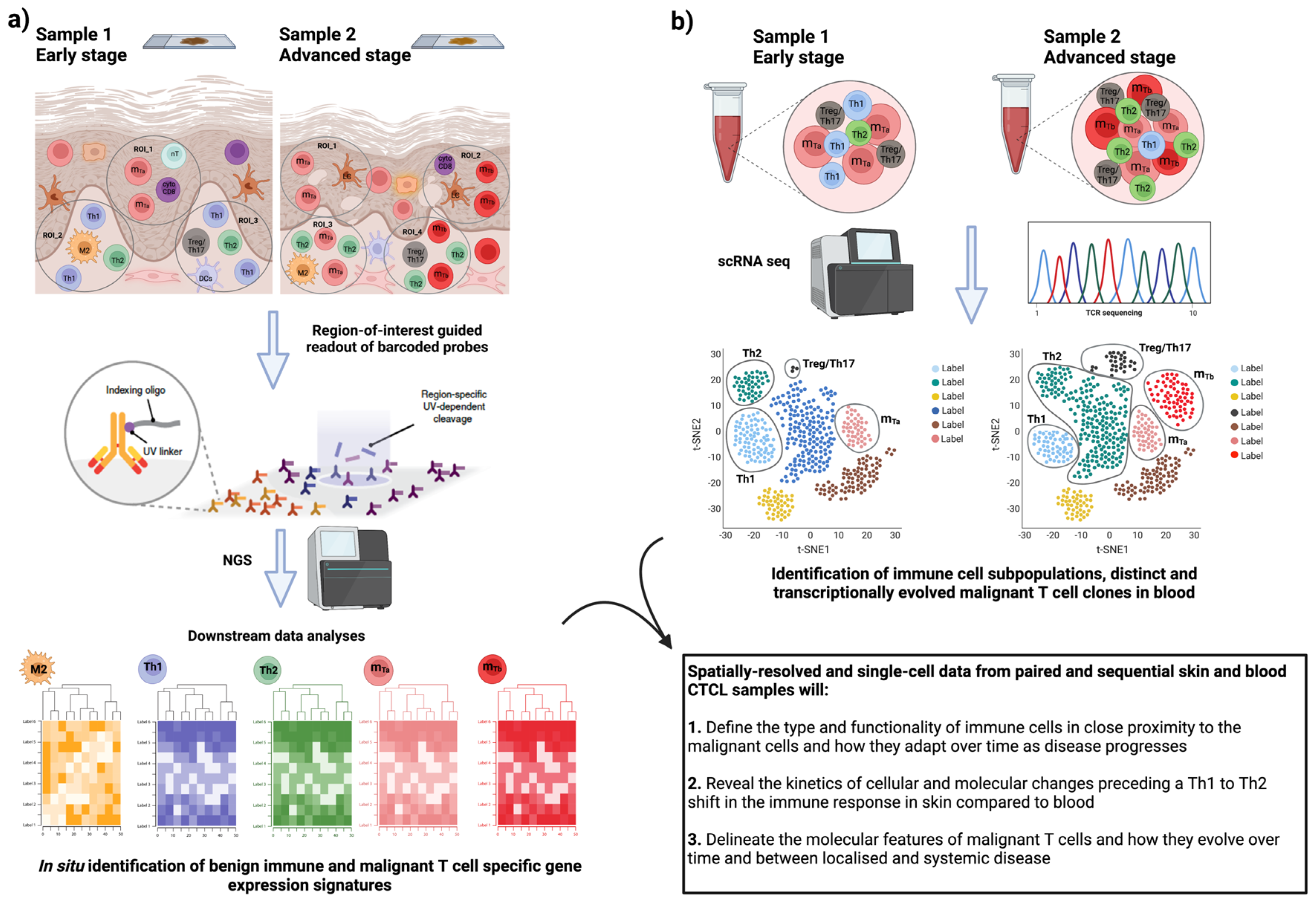
4. Moving Forward: Deep Immune Profiling of CTCL Heterogeneity by Spatially Resolved Omics Technologies
4.1. Introducing Spatially Resolved Transcriptomics (SRT)
4.2. SRT Application in Cancer Disease
4.3. SRT Application in CTCL
5. Conclusions—Future Research Directions
Author Contributions
Funding
Conflicts of Interest
References
- Willemze, R.; Cerroni, L.; Kempf, W. The 2018 update of the WHO-EORTC classification for primary cutaneous lymphomas. Blood 2019, 134, 1112. [Google Scholar] [CrossRef]
- Kempf, W.; Zimmermann, A.K.; Mitteldorf, C. Cutaneous lymphomas—An update 2019. Hematol. Oncol. 2019, 37, 43–47. [Google Scholar] [CrossRef]
- Kempf, W.; Kazakov, D.V.; Kerl, K. Cutaneous Lymphomas: An Update. Part 1: T-Cell and Natural Killer/T-Cell Lymphomas and Related Conditions. Am. J. Dermatopathol. 2014, 36, 105–123. [Google Scholar] [CrossRef] [PubMed]
- Gilson, D.; Whittaker, S.J.; Child, F.J.; Scarisbrick, J.J.; Illidge, T.M.; Parry, E.J.; Mustapa, M.F.M.; Exton, L.S.; Kanfer, E.; Rezvani, K.; et al. British Association of Dermatologists and UK Cutaneous Lymphoma Group guidelines for the management of primary cutaneous lymphomas 2018. Br. J. Dermatol. 2019, 180, 496–526. [Google Scholar] [CrossRef] [PubMed]
- Farabi, B.; Seminario-Vidal, L.; Jamgochian, M.; Akay, B.N.; Atak, M.F.; Rao, B.K.; Karagaiah, P.; Grabbe, S.; Goldust, M. Updated review on prognostic factors in mycosis fungoides and new skin lymphoma trials. J. Cosmet. Dermatol. 2022, 21, 2742–2748. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, R.A. Cutaneous T-cell lymphoma: 2016 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2016, 91, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Phyo, Z.H.; Shanbhag, S.; Rozati, S. Update on Biology of Cutaneous T-Cell Lymphoma. Front. Oncol. 2020, 10, 765. [Google Scholar] [CrossRef]
- Agar, N.S.; Wedgeworth, E.; Crichton, S.; Mitchell, T.J.; Cox, M.; Ferreira, S.; Robson, A.; Calonje, E.; Stefanato, C.M.; Wain, E.M.; et al. Survival Outcomes and Prognostic Factors in Mycosis Fungoides/Sezary Syndrome: Validation of the Revised International Society for Cutaneous Lymphomas/European Organisation for Research and Treatment of Cancer Staging Proposal. J. Clin. Oncol. 2010, 28, 4730–4739. [Google Scholar] [CrossRef]
- Benton, E.C.; Crichton, S.; Talpur, R.; Agar, N.S.; Fields, P.A.; Wedgeworth, E.; Mitchell, T.J.; Cox, M.; Ferreira, S.; Liu, P.; et al. A cutaneous lymphoma international prognostic index (CLIPi) for mycosis fungoides and Sezary syndrome. Eur. J. Cancer 2013, 49, 2859–2868. [Google Scholar] [CrossRef]
- Bobrowicz, M.; Fassnacht, C.; Ignatova, D.; Chang, Y.T.; Dimitriou, F.; Guenova, E. Pathogenesis and Therapy of Primary Cutaneous T-Cell Lymphoma: Collegium Internationale Allergologicum (CIA) Update 2020. Int. Arch. Allergy Immunol. 2020, 181, 733–745. [Google Scholar] [CrossRef]
- Brunner, P.M.; Jonak, C.; Knobler, R. Recent advances in understanding and managing cutaneous T-cell lymphomas. F1000Research 2020, 9, F1000 Faculty Rev-1331. [Google Scholar] [CrossRef] [PubMed]
- Geller, S.; Myskowski, P.L.; Pulitzer, M.; Horwitz, S.M.; Moskowitz, A.J. Cutaneous T-cell lymphoma (CTCL), rare subtypes: Five case presentations and review of the literature. Chin. Clin. Oncol. 2019, 8, 5. [Google Scholar] [CrossRef] [PubMed]
- Kempf, W.; Mitteldorf, C. Cutaneous T-cell lymphomas-An update 2021. Hematol. Oncol. 2021, 39, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Patil, K.; Kuttikrishnan, S.; Khan, A.Q.; Ahmad, F.; Alam, M.; Buddenkotte, J.; Ahmad, A.; Steinhoff, M.; Uddin, S. Molecular pathogenesis of Cutaneous T cell Lymphoma: Role of chemokines, cytokines, and dysregulated signaling pathways. Semin. Cancer Biol. 2022, 86 Pt 3, 382–399. [Google Scholar] [CrossRef]
- Hwang, S.T.; Janik, J.E.; Jaffe, E.S.; Wilson, W.H. Mycosis fungoides and Sézary syndrome. Lancet 2008, 371, 945–957. [Google Scholar] [CrossRef]
- Campbell, J.J.; Clark, R.A.; Watanabe, R.; Kupper, T.S. Sezary syndrome and mycosis fungoides arise from distinct T-cell subsets: A biologic rationale for their distinct clinical behaviors. Blood 2010, 116, 767–771. [Google Scholar] [CrossRef]
- Miyagaki, T. Diagnosis of Early Mycosis Fungoides. Diagnostics 2021, 11, 1721. [Google Scholar] [CrossRef]
- Photiou, L.; van der Weyden, C.; McCormack, C.; Miles Prince, H. Systemic Treatment Options for Advanced-Stage Mycosis Fungoides and Sézary Syndrome. Curr. Oncol. Rep. 2018, 20, 32. [Google Scholar] [CrossRef]
- Vural, S.; Akay, B.N.; Botsalı, A.; Atilla, E.; Parlak, N.; Okçu Heper, A.; Şanlı, H. Transformation of Mycosis Fungoides/Sezary Syndrome: Clinical Characteristics and Prognosis. Turk. J. Haematol. 2018, 35, 35–41. [Google Scholar] [CrossRef]
- Cerroni, L.; Rieger, E.; Hödl, S.; Kerl, H. Clinicopathologic and immunologic features associated with transformation of mycosis fungoides to large-cell lymphoma. Am. J. Surg. Pathol. 1992, 16, 543–552. [Google Scholar] [CrossRef]
- Diamandidou, E.; Colome-Grimmer, M.; Fayad, L.; Duvic, M.; Kurzrock, R. Transformation of Mycosis Fungoides/Sezary Syndrome: Clinical Characteristics and Prognosis. Blood 1998, 92, 1150–1159. [Google Scholar] [CrossRef] [PubMed]
- Nicolay, J.P.; Felcht, M.; Schledzewski, K.; Goerdt, S.; Geraud, C. Sezary syndrome: Old enigmas, new targets. J. Der Dtsch. Dermatol. Ges. 2016, 14, 256–264. [Google Scholar] [CrossRef]
- Stolearenco, V.; Namini, M.R.J.; Hasselager, S.S.; Gluud, M.; Buus, T.B.; Willerslev-Olsen, A.; Ødum, N.; Krejsgaard, T. Cellular Interactions and Inflammation in the Pathogenesis of Cutaneous T-Cell Lymphoma. Front. Cell Dev. Biol. 2020, 8, 851. [Google Scholar] [CrossRef]
- Odum, N.; Lindahl, L.M.; Wod, M.; Krejsgaard, T.; Skytthe, A.; Woetmann, A.; Iversen, L.; Christensen, K. Investigating heredity in cutaneous T-cell lymphoma in a unique cohort of Danish twins. Blood Cancer J. 2017, 7, e517. [Google Scholar] [CrossRef] [PubMed]
- Tensen, C.P.; Quint, K.D.; Vermeer, M.H. Genetic and epigenetic insights into cutaneous T-cell lymphoma. Blood 2022, 139, 15–33. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Lillington, D.; Scarisbrick, J.J.; Mitchell, T.; Czepulkowski, B.; Russell-Jones, R.; Young, B.; Whittaker, S.J. Molecular cytogenetic analysis of cutaneous T-cell lymphomas: Identification of common genetic alterations in Sezary syndrome and mycosis fungoides. Br. J. Dermatol. 2002, 147, 464–475. [Google Scholar] [CrossRef]
- van Doorn, R.; van Kester, M.S.; Dijkman, R.; Vermeer, M.H.; Mulder, A.A.; Szuhai, K.; Knijnenburg, J.; Boer, J.M.; Willemze, R.; Tensen, C.P. Oncogenomic analysis of mycosis fungoides reveals major differences with Sezary syndrome. Blood 2009, 113, 127–136. [Google Scholar] [CrossRef]
- McGregor, J.M.; Crook, T.; Fraser-Andrews, E.A.; Rozycka, M.; Crossland, S.; Brooks, L.; Whittaker, S.J. Spectrum of p53 gene mutations suggests a possible role for ultraviolet radiation in the pathogenesis of advanced cutaneous lymphomas. J. Investig. Dermatol. 1999, 112, 317–321. [Google Scholar] [CrossRef]
- Vaque, J.P.; Gomez-Lopez, G.; Monsalvez, V.; Varela, I.; Martinez, N.; Perez, C.; Dominguez, O.; Grana, O.; Rodriguez-Peralto, J.L.; Rodriguez-Pinilla, S.M.; et al. PLCG1 mutations in cutaneous T-cell lymphomas. Blood 2014, 123, 2034–2043. [Google Scholar] [CrossRef]
- Scarisbrick, J.J.; Prince, H.M.; Vermeer, M.H.; Quaglino, P.; Horwitz, S.; Porcu, P.; Stadler, R.; Wood, G.S.; Beylot-Barry, M.; Pham-Ledard, A.; et al. Cutaneous Lymphoma International Consortium Study of Outcome in Advanced Stages of Mycosis Fungoides and Sezary Syndrome: Effect of Specific Prognostic Markers on Survival and Development of a Prognostic Model. J. Clin. Oncol. 2015, 33, 3766–3773. [Google Scholar] [CrossRef]
- Miyagaki, T.; Sugaya, M. Immunological milieu in mycosis fungoides and Sezary syndrome. J. Dermatol. 2014, 41, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Krejsgaard, T.; Lindahl, L.M.; Mongan, N.P.; Wasik, M.A.; Litvinov, I.V.; Iversen, L.; Langhoff, E.; Woetmann, A.; Odum, N. Malignant inflammation in cutaneous TaEurocell lymphoma-a hostile takeover. Semin. Immunopathol. 2017, 39, 269–282. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.R.; Wu, X.S.; Hwang, S.; Liu, J. The Role of Tumor Microenvironment in Mycosis Fungoides and Sezary Syndrome. Ann. Dermatol. 2021, 33, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Vermeer, M.H.; van Doorn, R.; Dukers, D.; Bekkenk, M.W.; Meijer, C.; Willemze, R. CD8+T cells in cutaneous T-cell lymphoma: Expression of cytotoxic proteins, Fas ligand, and killing inhibitory receptors and their relationship with clinical behavior. J. Clin. Oncol. 2001, 19, 4322–4329. [Google Scholar] [CrossRef]
- Goteri, G.; Filosa, A.; Mannello, B.; Stramazzotti, D.; Rupoli, S.; Leoni, P.; Fabris, G. Density of neoplastic lymphoid infiltrate, CD8+T cells, and CD1a+dendritic cells in mycosis fungoides. J. Clin. Pathol. 2003, 56, 453–458. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gonzalez, B.R.; Zain, J.; Rosen, S.T.; Querfeld, C. Tumor microenvironment in mycosis fungoides and Sezary syndrome. Curr. Opin. Oncol. 2016, 28, 88–96. [Google Scholar] [CrossRef]
- Dummer, R.; Vermeer, M.H.; Scarisbrick, J.J.; Kim, Y.H.; Stonesifer, C.; Tensen, C.P.; Ceskin, L.J.; Quaglino, P.; Ramelyte, E. Cutaneous T cell lymphoma. Nat. Rev. Dis. Prim. 2021, 7, 61. [Google Scholar] [CrossRef]
- Miyashiro, D.; Souza, B.D.E.; Torrealba, M.P.; Manfrere, K.C.G.; Sato, M.N.; Sanches, J.A. The Role of Tumor Microenvironment in the Pathogenesis of Sezary Syndrome. Int. J. Mol. Sci. 2022, 23, 936. [Google Scholar] [CrossRef]
- Takahashi, N.; Sugaya, M.; Suga, H.; Oka, T.; Kawaguchi, M.; Miyagaki, T.; Fujita, H.; Sato, S. Thymic Stromal Chemokine TSLP Acts through Th2 Cytokine Production to Induce Cutaneous T-cell Lymphoma. Cancer Res. 2016, 76, 6241–6252. [Google Scholar] [CrossRef]
- Chong, B.F.; Wilson, A.J.; Gibson, H.M.; Hafner, M.S.; Luo, Y.; Hedgcock, C.J.; Wong, H.K. Immune function abnormalities in peripheral blood mononuclear cell cytokine expression differentiates stages of cutaneous T-Cell lymphoma/mycosis fungoides. Clin. Cancer Res. 2008, 14, 646–653. [Google Scholar] [CrossRef]
- Vowels, B.R.; Cassin, M.; Vonderheid, E.C.; Rook, A.H. Aberrant Cytokine Production By Sezary-Syndrome Patients—Cytokine Secretion Pattern Resembles Murine Th2-Cells. J. Investig. Dermatol. 1992, 99, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Geskin, L.J.; Viragova, S.; Stolz, D.B.; Fuschiotti, P. Interleukin-13 is overexpressed in cutaneous T-cell lymphoma cells and regulates their proliferation. Blood 2015, 125, 2798–2805. [Google Scholar] [CrossRef] [PubMed]
- Vowels, B.R.; Lessin, S.R.; Cassin, M.; Jaworsky, C.; Benoit, B.; Wolfe, J.T.; Rook, A.H. TH2 Cytokine Messenger-Rna Expression in Skin in Cutaneous T-Cell Lymphoma. J. Investig. Dermatol. 1994, 103, 669–673. [Google Scholar] [CrossRef] [PubMed]
- Cedeno-Laurent, F.; Watanabe, R.; Teague, J.E.; Kupper, T.S.; Clark, R.A.; Dimitroff, C.J. Galectin-1 inhibits the viability, proliferation, and Th1 cytokine production of nonmalignant T cells in patients with leukemic cutaneous T-cell lymphoma. Blood 2012, 119, 3534–3538. [Google Scholar] [CrossRef]
- Guenova, E.; Watanabe, R.; Teague, J.E.; Desimone, J.A.; Jiang, Y.; Dowlatshahi, M.; Schlapbach, C.; Schaekel, K.; Rook, A.H.; Tawa, M.; et al. TH2 Cytokines from Malignant Cells Suppress TH1 Responses and Enforce a Global TH2 Bias in Leukemic Cutaneous T-cell Lymphoma. Clin. Cancer Res. 2013, 19, 3755–3763. [Google Scholar] [CrossRef]
- Gjerdrum, L.M.; Woetmann, A.; Odum, N.; Burton, C.M.; Rossen, K.; Skovgaard, G.L.; Ryder, L.P.; Ralfkiaer, E. FOXP3+ regulatory T cells in cutaneous T-cell lymphomas: Association with stage and survival. Leukemia 2007, 21, 2512–2518. [Google Scholar] [CrossRef]
- Krejsgaard, T.; Gjerdrum, L.M.; Ralfkiaer, E.; Lauenborg, B.; Eriksen, K.W.; Mathiesen, A.M.; Bovin, L.F.; Gniadecki, R.; Geisler, C.; Ryder, L.P.; et al. Malignant Tregs express low molecular splice forms of FOXP3 in Sezary syndrome. Leukemia 2008, 22, 2230–2239. [Google Scholar] [CrossRef]
- Heid, J.; Schmidt, A.; Oberle, N.; Goerdt, S.; Krammer, P.; Suri-Payer, E.; Klemke, C.D. FOXP3+CD25-tumor cells with regulatory function in Sezary Syndrome. Exp. Dermatol. 2009, 18, 308. [Google Scholar] [CrossRef]
- Berger, C.L.; Tigelaar, R.; Cohen, J.; Mariwalla, K.; Trinh, J.; Wang, N.C.; Edelson, R.L. Cutaneous T-cell lymphoma: Malignant proliferation of T-regulatory cells. Blood 2005, 105, 1640–1647. [Google Scholar] [CrossRef]
- Gaydosik, A.M.; Tabib, T.; Geskin, L.J.; Bayan, C.A.; Conway, J.F.; Lafyatis, R.; Fuschiotti, P. Single-Cell Lymphocyte Heterogeneity in Advanced Cutaneous T-cell Lymphoma Skin Tumors. Clin. Cancer Res. 2019, 25, 4443–4454. [Google Scholar] [CrossRef]
- Rindler, K.; Jonak, C.; Alkon, N.; Thaler, F.M.; Kurz, H.; Shaw, L.E.; Stingl, G.; Weninger, W.; Halbritter, F.; Bauer, W.M.; et al. Single-cell RNA sequencing reveals markers of disease progression in primary cutaneous T-cell lymphoma. Mol. Cancer 2021, 20, 124. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Cai, Y.; Lv, Y.; Zhang, L.; Yang, H.; Liu, Q.; Hong, M.; Teng, Y.; Tang, W.; Ma, R.; et al. Single-cell RNA sequencing unveils the communications between malignant T and myeloid cells contributing to tumor growth and immunosuppression in cutaneous T-cell lymphoma. Cancer Lett. 2022, 551, 215972. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Wu, R.; Chen, L.; Peng, Q.; Li, S.; Zhang, Y.; Zhou, L.; Duan, L. S100A9 Regulates MDSCs-Mediated Immune Suppression via the RAGE and TLR4 Signaling Pathways in Colorectal Carcinoma. Front. Immunol. 2019, 10, 2243. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Wu, R.; Zhang, X.; Wang, D.; You, Y.; Zhang, Y.; Zhou, L.; Chen, W. HBx-induced S100A9 in NF-κB dependent manner promotes growth and metastasis of hepatocellular carcinoma cells article. Cell Death Dis. 2018, 9, 629. [Google Scholar] [CrossRef] [PubMed]
- Nakai, S.; Kiyohara, E.; Watanabe, R. Malignant and Benign T Cells Constituting Cutaneous T-Cell Lymphoma. Int. J. Mol. Sci. 2021, 22, 2933. [Google Scholar] [CrossRef] [PubMed]
- Roelens, M.; Delord, M.; Ram-Wolff, C.; Marie-Cardine, A.; Alberdi, A.; Maki, G.; Homyrda, L.; Bensussan, A.; Bagot, M.; Toubert, A.; et al. Circulating and skin-derived Sezary cells: Clonal but with phenotypic plasticity. Blood 2017, 130, 1468–1471. [Google Scholar] [CrossRef] [PubMed]
- Cristofoletti, C.; Bresin, A.; Picozza, M.; Picchio, M.C.; Monzo, F.; Citterich, M.H.; Passarelli, F.; Frezzolini, A.; Scala, E.; Monopoli, A.; et al. Blood and skin-derived Sezary cells: Differences in proliferation-index, activation of PI3K/AKT/mTORC1 pathway and its prognostic relevance. Leukemia 2019, 33, 1231–1242. [Google Scholar] [CrossRef]
- Mahnke, Y.D.; Brodie, T.M.; Sallusto, F.; Roederer, M.; Lugli, E. The who’s who of T-cell differentiation: Human memory T-cell subsets. Eur. J. Immunol. 2013, 43, 2797–2809. [Google Scholar] [CrossRef]
- Duvic, M.; Pinter-Brown, L.C.; Foss, F.M.; Sokol, L.; Jorgensen, J.L.; Challagundla, P.; Dwyer, K.M.; Zhang, X.P.; Kurman, M.R.; Ballerini, R.; et al. Phase 1/2 study of mogamulizumab, a defucosylated anti-CCR4 antibody, in previously treated patients with cutaneous T-cell lymphoma. Blood 2015, 125, 1883–1889. [Google Scholar] [CrossRef]
- Bitar, C.; Hile, G.; Brown, N.A.; Fullen, D.R.; Lowe, L.; Tejasvi, T.; Wilcox, R.A.; Harms, P.W.; Chan, M.P.; Bresler, S.C.; et al. Immunophenotypic switch in cutaneous T-cell lymphoma: A series of three cases and review of the literature. J. Cutan. Pathol. 2021, 48, 986–994. [Google Scholar] [CrossRef]
- Marks, E.; Shi, Y.; Wang, Y.H. Two cases of phenotypic switch of primary cutaneous T cell lymphoma after treatment with an aggressive course and review of the literature. Virchows Arch. 2019, 475, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Endo, C.; Naka, Y.; Miyagaki, T.; Fujita, H.; Sugaya, M.; Kawashima, M.; Tsunemi, Y. Immunophenotypic shift from CD4(+) to CD8(+) in mycosis fungoides. Br. J. Dermatol. 2016, 175, 830–833. [Google Scholar] [CrossRef] [PubMed]
- Braue, J.A.; Daniels, A.B.; Zwerner, J.P.; Kim, S.J.; Zic, J.A. Intraocular involvement of mycosis fungoides associated with immunophenotypic switch from CD4(+) to CD8(+). Blood 2018, 131, 932–935. [Google Scholar] [CrossRef]
- Nikolova, M.; Echchakir, H.; Wechsler, J.; Boumsell, L.; Bensussan, A.; Bagot, M. Isolation of a CD8 alpha alpha+CD4-tumour T-cell clone with cytotoxic activity from a CD4+CD8-cutaneous T-cell lymphoma. Br. J. Dermatol. 2003, 148, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Kreuter, A.; Altmeyer, P. Rapid onset of CD8(+) aggressive T-cell lymphoma during bexarotene therapy in a patient with Sezary syndrome. J. Am. Acad. Dermatol. 2005, 53, 1093–1095. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.T.; Leeman-Neill, R.J.; Patel, P.; Ho, J.; Grandinetti, L.M.; Jedrych, J.; Craig, F.E. Fatal Case of Primary Cutaneous Aggressive T-Cell Lymphoma Switching From a CD4(+) to a CD8(+) Phenotype: Progressive Disease With Bexarotene and Romidepsin Treatment. Am. J. Dermatopathol. 2016, 38, 832–837. [Google Scholar] [CrossRef]
- Vermeer, M.H.; Geelen, F.; Kummer, J.A.; Meijer, C.; Willemze, R. Expression of cytotoxic proteins by neoplastic T cells in mycosis fungoides increases with progression from plaque stage to tumor stage disease. Am. J. Pathol. 1999, 154, 1203–1210. [Google Scholar] [CrossRef]
- Iyer, A.; Hennessey, D.; O’Keefe, S.; Patterson, J.; Wang, W.W.; Wong, G.K.S.; Gniadecki, R. Branched evolution and genomic intratumor heterogeneity in the pathogenesis of cutaneous T-cell lymphoma. Blood Adv. 2020, 4, 2489–2500. [Google Scholar] [CrossRef]
- Herrera, A.; Cheng, A.; Mimitou, E.P.; Seffens, A.; George, D.; Bar-Natan, M.; Heguy, A.; Ruggles, K.V.; Scher, J.U.; Hymes, K.; et al. Multimodal single-cell analysis of cutaneous T-cell lymphoma reveals distinct subclonal tissue-dependent signatures. Blood 2021, 138, 1456–1464. [Google Scholar] [CrossRef]
- Song, X.F.; Chang, S.; Seminario-Vidal, L.; Pulido, A.D.; Tordesillas, L.; Song, X.Z.; Reed, R.A.; Harkins, A.; Whiddon, S.; Nguyen, J.V.; et al. Genomic and Single-Cell Landscape Reveals Novel Drivers and Therapeutic Vulnerabilities of Transformed Cutaneous T-cell Lymphoma. Cancer Discov. 2022, 12, 1294–1313. [Google Scholar] [CrossRef]
- Borcherding, N.; Voigt, A.P.; Liu, V.; Link, B.K.; Zhang, W.Z.; Jabbari, A. Single-Cell Profiling of Cutaneous T-Cell Lymphoma Reveals Underlying Heterogeneity Associated with Disease Progression. Clin. Cancer Res. 2019, 25, 2996–3005. [Google Scholar] [CrossRef] [PubMed]
- Borcherding, N.; Severson, K.J.; Henderson, N.T.; Dos Santos Ortolan, L.; Rosenthal, A.C.; Bellizzi, A.M.; Liu, V.; Link, B.K.; Mangold, A.R.; Jabbari, A. Single-cell analysis of Sézary syndrome reveals novel markers and shifting gene profiles associated with treatment. Blood Adv. 2022, 7, 321–335. [Google Scholar] [CrossRef]
- Ren, J.; Qu, R.; Rahman, N.-T.; Lewis, J.M.; King, A.L.O.; Liao, X.; Mirza, F.N.; Carlson, K.R.; Huang, Y.; Gigante, S.; et al. Integrated transcriptome and trajectory analysis of cutaneous T-cell lymphoma identifies putative precancer populations. Blood Adv. 2023, 7, 445–457. [Google Scholar] [CrossRef]
- Litvinov, I.V.; Tetzlaff, M.T.; Thibault, P.; Gangar, P.; Moreau, L.; Watters, A.K.; Netchiporouk, E.; Pehr, K.; Prieto, V.G.; Rahme, E.; et al. Gene expression analysis in Cutaneous T-Cell Lymphomas (CTCL) highlights disease heterogeneity and potential diagnostic and prognostic indicators. Oncoimmunology 2017, 6, e1306618. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Berthelot, C.; Li, Y.F.; Glass, D.A.; George, D.; Pandya, A.; Kurzrock, R.; Duvic, M. Poor prognosis in non-Caucasian patients with early-onset mycosis fungoides. J. Am. Acad. Dermatol. 2009, 60, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Litvinov, I.V.; Netchiporouk, E.; Cordeiro, B.; Dore, M.A.; Moreau, L.; Pehr, K.; Gilbert, M.; Zhou, Y.W.; Sasseville, D.; Kupper, T.S. The Use of Transcriptional Profiling to Improve Personalized Diagnosis and Management of Cutaneous T-cell Lymphoma (CTCL). Clin. Cancer Res. 2015, 21, 2820–2829. [Google Scholar] [CrossRef]
- Liu, X.J.; Jin, S.Z.; Hu, S.M.; Li, R.Y.; Pan, H.H.; Liu, Y.; Lai, P.; Xu, D.S.; Sun, J.R.; Liu, Z.Y.; et al. Single-cell transcriptomics links malignant T cells to the tumor immune landscape in cutaneous T cell lymphoma. Nat. Commun. 2022, 13, 1158. [Google Scholar] [CrossRef]
- Tan, S.Y.; Chuang, S.S.; Tang, T.; Tan, L.; Ko, Y.H.; Chuah, K.L.; Ng, S.B.; Chng, W.J.; Gatter, K.; Loong, F.; et al. Type II EATL (epitheliotropic intestinal T-cell lymphoma): A neoplasm of intra-epithelial T-cells with predominant CD8 alpha alpha phenotype. Leukemia 2013, 27, 1688–1696. [Google Scholar] [CrossRef]
- Caballero, A.; Novelli, S.; Mozos, A.; Muret, P.G.; Sierra, J.; Briones, J. Silent T-cell receptor cutaneous T-cell lymphoma associated to a clonal plasma cell proliferation. Hematol. Rep. 2019, 11, 6–8. [Google Scholar] [CrossRef]
- Blank, C.U.; Haining, W.N.; Held, W.; Hogan, P.G.; Kallies, A.; Lugli, E.; Lynn, R.C.; Philip, M.; Rao, A.; Restifo, N.P.; et al. Defining ‘T cell exhaustion’. Nat. Rev. Immunol. 2019, 19, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Phillips, D.; Matusiak, M.; Gutierrez, B.R.; Bhate, S.S.; Barlow, G.L.; Jiang, S.Z.; Demeter, J.; Smythe, K.S.; Pierce, R.H.; Fling, S.P.; et al. Immune cell topography predicts response to PD-1 blockade in cutaneous T cell lymphoma. Nat. Commun. 2021, 12, 6726. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.C.C.; Nerurkar, S.N.; Cai, H.Y.; Ng, H.H.M.; Wu, D.D.; Wee, Y.T.F.; Lim, J.C.T.; Yeong, J.; Lim, T.K.H. Overview of multiplex immunohistochemistry/immunofluorescence techniques in the era of cancer immunotherapy. Cancer Commun. 2020, 40, 135–153. [Google Scholar] [CrossRef] [PubMed]
- Asp, M.; Bergenstrahle, J.; Lundeberg, J. Spatially Resolved Transcriptomes-Next Generation Tools for Tissue Exploration. Bioessays 2020, 42, 1900221. [Google Scholar] [CrossRef] [PubMed]
- Marx, V. Method of the Year: Spatially resolved transcriptomics. Nat. Methods 2021, 18, 9–14. [Google Scholar] [CrossRef]
- Maniatis, S.; Petrescu, J.; Phatnani, H. Spatially resolved transcriptomics and its applications in cancer. Curr. Opin. Genet. Dev. 2021, 66, 70–77. [Google Scholar] [CrossRef]
- Ståhl, P.L.; Salmén, F.; Vickovic, S.; Lundmark, A.; Navarro, J.F.; Magnusson, J.; Giacomello, S.; Asp, M.; Westholm, J.O.; Huss, M.; et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 2016, 353, 78–82. [Google Scholar] [CrossRef]
- Merritt, C.R.; Ong, G.T.; Church, S.E.; Barker, K.; Danaher, P.; Geiss, G.; Hoang, M.; Jung, J.; Liang, Y.; McKay-Fleisch, J.; et al. Multiplex digital spatial profiling of proteins and RNA in fixed tissue. Nat. Biotechnol. 2020, 38, 586–599. [Google Scholar] [CrossRef]
- McGinnis, L.M.; Ibarra-Lopez, V.; Rost, S.; Ziai, J. Clinical and research applications of multiplexed immunohistochemistry and in situ hybridization. J. Pathol. 2021, 254, 405–417. [Google Scholar] [CrossRef]
- Zugazagoitia, J.; Gupta, S.; Liu, Y.T.; Fuhrman, K.; Gettinger, S.; Herbst, R.S.; Schalper, K.A.; Rimm, D.L. Biomarkers Associated with Beneficial PD-1 Checkpoint Blockade in Non-Small Cell Lung Cancer (NSCLC) Identified Using High-Plex Digital Spatial Profiling. Clin. Cancer Res. 2020, 26, 4360–4368. [Google Scholar] [CrossRef]
- Toki, M.I.; Merritt, C.R.; Wong, P.F.; Smithy, J.W.; Kluger, H.M.; Syrigos, K.N.; Ong, G.T.; Warren, S.E.; Beechem, J.M.; Rimm, D.L. High-Plex Predictive Marker Discovery for Melanoma Immunotherapy-Treated Patients Using Digital Spatial Profiling. Clin. Cancer Res. 2019, 25, 5503–5512. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalliara, E.; Belfrage, E.; Gullberg, U.; Drott, K.; Ek, S. Spatially Guided and Single Cell Tools to Map the Microenvironment in Cutaneous T-Cell Lymphoma. Cancers 2023, 15, 2362. https://doi.org/10.3390/cancers15082362
Kalliara E, Belfrage E, Gullberg U, Drott K, Ek S. Spatially Guided and Single Cell Tools to Map the Microenvironment in Cutaneous T-Cell Lymphoma. Cancers. 2023; 15(8):2362. https://doi.org/10.3390/cancers15082362
Chicago/Turabian StyleKalliara, Eirini, Emma Belfrage, Urban Gullberg, Kristina Drott, and Sara Ek. 2023. "Spatially Guided and Single Cell Tools to Map the Microenvironment in Cutaneous T-Cell Lymphoma" Cancers 15, no. 8: 2362. https://doi.org/10.3390/cancers15082362
APA StyleKalliara, E., Belfrage, E., Gullberg, U., Drott, K., & Ek, S. (2023). Spatially Guided and Single Cell Tools to Map the Microenvironment in Cutaneous T-Cell Lymphoma. Cancers, 15(8), 2362. https://doi.org/10.3390/cancers15082362