

TIGD1 Function as a Potential Cuproptosis Regulator Following a Novel Cuproptosis-Related Gene Risk Signature in Colorectal Cancer
 ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Acquisition
2.2. Differential Expression Analysis
2.3. Identification of Cuproptosis-Related Genes by WGCNA
2.4. Construction of the Cuproptosis-Related Prognostic Signature
2.5. Gene Set Enrichment Analyses
2.6. Functional Enrichment Analysis
2.7. Assessment of Immune Cell Infiltration and Immune Microenvironment
2.8. Drug Sensitivity Prediction
2.9. Tissue Sample Collection, RNA Extraction, and Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
2.10. Cell Viability and Colony Formation Assays
2.11. Chemical Reagent Assay
2.12. Western Blotting
2.13. Detection of Viable and Dead Cells Using Calcein-Methyl 4-Acetoxybenzoate (Calcein-AM)/Prodium Iodide (PI)
2.14. Measurement of the Copper Content
2.15. Statistical Analysis
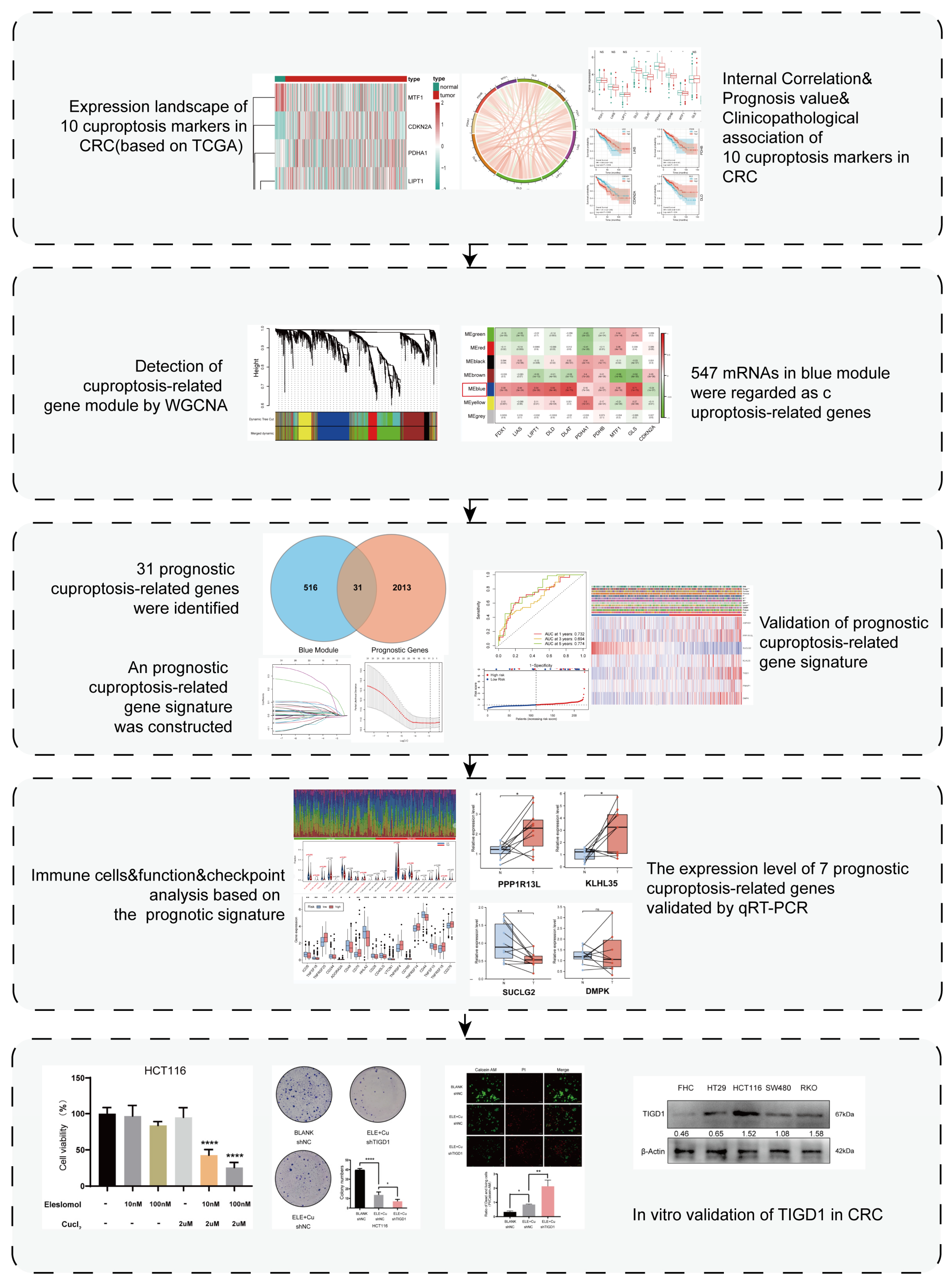
3. Results
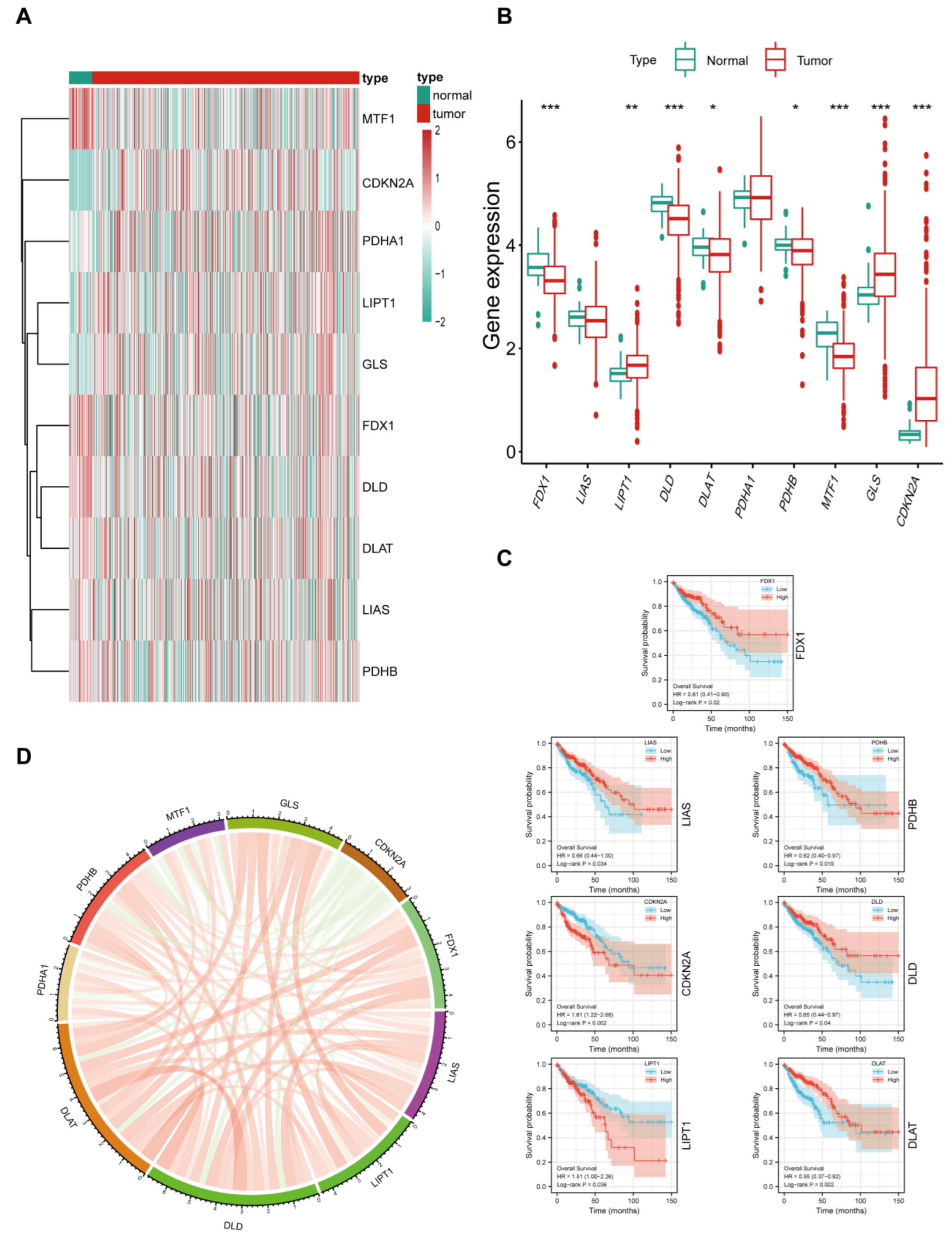
3.1. Expression Landscape of Cuproptosis Markers in CRC
3.2. Relationship between Cuproptosis Marker Expression Levels and Clinicopathological Features of Patients with CRC
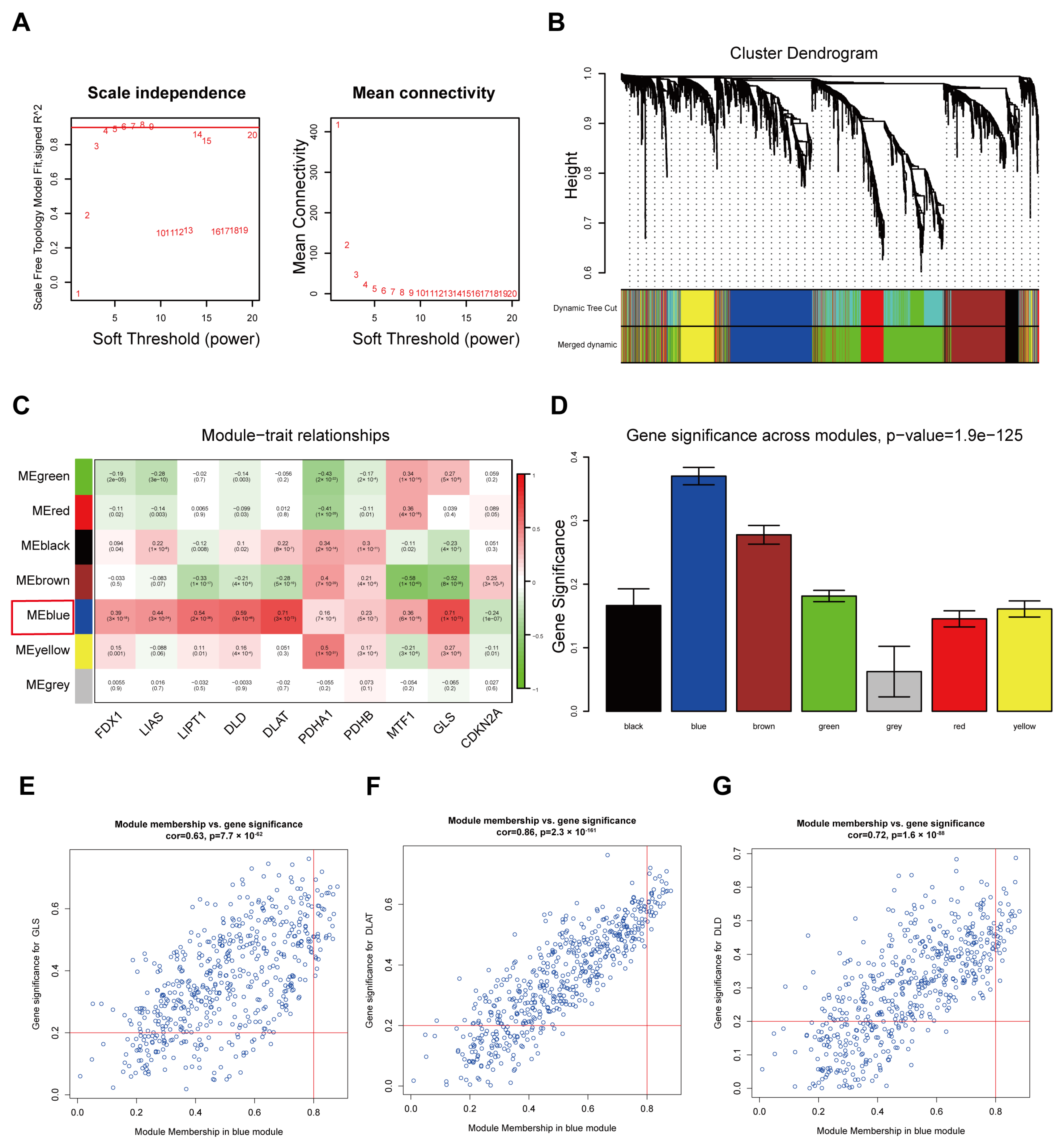
3.3. Detection of Cuproptosis-Related mRNAs Using WGCNA
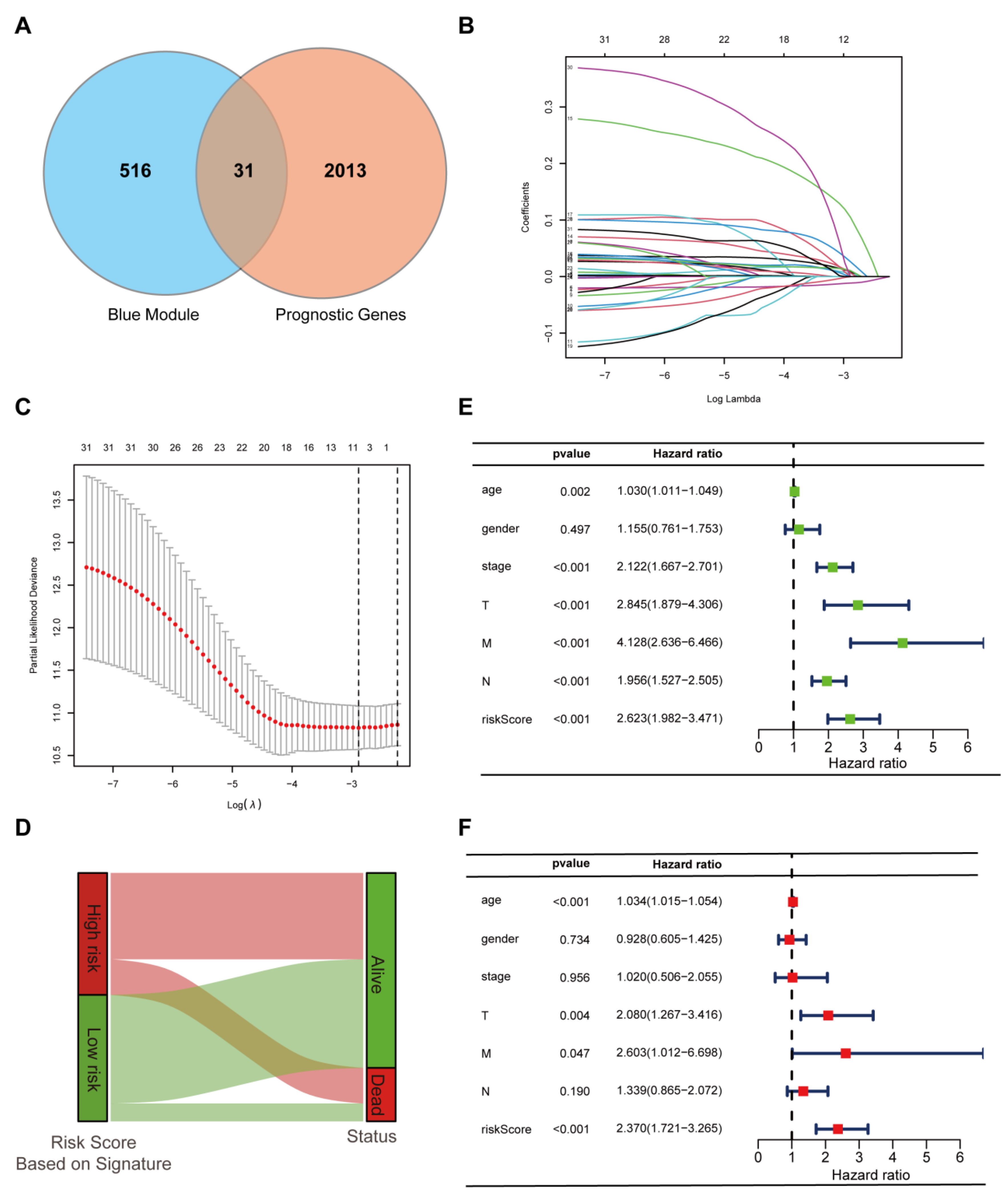
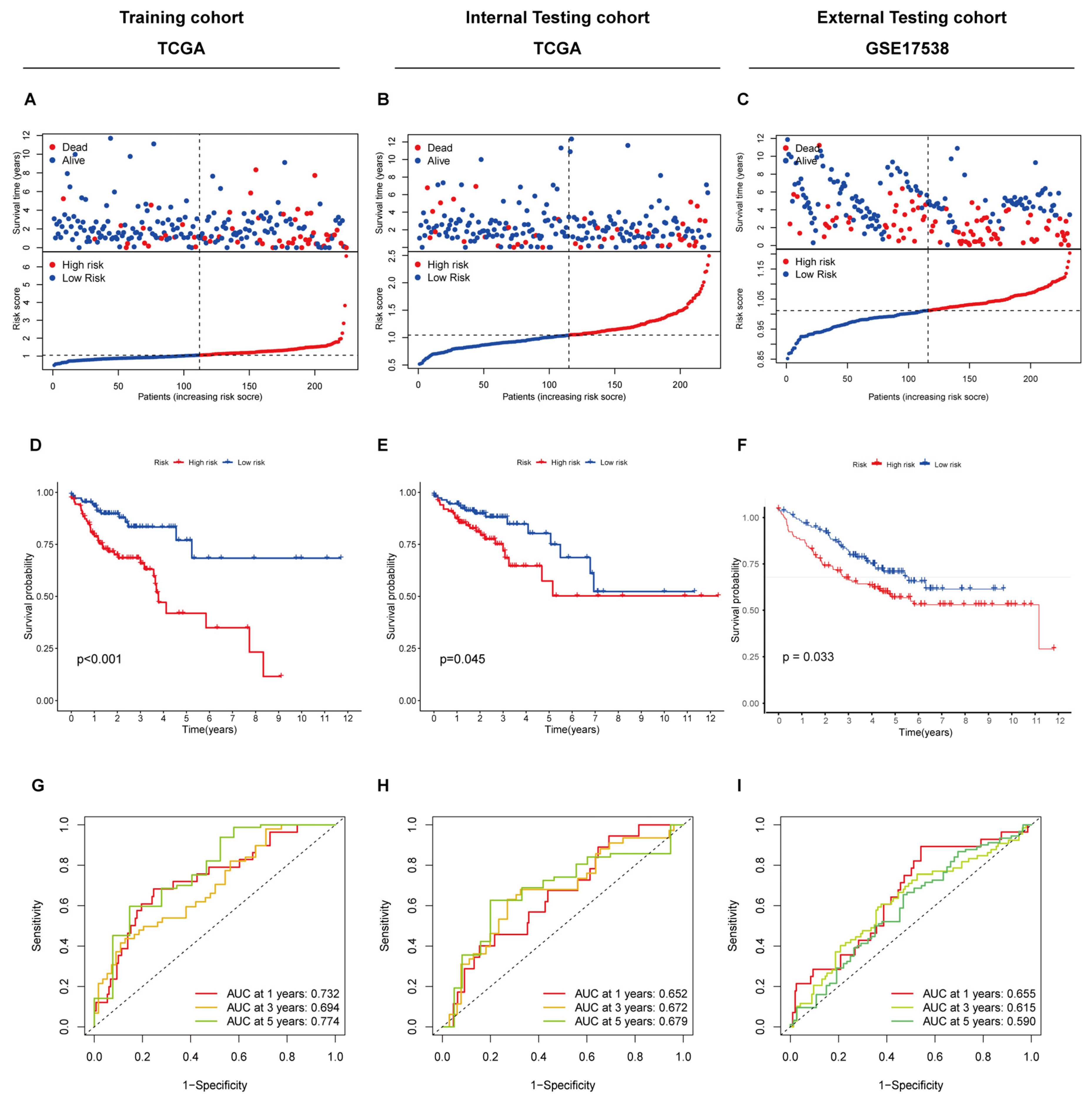
3.4. Construction and Validation of the Prognostic Cuproptosis-Related Gene Signature
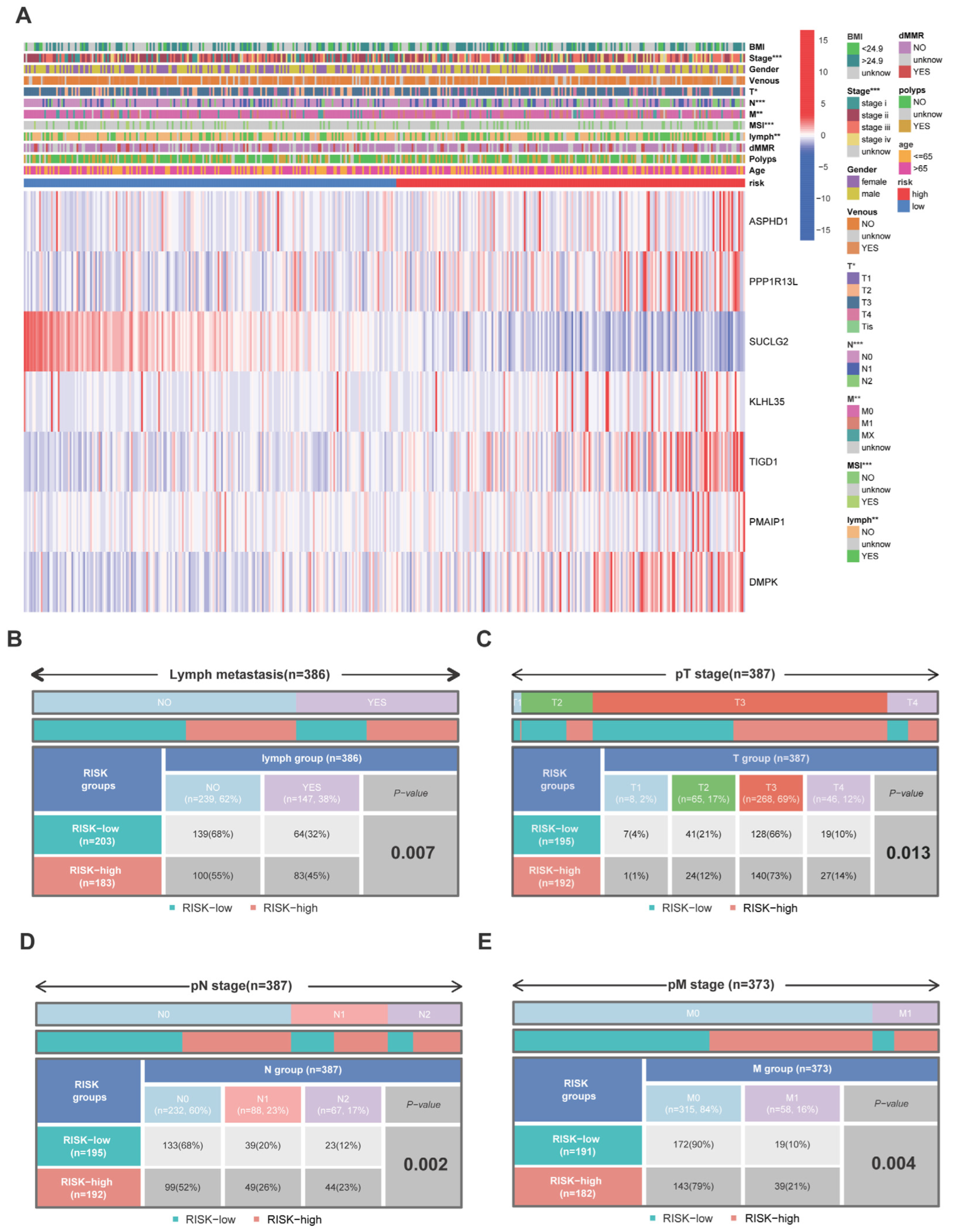
3.5. Relationship between the 7-PCRG Signature and the Clinicopathological Characteristics of Patients with CRC
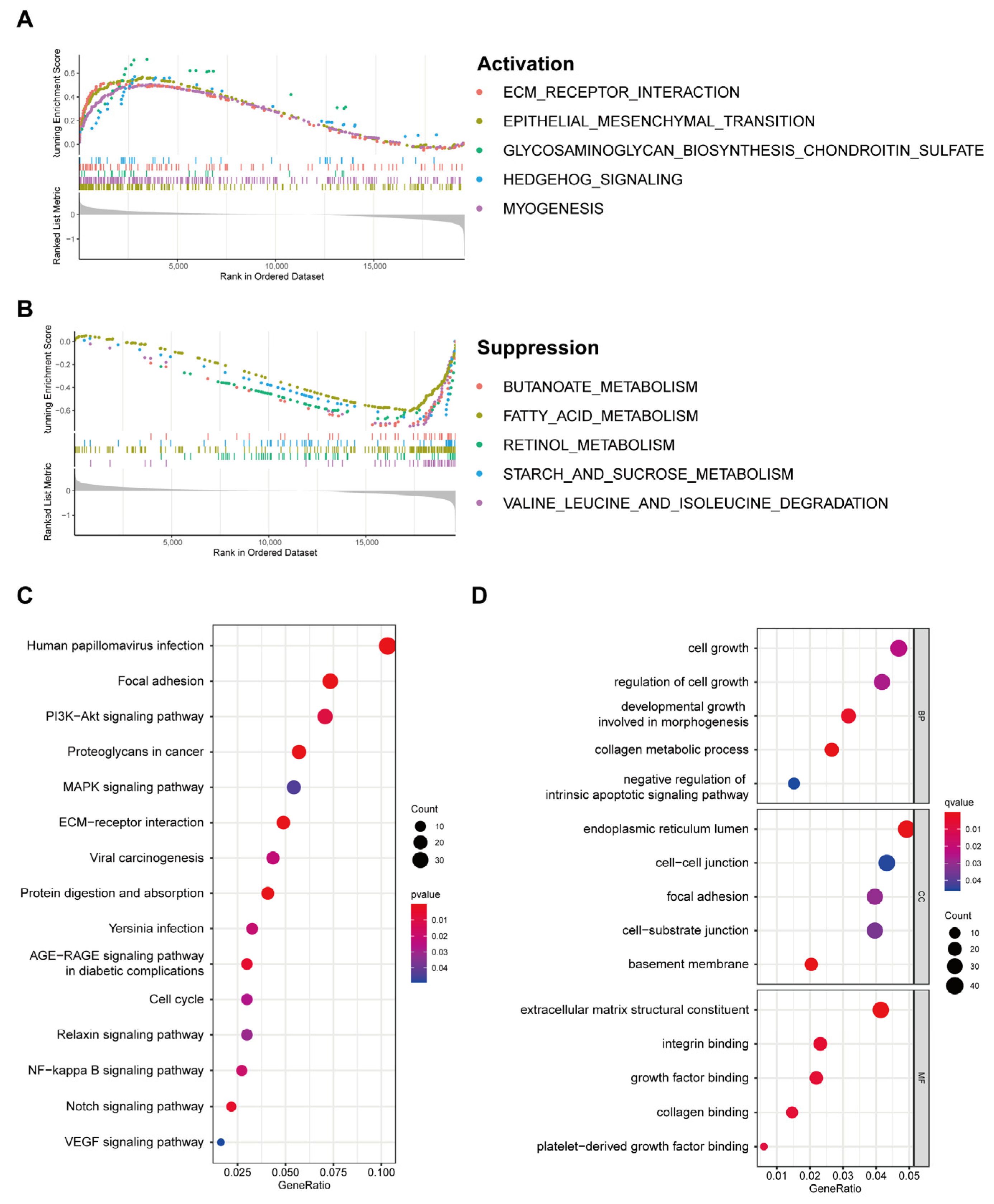
3.6. Discovery of Pathways and Molecular Functions of a Cuproptosis-Related Gene Signature via Enrichment Analysis
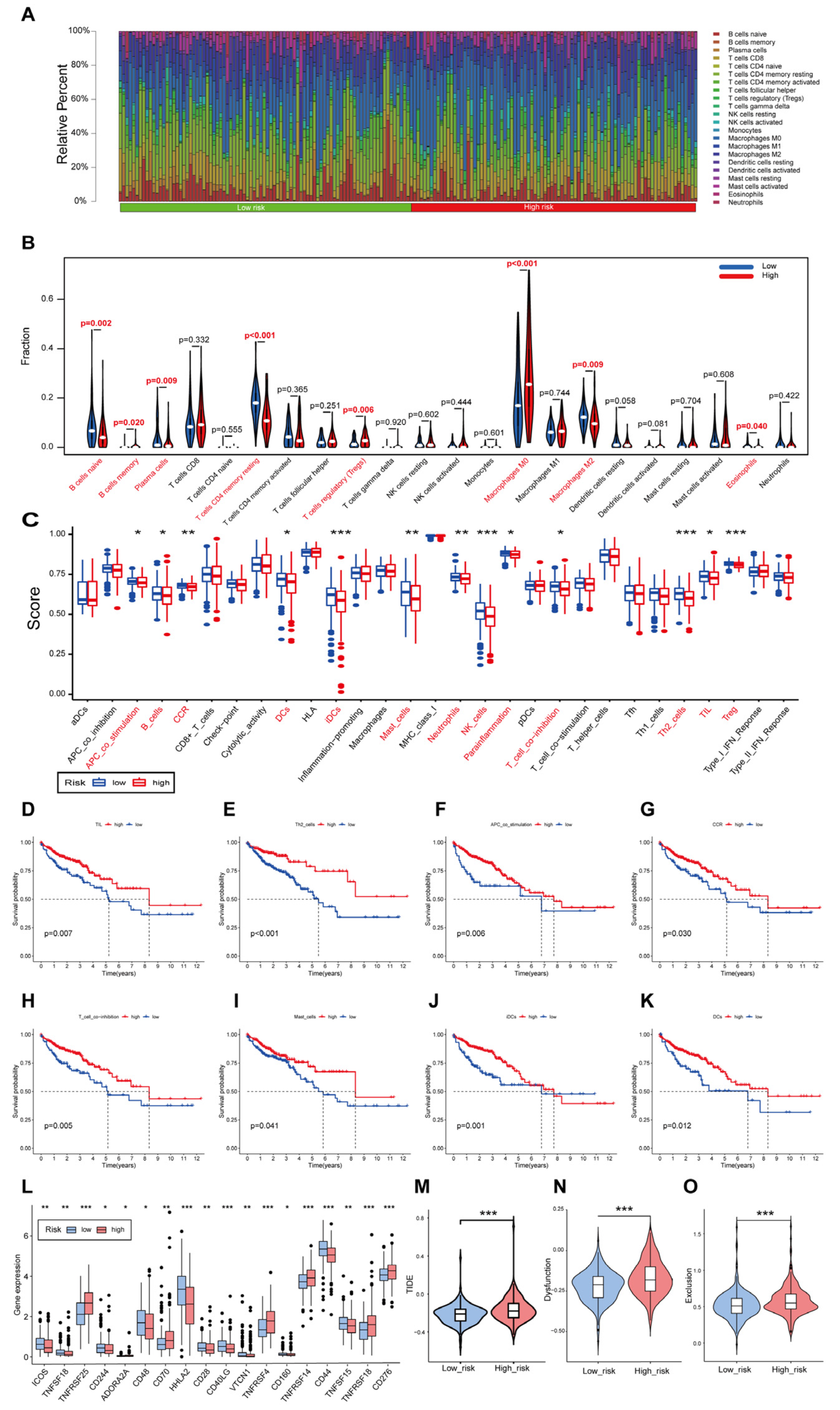
3.7. Immune Landscape of Patients with CRC Using the Cuproptosis-Related Gene Signature
3.8. Immunotherapy and Drug Sensitivity Analysis Based on the 7-PCRG Signature
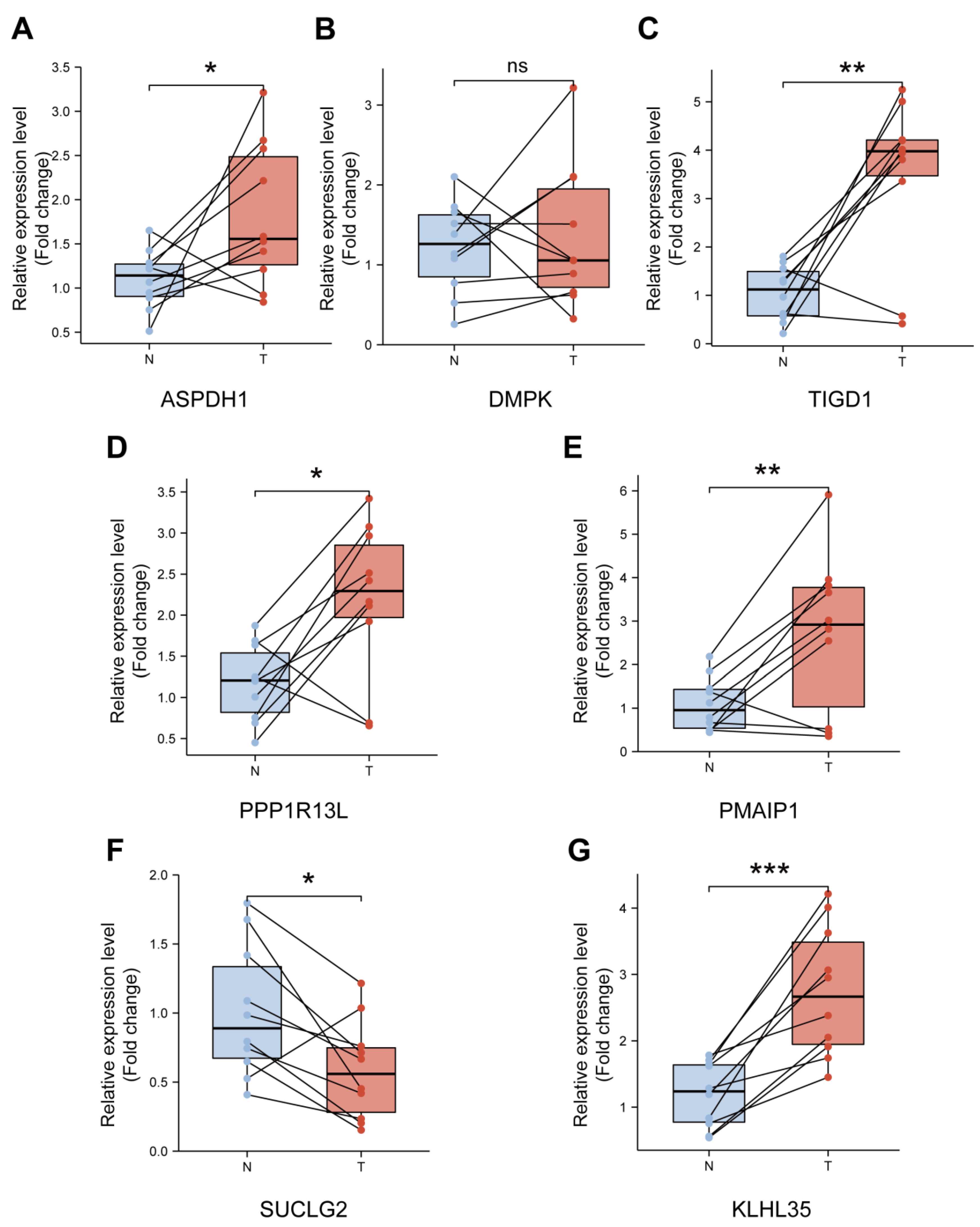
3.9. Validation of the Expression Levels of Seven PCRGs in CRC Samples
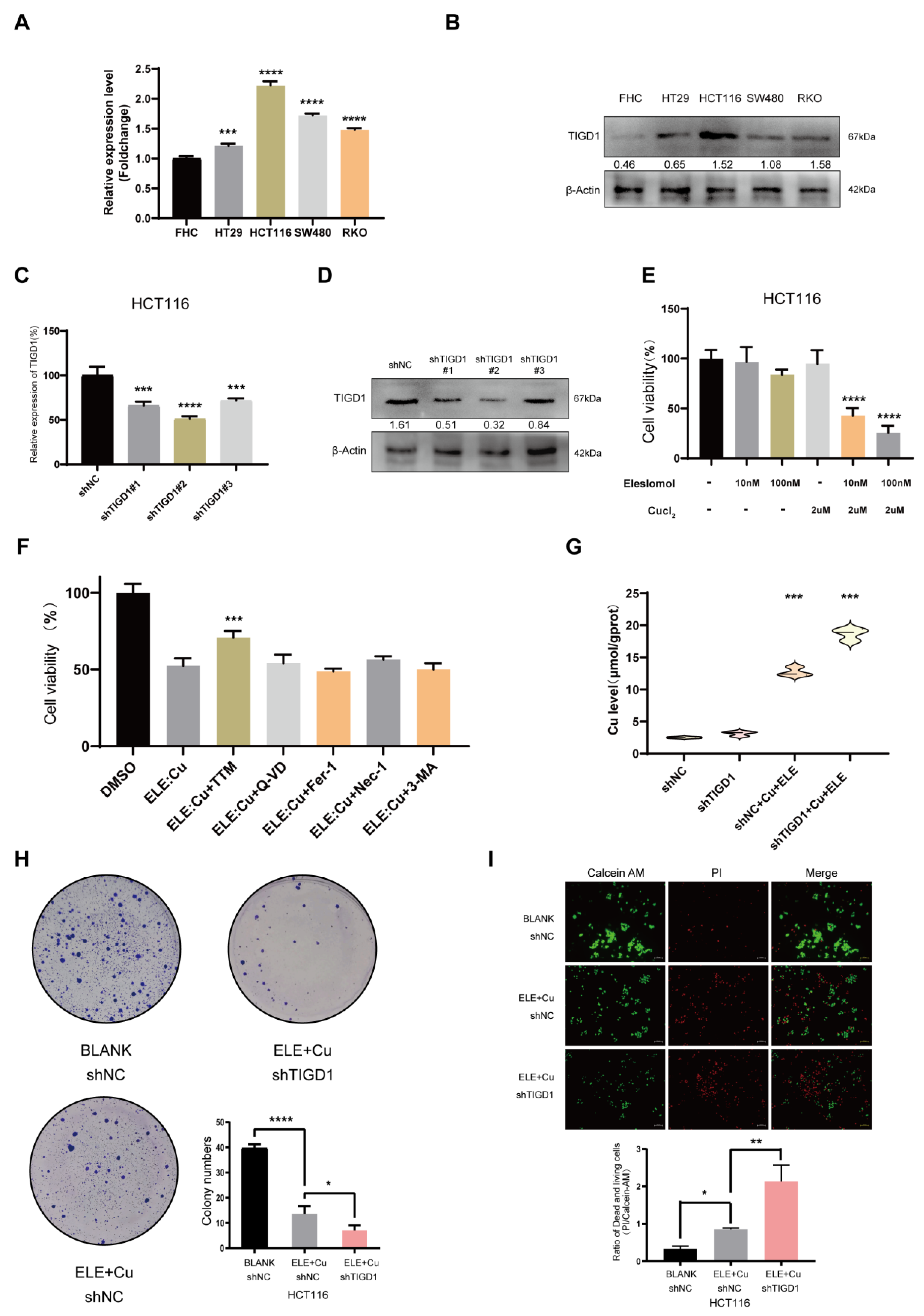
3.10. Elesclomol-Induced Cuproptosis and the Identification of TIGD1 in Cuproptosis Regulation in CRC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef]
- Global Burden of Disease Cancer C; Fitzmaurice, C.; Akinyemiju, T.F.; Al Lami, F.H.; Alam, T.; Alizadeh-Navaei, R.; Allen, C.; Alsharif, U.; Alvis-Guzman, N.; Amini, E.; et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived With Disability, and Disability-Adjusted Life-Years for 29 Cancer Groups, 1990 to 2016: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2018, 4, 1553–1568. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.; Pantel, K.; Kang, Y. Tumor metastasis: Moving new biological insights into the clinic. Nat. Med. 2013, 19, 1450–1464. [Google Scholar] [CrossRef] [PubMed]
- Hissong, E.; Pittman, M.E. Colorectal carcinoma screening: Established methods and emerging technology. Crit. Rev. Clin. Lab. Sci. 2020, 57, 22–36. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.S. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer Res. 2011, 30, 87. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef]
- Su, Z.; Yang, Z.; Xu, Y.; Chen, Y.; Yu, Q. Apoptosis, autophagy, necroptosis, and cancer metastasis. Mol. Cancer 2015, 14, 48. [Google Scholar] [CrossRef]
- Yang, C.; Song, J.; Hwang, S.; Choi, J.; Song, G.; Lim, W. Apigenin enhances apoptosis induction by 5-fluorouracil through regulation of thymidylate synthase in colorectal cancer cells. Redox Biol. 2021, 47, 102144. [Google Scholar] [CrossRef]
- Chen, X.; Zeh, H.J.; Kang, R.; Kroemer, G.; Tang, D. Cell death in pancreatic cancer: From pathogenesis to therapy. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 804–823. [Google Scholar] [CrossRef]
- Koren, E.; Fuchs, Y. Modes of Regulated Cell Death in Cancer. Cancer Discov. 2021, 11, 245–265. [Google Scholar] [CrossRef]
- Luo, W.; Xiang, W.; Gan, L.; Che, J.; Li, J.; Wang, Y.; Han, L.; Gu, R.; Ye, L.; Wang, R.; et al. Bulk and single-cell transcriptome profiling reveal necroptosis-based molecular classification, tumor microenvironment infiltration characterization, and prognosis prediction in colorectal cancer. J. Transl. Med. 2022, 20, 235. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.; Xu, J.; Zhang, B.; Liu, J.; Liang, C.; Hua, J.; Meng, Q.; Yu, X.; Shi, S. Ferroptosis, necroptosis, and pyroptosis in anticancer immunity. J. Hematol. Oncol. 2020, 13, 110. [Google Scholar] [CrossRef] [PubMed]
- Fan, F.; Liu, P.; Bao, R.; Chen, J.; Zhou, M.; Mo, Z.; Ma, Y.; Liu, H.; Zhou, Y.; Cai, X.; et al. A Dual PI3K/HDAC Inhibitor Induces Immunogenic Ferroptosis to Potentiate Cancer Immune Checkpoint Therapy. Cancer Res. 2021, 81, 6233–6245. [Google Scholar] [CrossRef]
- Tang, D.; Chen, X.; Kroemer, G. Cuproptosis: A copper-triggered modality of mitochondrial cell death. Cell. Res. 2022, 32, 417–418. [Google Scholar] [CrossRef]
- Tsvetkov, P.; Coy, S.; Petrova, B.; Dreishpoon, M.; Verma, A.; Abdusamad, M.; Rossen, J.; Joesch-Cohen, L.; Humeidi, R.; Spangler, R.D.; et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 2022, 375, 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- Brady, D.C.; Crowe, M.S.; Turski, M.L.; Hobbs, G.A.; Yao, X.; Chaikuad, A.; Knapp, S.; Xiao, K.; Campbell, S.L.; Thiele, D.J.; et al. Copper is required for oncogenic BRAF signalling and tumorigenesis. Nature 2014, 509, 492–496. [Google Scholar] [CrossRef]
- Turski, M.L.; Brady, D.C.; Kim, H.J.; Kim, B.E.; Nose, Y.; Counter, C.M.; Winge, D.R.; Thiele, D.J. A novel role for copper in Ras/mitogen-activated protein kinase signaling. Mol. Cell. Biol. 2012, 32, 1284–1295. [Google Scholar] [CrossRef]
- Wu, Z.; Lu, Z.; Li, L.; Ma, M.; Long, F.; Wu, R.; Huang, L.; Chou, J.; Yang, K.; Zhang, Y.; et al. Identification and Validation of Ferroptosis-Related LncRNA Signatures as a Novel Prognostic Model for Colon Cancer. Front. Immunol. 2021, 12, 783362. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Wang, P.; Zhao, Y.; Liu, K.; Liu, X.; Liang, J.; Zhou, H.; Wang, Z.; Zhou, Z.; Xu, N. Wip1 cooperates with KPNA2 to modulate the cell proliferation and migration of colorectal cancer via a p53-dependent manner. J. Cell. Biochem. 2019, 120, 15709–15718. [Google Scholar] [CrossRef]
- Tang, J.; Kong, D.; Cui, Q.; Wang, K.; Zhang, D.; Gong, Y.; Wu, G. Prognostic Genes of Breast Cancer Identified by Gene Co-expression Network Analysis. Front. Oncol. 2018, 8, 374. [Google Scholar] [CrossRef] [PubMed]
- Kamarudin, A.N.; Cox, T.; Kolamunnage-Dona, R. Time-dependent ROC curve analysis in medical research: Current methods and applications. BMC Med. Res. Methodol. 2017, 17, 53. [Google Scholar] [CrossRef]
- Zhao, W.J.; Ou, G.Y.; Lin, W.W. Integrative Analysis of Neuregulin Family Members-Related Tumor Microenvironment for Predicting the Prognosis in Gliomas. Front. Immunol. 2021, 12, 682415. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Fan, J.; Wang, B.; Traugh, N.; Chen, Q.; Liu, J.S.; Li, B.; Liu, X.S. TIMER: A Web Server for Comprehensive Analysis of Tumor-Infiltrating Immune Cells. Cancer Res. 2017, 77, e108–e110. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Khodadoust, M.S.; Liu, C.L.; Newman, A.M.; Alizadeh, A.A. Profiling Tumor Infiltrating Immune Cells with CIBERSORT. Methods Mol. Biol. 2018, 1711, 243–259. [Google Scholar] [PubMed]
- Jiang, P.; Gu, S.; Pan, D.; Fu, J.; Sahu, A.; Hu, X.; Li, Z.; Traugh, N.; Bu, X.; Li, B.; et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018, 24, 1550–1558. [Google Scholar] [CrossRef]
- Geeleher, P.; Cox, N.; Huang, R.S. pRRophetic: An R package for prediction of clinical chemotherapeutic response from tumor gene expression levels. PLoS ONE 2014, 9, e107468. [Google Scholar] [CrossRef]
- Kanth, P.; Inadomi, J.M. Screening and prevention of colorectal cancer. BMJ 2021, 374, n1855. [Google Scholar] [CrossRef]
- Sui, X.; Zhang, R.; Liu, S.; Duan, T.; Zhai, L.; Zhang, M.; Han, X.; Xiang, Y.; Huang, X.; Lin, H.; et al. RSL3 Drives Ferroptosis Through GPX4 Inactivation and ROS Production in Colorectal Cancer. Front. Pharmacol. 2018, 9, 1371. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, F.; Wang, L.; Lou, Y. A438079 affects colorectal cancer cell proliferation, migration, apoptosis, and pyroptosis by inhibiting the P2X7 receptor. Biochem. Biophys. Res. Commun. 2021, 558, 147–153. [Google Scholar] [CrossRef]
- Guo, J.; Zheng, J.; Mu, M.; Chen, Z.; Xu, Z.; Zhao, C.; Yang, K.; Qin, X.; Sun, X.; Yu, J. GW4064 enhances the chemosensitivity of colorectal cancer to oxaliplatin by inducing pyroptosis. Biochem. Biophys. Res. Commun. 2021, 548, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Shimura, T.; Banwait, J.K.; Goel, A. Andrographis-mediated chemosensitization through activation of ferroptosis and suppression of beta-catenin/Wnt-signaling pathways in colorectal cancer. Carcinogenesis 2020, 41, 1385–1394. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Green, M.; Choi, J.E.; Gijon, M.; Kennedy, P.D.; Johnson, J.K.; Liao, P.; Lang, X.; Kryczek, I.; Sell, A.; et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 2019, 569, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Zhang, T.; Ma, X.; Yang, Q.C.; Yang, L.L.; Yang, S.C.; Liang, M.; Xu, Z.; Sun, Z.J. Microenvironment-Responsive Prodrug-Induced Pyroptosis Boosts Cancer Immunotherapy. Adv. Sci. 2021, 8, e2101840. [Google Scholar] [CrossRef]
- Huo, K.; Shangguan, X.; Xia, Y.; Shen, Z.; Chen, C. Excess copper inhibits the growth of rice seedlings by decreasing uptake of nitrate. Ecotoxicol. Environ. Saf. 2020, 190, 110105. [Google Scholar] [CrossRef]
- Li, Y.; Kang, Z.L.; Qiao, N.; Hu, L.M.; Ma, Y.J.; Liang, X.H.; Liu, J.L.; Yang, Z.M. Effects of Excess Copper Ions on Decidualization of Human Endometrial Stromal Cells. Biol. Trace Elem. Res. 2017, 177, 10–15. [Google Scholar] [CrossRef]
- Shanbhag, V.C.; Gudekar, N.; Jasmer, K.; Papageorgiou, C.; Singh, K.; Petris, M.J. Copper metabolism as a unique vulnerability in cancer. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118893. [Google Scholar] [CrossRef]
- Li, Y. Copper homeostasis: Emerging target for cancer treatment. IUBMB Life 2020, 72, 1900–1908. [Google Scholar] [CrossRef]
- Roumaud, P.; Rwigemera, A.; Martin, L.J. Transcription factors SF1 and cJUN cooperate to activate the Fdx1 promoter in MA-10 Leydig cells. J. Steroid. Biochem. Mol. Biol. 2017, 171, 121–132. [Google Scholar] [CrossRef]
- Lubecka, K.; Kaufman-Szymczyk, A.; Fabianowska-Majewska, K. Inhibition of breast cancer cell growth by the combination of clofarabine and sulforaphane involves epigenetically mediated CDKN2A upregulation. Nucleosides Nucleotides Nucleic Acids 2018, 37, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Liggett, W.H.; Sidransky, D., Jr. Role of the p16 tumor suppressor gene in cancer. J. Clin. Oncol. 1998, 16, 1197–1206. [Google Scholar] [CrossRef]
- Goh, W.Q.; Ow, G.S.; Kuznetsov, V.A.; Chong, S.; Lim, Y.P. DLAT subunit of the pyruvate dehydrogenase complex is upregulated in gastric cancer-implications in cancer therapy. Am. J. Transl. Res. 2015, 7, 1140–1151. [Google Scholar]
- Wang, L.; Cao, Y.; Guo, W.; Xu, J. High expression of cuproptosis-related gene FDX1 in relation to good prognosis and immune cells infiltration in colon adenocarcinoma (COAD). J. Cancer Res. Clin. Oncol. 2023, 149, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.K.; Li, Y.H.; Bai, X.S.; Lin, G.L. The Cell Cycle-Associated Protein CDKN2A May Promotes Colorectal Cancer Cell Metastasis by Inducing Epithelial-Mesenchymal Transition. Front. Oncol. 2022, 12, 834235. [Google Scholar] [CrossRef] [PubMed]
- Chung, T.W.; Lee, J.H.; Choi, H.J.; Park, M.J.; Kim, E.Y.; Han, J.H.; Jang, S.B.; Lee, S.O.; Lee, S.W.; Hang, J.; et al. Anemone rivularis inhibits pyruvate dehydrogenase kinase activity and tumor growth. J. Ethnopharmacol. 2017, 203, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.S.; Kim, J.G.; Kang, B.W.; Lee, S.J.; Jeon, H.S.; Park, J.S.; Choi, G.S.; Lee, W.K. PPP1R13L variant associated with prognosis for patients with rectal cancer. J. Cancer Res. Clin. Oncol. 2013, 139, 465–473. [Google Scholar] [CrossRef]
- Jeong, S.; Yun, H.K.; Jeong, Y.A.; Jo, M.J.; Kang, S.H.; Kim, J.L.; Kim, D.Y.; Park, S.H.; Kim, B.R.; Na, Y.J.; et al. Cannabidiol-induced apoptosis is mediated by activation of Noxa in human colorectal cancer cells. Cancer Lett. 2019, 447, 12–23. [Google Scholar] [CrossRef]
- Zhou, Z.; He, H.; Wang, K.; Shi, X.; Wang, Y.; Su, Y.; Wang, Y.; Li, D.; Liu, W.; Zhang, Y.; et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science 2020, 368, eaaz7548. [Google Scholar] [CrossRef]
- Feng, M.; Zhao, Z.; Yang, M.; Ji, J.; Zhu, D. T-cell-based immunotherapy in colorectal cancer. Cancer Lett. 2021, 498, 201–209. [Google Scholar] [CrossRef]
- Wang, W.; Zhong, Y.; Zhuang, Z.; Xie, J.; Lu, Y.; Huang, C.; Sun, Y.; Wu, L.; Yin, J.; Yu, H.; et al. Multiregion single-cell sequencing reveals the transcriptional landscape of the immune microenvironment of colorectal cancer. Clin. Transl. Med. 2021, 11, e253. [Google Scholar] [CrossRef]
- Zhao, S.; Mi, Y.; Guan, B.; Zheng, B.; Wei, P.; Gu, Y.; Zhang, Z.; Cai, S.; Xu, Y.; Li, X.; et al. Tumor-derived exosomal miR-934 induces macrophage M2 polarization to promote liver metastasis of colorectal cancer. J. Hematol. Oncol. 2020, 13, 156. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Vlaming, M.; van Meerten, T.; Bremer, E. The Implementation of TNFRSF Co-Stimulatory Domains in CAR-T Cells for Optimal Functional Activity. Cancers 2022, 14, 299. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, Y.; Jia, L.; Kim, J.K.; Li, J.; Deng, P.; Zhang, W.; Krebsbach, P.H.; Wang, C.Y. CD276 expression enables squamous cell carcinoma stem cells to evade immune surveillance. Cell Stem Cell 2021, 28, 1597–1613.e1597. [Google Scholar] [CrossRef] [PubMed]
- Honeywell, R.J.; Kathmann, I.; Giovannetti, E.; Tibaldi, C.; Smit, E.F.; Rovithi, M.N.; Verheul, H.M.W.; Peters, G.J. Epithelial Transfer of the Tyrosine Kinase Inhibitors Erlotinib, Gefitinib, Afatinib, Crizotinib, Sorafenib, Sunitinib, and Dasatinib: Implications for Clinical Resistance. Cancers 2020, 12, 3322. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yin, D.; Wu, L. Identification of cuproptosis-related subtypes and development of a prognostic signature in colorectal cancer. Sci. Rep. 2022, 12, 17348. [Google Scholar] [CrossRef]
- Xu, M.; Mu, J.; Wang, J.; Zhou, Q.; Wang, J. Construction and validation of a cuproptosis-related lncRNA signature as a novel and robust prognostic model for colon adenocarcinoma. Front. Oncol. 2022, 12, 961213. [Google Scholar] [CrossRef]
- He, R.; Zhang, H.; Zhao, H.; Yin, X.; Lu, J.; Gu, C.; Gao, J.; Xu, Q. Multiomics Analysis Reveals Cuproptosis-Related Signature for Evaluating Prognosis and Immunotherapy Efficacy in Colorectal Cancer. Cancers 2023, 15, 387. [Google Scholar] [CrossRef]
- Liang, G.Z.; Wen, X.F.; Song, Y.W.; Zhang, Z.J.; Chen, J.; Chen, Y.L.; Pan, W.D.; He, X.W.; Hu, T.; Xian, Z.Y. Construction and Validation of a Novel Prognosis Model in Colon Cancer Based on Cuproptosis-Related Long Non-Coding RNAs. J. Clin. Med. 2023, 12, 1528. [Google Scholar] [CrossRef]
- Qiu, Y.; Cao, Y.; Cao, W.; Jia, Y.; Lu, N. The Application of Ferroptosis in Diseases. Pharmacol. Res. 2020, 159, 104919. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Training Group | Testing Group | p-Value | ||
|---|---|---|---|---|---|
| No. | % | No. | % | ||
| Age | — | — | — | ||
| ≤65 | 93 | 83 | >0.05 | ||
| >65 | 119 | 129 | — | ||
| Gender | — | — | — | ||
| Male | 121 | 107 | >0.05 | ||
| Female | 91 | 105 | — | ||
| AJCC Stage | — | — | — | ||
| I | 36 | 37 | >0.05 | ||
| II | 83 | 82 | — | ||
| III | 53 | 64 | — | ||
| IV | 35 | 23 | — | ||
| T stage | — | — | — | ||
| T1 | 4 | 6 | >0.05 | ||
| T2 | 38 | 36 | — | ||
| T3 | 141 | 149 | — | ||
| T4 | 29 | 20 | — | ||
| N stage | — | — | — | ||
| N0 | 127 | 126 | >0.05 | ||
| N1 | 44 | 53 | — | ||
| N2 | 41 | 33 | — | ||
| M stage | — | — | — | ||
| M0 | 155 | 160 | >0.05 | ||
| M1 | 35 | 23 | — |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Z.; Lin, C.; Zhang, F.; Lu, Z.; Wang, Y.; Liu, Y.; Zhou, Z.; Li, L.; Song, L. TIGD1 Function as a Potential Cuproptosis Regulator Following a Novel Cuproptosis-Related Gene Risk Signature in Colorectal Cancer. Cancers 2023, 15, 2286. https://doi.org/10.3390/cancers15082286
Wu Z, Lin C, Zhang F, Lu Z, Wang Y, Liu Y, Zhou Z, Li L, Song L. TIGD1 Function as a Potential Cuproptosis Regulator Following a Novel Cuproptosis-Related Gene Risk Signature in Colorectal Cancer. Cancers. 2023; 15(8):2286. https://doi.org/10.3390/cancers15082286
Chicago/Turabian StyleWu, Zhiwei, Changwei Lin, Fan Zhang, Zhixing Lu, Yaohui Wang, Yang Liu, Zhijiao Zhou, Liang Li, and Liying Song. 2023. "TIGD1 Function as a Potential Cuproptosis Regulator Following a Novel Cuproptosis-Related Gene Risk Signature in Colorectal Cancer" Cancers 15, no. 8: 2286. https://doi.org/10.3390/cancers15082286
APA StyleWu, Z., Lin, C., Zhang, F., Lu, Z., Wang, Y., Liu, Y., Zhou, Z., Li, L., & Song, L. (2023). TIGD1 Function as a Potential Cuproptosis Regulator Following a Novel Cuproptosis-Related Gene Risk Signature in Colorectal Cancer. Cancers, 15(8), 2286. https://doi.org/10.3390/cancers15082286