
Microsatellite Instability: A Review of Molecular Epidemiology and Implications for Immune Checkpoint Inhibitor Therapy
 , , , and
, , , and
Abstract
Simple Summary
Abstract
1. Introduction
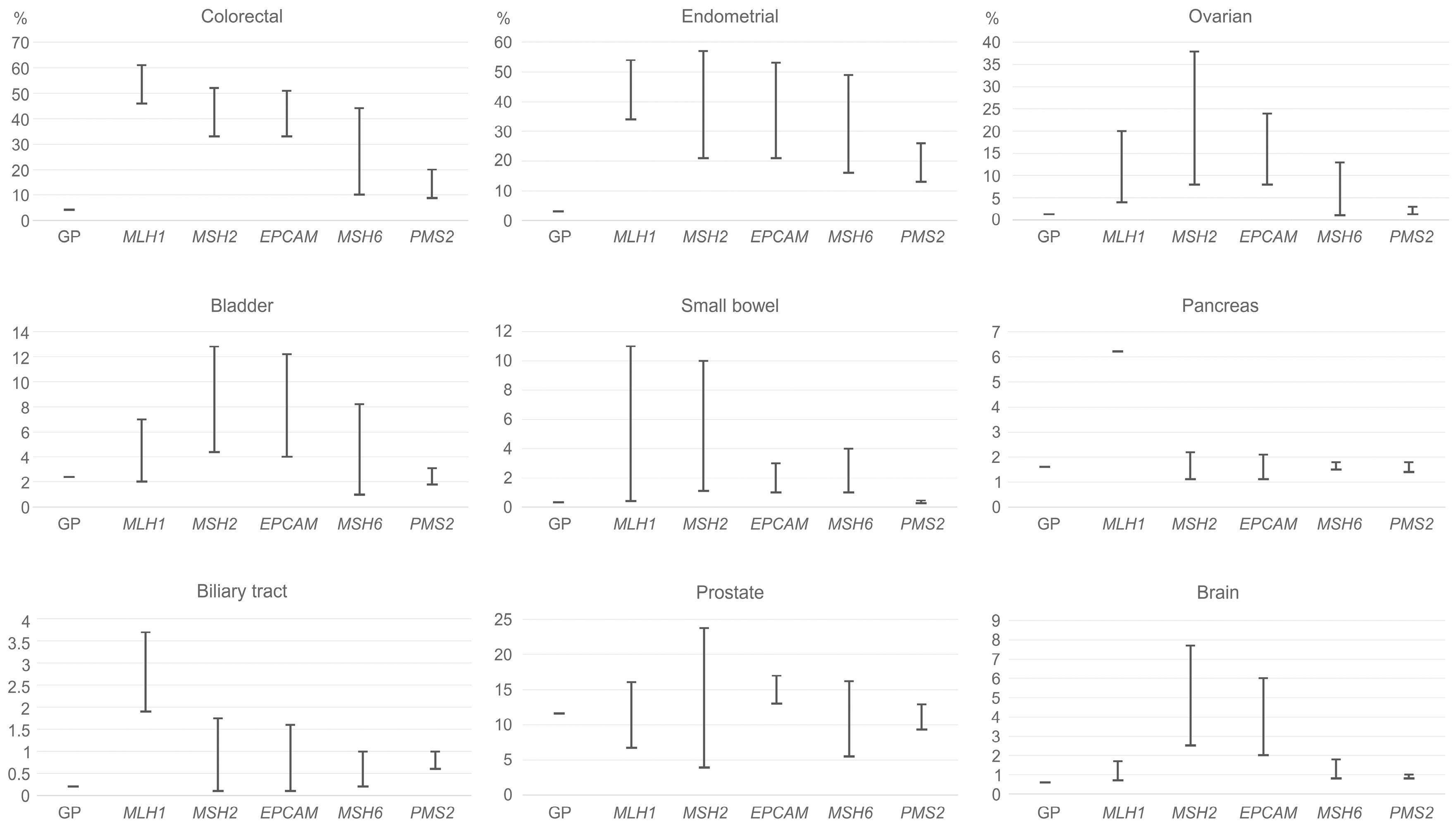
2. Lynch Syndrome-Associated Cancer Risks
3. MSI: Molecular Epidemiology across Cancer Types
4. MSI and Chemotherapy
5. MSI as a Predictive Biomarker for ICI Efficacy
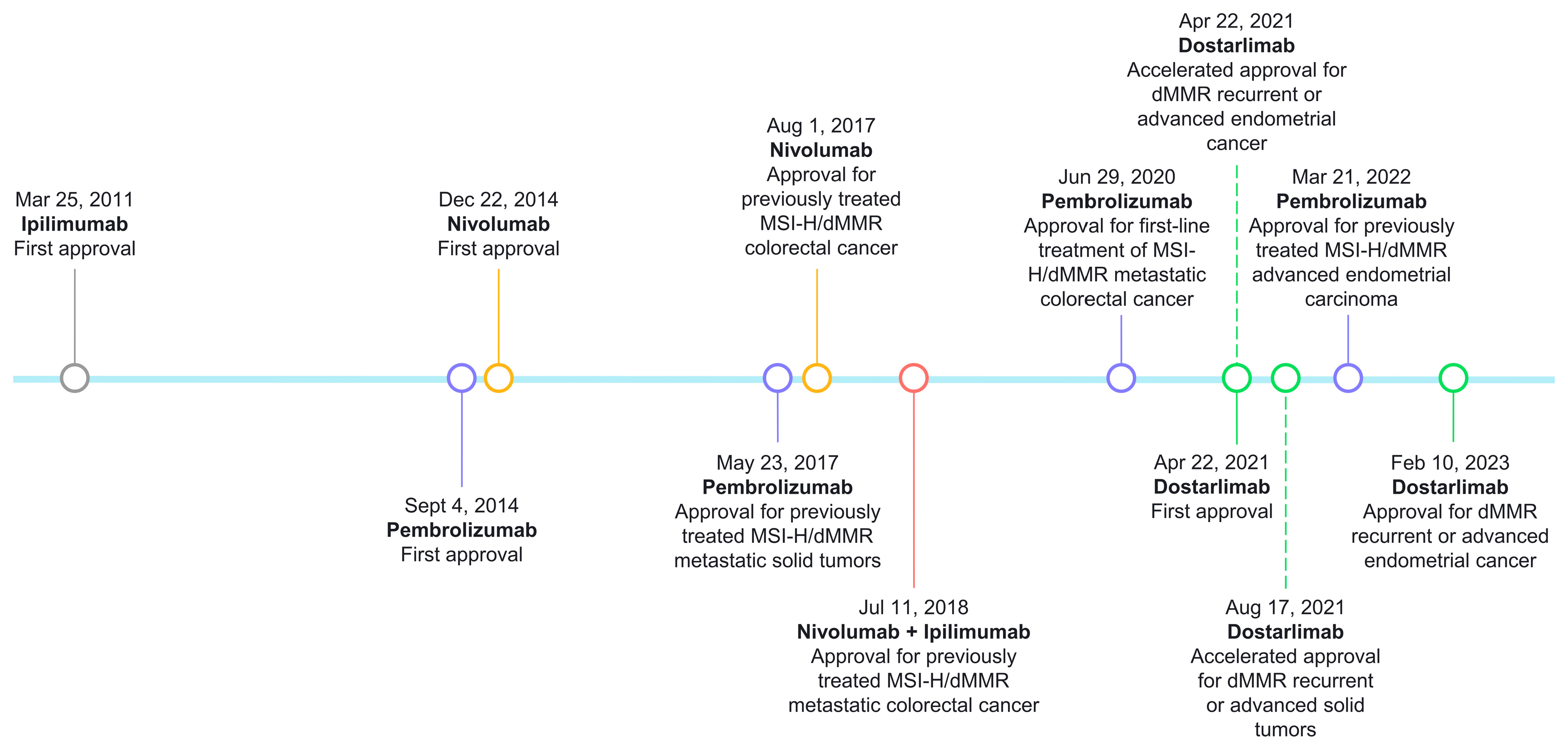
5.1. Site-Agnostic Indications and Clinical Evidence
5.2. Site-Specific Indications and Clinical Evidence
5.2.1. Colorectal Cancer
- Pembrolizumab
- 2.
- Nivolumab and Ipilimumab
- 3.
- Dostarlimab
- 4.
- Other currently not FDA-approved ICIs for MSI CRC
5.2.2. Tumor Types other Than Colorectal
6. Mechanisms of Resistance to ICI
7. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Baretti, M.; Le, D.T. DNA Mismatch Repair in Cancer. Pharmacol. Ther. 2018, 189, 45–62. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Setton, J.; Lee, N.Y.; Riaz, N.; Powell, S.N. The Therapeutic Significance of Mutational Signatures from DNA Repair Deficiency in Cancer. Nat. Commun. 2018, 9, 3292. [Google Scholar] [CrossRef] [PubMed]
- Bass, A.J.; Thorsson, V.; Shmulevich, I.; Reynolds, S.M.; Miller, M.; Bernard, B.; Hinoue, T.; Laird, P.W.; Curtis, C.; Shen, H.; et al. Comprehensive Molecular Characterization of Gastric Adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef]
- Lepore Signorile, M.; Disciglio, V.; di Carlo, G.; Pisani, A.; Simone, C.; Ingravallo, G. From Genetics to Histomolecular Characterization: An Insight into Colorectal Carcinogenesis in Lynch Syndrome. Int. J. Mol. Sci. 2021, 22, 6767. [Google Scholar] [CrossRef] [PubMed]
- Lower, S.S.; McGurk, M.P.; Clark, A.G.; Barbash, D.A. Satellite DNA Evolution: Old Ideas, New Approaches. Curr. Opin. Genet. Dev. 2018, 49, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Kolodner, R.D.; Marsischky, G.T. Eukaryotic DNA Mismatch Repair. Curr. Opin. Genet. Dev. 1999, 9, 89–96. [Google Scholar] [CrossRef]
- Kunkel, T.A.; Erie, D.A. DNA Mismatch Repair. Annu. Rev. Biochem. 2005, 74, 681–710. [Google Scholar] [CrossRef]
- Modrich, P.; Lahue, R. Mismatch Repair in Replication Fidelity, Genetic Recombination, and Cancer Biology. Annu. Rev. Biochem. 1996, 65, 101–133. [Google Scholar] [CrossRef]
- Bronner, C.E.; Baker, S.M.; Morrison, P.T.; Warren, G.; Smith, L.G.; Lescoe, M.K.; Kane, M.; Earabino, C.; Lipford, J.; Lindblom, A.; et al. Mutation in the DNA Mismatch Repair Gene Homologue HMLH1 Is Associated with Hereditary Non-Polyposis Colon Cancer. Nature 1994, 368, 258–261. [Google Scholar] [CrossRef]
- Papadopoulos, N.; Nicolaides, N.C.; Wei, Y.F.; Ruben, S.M.; Carter, K.C.; Rosen, C.A.; Haseltine, W.A.; Fleischmann, R.D.; Fraser, C.M.; Adams, M.D.; et al. Mutation of a MutL Homolog in Hereditary Colon Cancer. Science 1994, 263, 1625–1629. [Google Scholar] [CrossRef]
- Fishel, R.; Lescoe, M.K.; Rao, M.R.S.; Copeland, N.G.; Jenkins, N.A.; Garber, J.; Kane, M.; Kolodner, R. The Human Mutator Gene Homolog MSH2 and Its Association with Hereditary Nonpolyposis Colon Cancer. Cell 1993, 75, 1027–1038. [Google Scholar] [CrossRef] [PubMed]
- Nicolaides, N.C.; Papadopoulos, N.; Liu, B.; Weit, Y.F.; Carter, K.C.; Ruben, S.M.; Rosen, C.A.; Haseltine, W.A.; Fleischmann, R.D.; Fraser, C.M.; et al. Mutations of Two PMS Homologues in Hereditary Nonpolyposis Colon Cancer. Nature 1994, 371, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Jiricny, J. Postreplicative Mismatch Repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012633. [Google Scholar] [CrossRef] [PubMed]
- Gilson, P.; Merlin, J.L.; Harlé, A. Detection of Microsatellite Instability: State of the Art and Future Applications in Circulating Tumour DNA (CtDNA). Cancers 2021, 13, 1491. [Google Scholar] [CrossRef] [PubMed]
- Iyer, R.R.; Pluciennik, A. DNA Mismatch Repair and Its Role in Huntington’s Disease. J. Huntingtons Dis. 2021, 10, 75–94. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Pearlman, A.H.; Hsieh, P. DNA Mismatch Repair and the DNA Damage Response. DNA Repair 2016, 38, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Kadyrov, F.A.; Dzantiev, L.; Constantin, N.; Modrich, P. Endonucleolytic Function of MutLalpha in Human Mismatch Repair. Cell 2006, 126, 297–308. [Google Scholar] [CrossRef]
- Ortega, J.; Lee, G.S.; Gu, L.; Yang, W.; Li, G.M. Mispair-Bound Human MutS-MutL Complex Triggers DNA Incisions and Activates Mismatch Repair. Cell Res. 2021, 31, 542–553. [Google Scholar] [CrossRef]
- Zhang, Y.; Yuan, F.; Presnell, S.R.; Tian, K.; Gao, Y.; Tomkinson, A.E.; Gu, L.; Li, G.M. Reconstitution of 5′-Directed Human Mismatch Repair in a Purified System. Cell 2005, 122, 693–705. [Google Scholar] [CrossRef]
- Guan, J.; Lu, C.; Jin, Q.; Lu, H.; Chen, X.; Tian, L.; Zhang, Y.; Ortega, J.; Zhang, J.; Siteni, S.; et al. MLH1 Deficiency-Triggered DNA Hyperexcision by Exonuclease 1 Activates the CGAS-STING Pathway. Cancer Cell 2021, 39, 109–121.e5. [Google Scholar] [CrossRef]
- Olave, M.C.; Graham, R.P. Mismatch Repair Deficiency: The What, How and Why It Is Important. Genes Chromosomes Cancer 2022, 61, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Li, G.M. Mechanisms and Functions of DNA Mismatch Repair. Cell Res. 2008, 18, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Diao, Z.; Han, Y.; Chen, Y.; Zhang, R.; Li, J. The Clinical Utility of Microsatellite Instability in Colorectal Cancer. Crit. Rev. Oncol. Hematol. 2021, 157, 103171. [Google Scholar] [CrossRef] [PubMed]
- Cheah, P.-L.; Looi, L.-M.; Koh, C.-C.; Lau, T.-P.; Chang, S.-W.; Teoh, K.-H.; Mun, K.-S.; Nazarina, A.R. Screening for Microsatellite Instability in Colorectal Carcinoma: Practical Utility of Immunohistochemistry and PCR with Fragment Analysis in a Diagnostic Histopathology Setting. Malays. J. Pathol. 2019, 41, 91–100. [Google Scholar] [PubMed]
- Diaz-Padilla, I.; Romero, N.; Amir, E.; Matias-Guiu, X.; Vilar, E.; Muggia, F.; Garcia-Donas, J. Mismatch Repair Status and Clinical Outcome in Endometrial Cancer: A Systematic Review and Meta-Analysis. Crit. Rev. Oncol. Hematol. 2013, 88, 154–167. [Google Scholar] [CrossRef] [PubMed]
- Black, D.; Soslow, R.A.; Levine, D.A.; Tornos, C.; Chen, S.C.; Hummer, A.J.; Bogomolniy, F.; Olvera, N.; Barakat, R.R.; Boyd, J. Clinicopathologic Significance of Defective DNA Mismatch Repair in Endometrial Carcinoma. J. Clin. Oncol. 2006, 24, 1745–1753. [Google Scholar] [CrossRef]
- Cortes-Ciriano, I.; Lee, S.; Park, W.Y.; Kim, T.M.; Park, P.J. A Molecular Portrait of Microsatellite Instability across Multiple Cancers. Nat. Commun. 2017, 8, 15180. [Google Scholar] [CrossRef]
- Polom, K.; Marano, L.; Marrelli, D.; de Luca, R.; Roviello, G.; Savelli, V.; Tan, P.; Roviello, F. Meta-Analysis of Microsatellite Instability in Relation to Clinicopathological Characteristics and Overall Survival in Gastric Cancer. Br. J. Surg. 2018, 105, 159–167. [Google Scholar] [CrossRef]
- Schrock, A.B.; Devoe, C.E.; McWilliams, R.; Sun, J.; Aparicio, T.; Stephens, P.J.; Ross, J.S.; Wilson, R.; Miller, V.A.; Ali, S.M.; et al. Genomic Profiling of Small-Bowel Adenocarcinoma. JAMA Oncol. 2017, 3, 1546–1553. [Google Scholar] [CrossRef]
- Luchini, C.; Bibeau, F.; Ligtenberg, M.J.L.; Singh, N.; Nottegar, A.; Bosse, T.; Miller, R.; Riaz, N.; Douillard, J.Y.; Andre, F.; et al. ESMO Recommendations on Microsatellite Instability Testing for Immunotherapy in Cancer, and Its Relationship with PD-1/PD-L1 Expression and Tumour Mutational Burden: A Systematic Review-Based Approach. Ann. Oncol. 2019, 30, 1232–1243. [Google Scholar] [CrossRef]
- Porkka, N.; Valo, S.; Nieminen, T.T.; Olkinuora, A.; Mäki-Nevala, S.; Eldfors, S.; Peltomäki, P. Sequencing of Lynch Syndrome Tumors Reveals the Importance of Epigenetic Alterations. Oncotarget 2017, 8, 108020–108030. [Google Scholar] [CrossRef] [PubMed]
- Lynch, H.T.; Lynch, P.M.; Lanspa, S.J.; Snyder, C.L.; Lynch, J.F.; Boland, C.R. Review of the Lynch Syndrome: History, Molecular Genetics, Screening, Differential Diagnosis, and Medicolegal Ramifications. Clin. Genet. 2009, 76, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The Blockade of Immune Checkpoints in Cancer Immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Sadreddini, S.; Baradaran, B.; Aghebati-Maleki, A.; Sadreddini, S.; Shanehbandi, D.; Fotouhi, A.; Aghebati-Maleki, L. Immune Checkpoint Blockade Opens a New Way to Cancer Immunotherapy. J. Cell Physiol. 2019, 234, 8541–8549. [Google Scholar] [CrossRef] [PubMed]
- de Giglio, A.; di Federico, A.; Nuvola, G.; Deiana, C.; Gelsomino, F. The Landscape of Immunotherapy in Advanced NSCLC: Driving beyond PD-1/PD-L1 Inhibitors (CTLA-4, LAG3, IDO, OX40, TIGIT, Vaccines). Curr. Oncol. Rep. 2021, 23, 126. [Google Scholar] [CrossRef]
- Umar, A.; Boland, C.R.; Terdiman, J.P.; Syngal, S.; de la Chapelle, A.; Rüschoff, J.; Fishel, R.; Lindor, N.M.; Burgart, L.J.; Hamelin, R.; et al. Revised Bethesda Guidelines for Hereditary Nonpolyposis Colorectal Cancer (Lynch Syndrome) and Microsatellite Instability. J. Natl. Cancer Inst. 2004, 96, 261–268. [Google Scholar] [CrossRef]
- Goel, A.; Nagasaka, T.; Hamelin, R.; Boland, C.R. An Optimized Pentaplex PCR for Detecting DNA Mismatch Repair-Deficient Colorectal Cancers. PLoS ONE 2010, 5, 9393. [Google Scholar] [CrossRef]
- Ratovomanana, T.; Cohen, R.; Svrcek, M.; Renaud, F.; Cervera, P.; Siret, A.; Letourneur, Q.; Buhard, O.; Bourgoin, P.; Guillerm, E.; et al. Performance of Next-Generation Sequencing for the Detection of Microsatellite Instability in Colorectal Cancer with Deficient DNA Mismatch Repair. Gastroenterology 2021, 161, 814–826.e7. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration Website. KEYTRUDA (Pembrolizumab). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/125514s133lbl.pdf (accessed on 7 February 2023).
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; di Giacomo, A.M.; de Jesus-Acosta, A.; Delord, J.P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients with Noncolorectal High Microsatellite Instability/Mismatch Repair-Deficient Cancer: Results from the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 2020, 38, 1–10. [Google Scholar] [CrossRef]
- Lenz, H.J.; van Cutsem, E.; Limon, M.L.; Wong, K.Y.M.; Hendlisz, A.; Aglietta, M.; García-Alfonso, P.; Neyns, B.; Luppi, G.; Cardin, D.B.; et al. First-Line Nivolumab Plus Low-Dose Ipilimumab for Microsatellite Instability-High/Mismatch Repair-Deficient Metastatic Colorectal Cancer: The Phase II CheckMate 142 Study. J. Clin. Oncol. 2022, 40, 161–170. [Google Scholar] [CrossRef]
- Cercek, A.; Lumish, M.; Sinopoli, J.; Weiss, J.; Shia, J.; Lamendola-Essel, M.; el Dika, I.H.; Segal, N.; Shcherba, M.; Sugarman, R.; et al. PD-1 Blockade in Mismatch Repair-Deficient, Locally Advanced Rectal Cancer. N. Engl. J. Med. 2022, 386, 2363–2376. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Lian, J.; Wang, X.; Pang, X.; Xu, B.; Tang, S.; Shao, J.; Lu, H. Intrinsic Resistance and Efficacy of Immunotherapy in Microsatellite Instability-High Colorectal Cancer: A Systematic Review and Meta-Analysis. Bosn. J. Basic Med. Sci. 2022, 23, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Maio, M.; Ascierto, P.A.; Manzyuk, L.; Motola-Kuba, D.; Penel, N.; Cassier, P.A.; Bariani, G.M.; de Jesus Acosta, A.; Doi, T.; Longo, F.; et al. Pembrolizumab in Microsatellite Instability High or Mismatch Repair Deficient Cancers: Updated Analysis from the Phase II KEYNOTE-158 Study. Ann. Oncol. 2022, 33, 929–938. [Google Scholar] [CrossRef] [PubMed]
- Taieb, J.; Svrcek, M.; Cohen, R.; Basile, D.; Tougeron, D.; Phelip, J.M. Deficient Mismatch Repair/Microsatellite Unstable Colorectal Cancer: Diagnosis, Prognosis and Treatment. Eur. J. Cancer 2022, 175, 136–157. [Google Scholar] [CrossRef]
- Grasso, C.S.; Giannakis, M.; Wells, D.K.; Hamada, T.; Mu, X.J.; Quist, M.; Nowak, J.A.; Nishihara, R.; Qian, Z.R.; Inamura, K.; et al. Genetic Mechanisms of Immune Evasion in Colorectal Cancer. Cancer Discov. 2018, 8, 730–749. [Google Scholar] [CrossRef]
- Zhang, C.; Li, D.; Xiao, B.; Zhou, C.; Jiang, W.; Tang, J.; Li, Y.; Zhang, R.; Han, K.; Hou, Z.; et al. B2M and JAK1/2-Mutated MSI-H Colorectal Carcinomas Can Benefit from Anti-PD-1 Therapy. J. Immunother. 2022, 45, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Abushukair, H.; Ababneh, O.; Zaitoun, S.; Saeed, A. Primary and Secondary Immune Checkpoint Inhibitors Resistance in Colorectal Cancer: Key Mechanisms and Ways to Overcome Resistance. Cancer Treat. Res. Commun. 2022, 33, 100643. [Google Scholar] [CrossRef] [PubMed]
- Bouferraa, Y.; Chedid, A.; Amhaz, G.; el Lakkiss, A.; Mukherji, D.; Temraz, S.; Shamseddine, A. The Role of Gut Microbiota in Overcoming Resistance to Checkpoint Inhibitors in Cancer Patients: Mechanisms and Challenges. Int. J. Mol. Sci. 2021, 22, 8036. [Google Scholar] [CrossRef]
- Ballhausen, A.; Przybilla, M.J.; Jendrusch, M.; Haupt, S.; Pfaffendorf, E.; Seidler, F.; Witt, J.; Hernandez Sanchez, A.; Urban, K.; Draxlbauer, M.; et al. The Shared Frameshift Mutation Landscape of Microsatellite-Unstable Cancers Suggests Immunoediting during Tumor Evolution. Nat. Commun. 2020, 11, 4740. [Google Scholar] [CrossRef]
- Bucksch, K.; Zachariae, S.; Aretz, S.; Büttner, R.; Holinski-Feder, E.; Holzapfel, S.; Hüneburg, R.; Kloor, M.; von Knebel Doeberitz, M.; Morak, M.; et al. Cancer Risks in Lynch Syndrome, Lynch-like Syndrome, and Familial Colorectal Cancer Type X: A Prospective Cohort Study. BMC Cancer 2020, 20, 460. [Google Scholar] [CrossRef]
- Lynch, H.T.; de la Chapelle, A. Hereditary Colorectal Cancer. N. Engl. J. Med. 2003, 348, 919–932. [Google Scholar] [CrossRef] [PubMed]
- Lynch, H.T.; Snyder, C.L.; Shaw, T.G.; Heinen, C.D.; Hitchins, M.P. Milestones of Lynch Syndrome: 1895–2015. Nat. Rev. Cancer 2015, 15, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Ligtenberg, M.J.L.; Kuiper, R.P.; Chan, T.L.; Goossens, M.; Hebeda, K.M.; Voorendt, M.; Lee, T.Y.H.; Bodmer, D.; Hoenselaar, E.; Hendriks-Cornelissen, S.J.B.; et al. Heritable Somatic Methylation and Inactivation of MSH2 in Families with Lynch Syndrome Due to Deletion of the 3′ Exons of TACSTD1. Nat. Genet. 2009, 41, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Modrich, P. Mismatch Repair, Genetic Stability, and Cancer. Science 1994, 266, 1959–1960. [Google Scholar] [CrossRef] [PubMed]
- Leach, F.S.; Nicolaides, N.C.; Papadopoulos, N.; Liu, B.; Jen, J.; Parsons, R.; Peltomäki, P.; Sistonen, P.; Aaltonen, L.A.; Nyström-Lahti, M.; et al. Mutations of a MutS Homolog in Hereditary Nonpolyposis Colorectal Cancer. Cell 1993, 75, 1215–1225. [Google Scholar] [CrossRef]
- Win, A.K.; Jenkins, M.A.; Dowty, J.G.; Antoniou, A.C.; Lee, A.; Giles, G.G.; Buchanan, D.D.; Clendenning, M.; Rosty, C.; Ahnen, D.J.; et al. Prevalence and Penetrance of Major Genes and Polygenes for Colorectal Cancer. Cancer Epidemiol. Biomark. Prev. 2017, 26, 404–412. [Google Scholar] [CrossRef]
- Poulogiannis, G.; Frayling, I.M.; Arends, M.J. DNA Mismatch Repair Deficiency in Sporadic Colorectal Cancer and Lynch Syndrome. Histopathology 2010, 56, 167–179. [Google Scholar] [CrossRef]
- Rossi, L.; le Frere-Belda, M.A.; Laurent-Puig, P.; Buecher, B.; de Pauw, A.; Stoppa-Lyonnet, D.; Canlorbe, G.; Caron, O.; Borghese, B.; Colas, C.; et al. Clinicopathologic Characteristics of Endometrial Cancer in Lynch Syndrome: A French Multicenter Study. Int. J. Gynecol. Cancer. 2017, 27, 953–960. [Google Scholar] [CrossRef]
- Evrard, C.; Alexandre, J. Predictive and Prognostic Value of Microsatellite Instability in Gynecologic Cancer (Endometrial and Ovarian). Cancers 2021, 13, 2434. [Google Scholar] [CrossRef]
- Tiwari, A.K.; Roy, H.K.; Lynch, H.T. Lynch Syndrome in the 21st Century: Clinical Perspectives. QJM Int. J. Med. 2016, 109, 151–158. [Google Scholar] [CrossRef]
- Kempers, M.J.E.; Kuiper, R.P.; Ockeloen, C.W.; Chappuis, P.O.; Hutter, P.; Rahner, N.; Schackert, H.K.; Steinke, V.; Holinski-Feder, E.; Morak, M.; et al. Risk of Colorectal and Endometrial Cancers in EPCAM Deletion-Positive Lynch Syndrome: A Cohort Study. Lancet Oncol. 2011, 12, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Bonadona, V.; Bonaïti, B.; Olschwang, S.; Grandjouan, S.; Huiart, L.; Longy, M.; Guimbaud, R.; Buecher, B.; Bignon, Y.J.; Caron, O.; et al. Cancer Risks Associated with Germline Mutations in MLH1, MSH2, and MSH6 Genes in Lynch Syndrome. JAMA 2011, 305, 2304–2310. [Google Scholar] [CrossRef] [PubMed]
- Møller, P.; Seppälä, T.T.; Bernstein, I.; Holinski-Feder, E.; Sala, P.; Evans, D.G.; Lindblom, A.; Macrae, F.; Blanco, I.; Sijmons, R.H.; et al. Cancer Risk and Survival in Path_MMR Carriers by Gene and Gender up to 75 Years of Age: A Report from the Prospective Lynch Syndrome Database. Gut 2018, 67, 1306–1316. [Google Scholar] [CrossRef]
- Ryan, N.A.J.; Morris, J.; Green, K.; Lalloo, F.; Woodward, E.R.; Hill, J.; Crosbie, E.J.; Gareth Evans, D. Association of Mismatch Repair Mutation with Age at Cancer Onset in Lynch Syndrome: Implications for Stratified Surveillance Strategies. JAMA Oncol. 2017, 3, E1–E5. [Google Scholar] [CrossRef] [PubMed]
- Møller, P.; Seppälä, T.; Bernstein, I.; Holinski-Feder, E.; Sala, P.; Evans, D.G.; Lindblom, A.; Macrae, F.; Blanco, I.; Sijmons, R.; et al. Cancer Incidence and Survival in Lynch Syndrome Patients Receiving Colonoscopic and Gynaecological Surveillance: First Report from the Prospective Lynch Syndrome Database. Gut 2017, 66, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Engel, C.; Loeffler, M.; Steinke, V.; Rahner, N.; Holinski-Feder, E.; Dietmaier, W.; Schackert, H.K.; Goergens, H.; von Knebel Doeberitz, M.; Goecke, T.O.; et al. Risks of Less Common Cancers in Proven Mutation Carriers with Lynch Syndrome. J. Clin. Oncol. 2012, 30, 4409–4415. [Google Scholar] [CrossRef]
- Capelle, L.G.; van Grieken, N.C.T.; Lingsma, H.F.; Steyerberg, E.W.; Klokman, W.J.; Bruno, M.J.; Vasen, H.F.A.; Kuipers, E.J. Risk and Epidemiological Time Trends of Gastric Cancer in Lynch Syndrome Carriers in the Netherlands. Gastroenterology 2010, 138, 487–492. [Google Scholar] [CrossRef]
- Dominguez-Valentin, M.; Joost, P.; Therkildsen, C.; Jonsson, M.; Rambech, E.; Nilbert, M. Frequent Mismatch-Repair Defects Link Prostate Cancer to Lynch Syndrome. BMC Urol. 2016, 16, 15. [Google Scholar] [CrossRef]
- Joost, P.; Therkildsen, C.; Dominguez-Valentin, M.; Jönsson, M.; Nilbert, M. Urinary Tract Cancer in Lynch Syndrome; Increased Risk in Carriers of MSH2 Mutations. Urology 2015, 86, 1212–1217. [Google Scholar] [CrossRef]
- Watson, P.; Vasen, H.F.A.; Mecklin, J.P.; Bernstein, I.; Aarnio, M.; Järvinen, H.J.; Myrhoj, T.; Sunde, L.; Wijnen, J.T.; Lynch, H.T. The Risk of Extra-Colonic, Extra-Endometrial Cancer in the Lynch Syndrome. Int. J. Cancer 2008, 123, 444–449. [Google Scholar] [CrossRef]
- Adan, F.; Crijns, M.B.; Zandstra, W.S.E.; Bekkenk, M.W.; Bleeker, F.E.; Dekker, E.; van Leerdam, M.E. Cumulative Risk of Skin Tumours in Patients with Lynch Syndrome. Br. J. Dermatol. 2018, 179, 522–523. [Google Scholar] [CrossRef] [PubMed]
- South, C.D.; Hampel, H.; Comeras, I.; Westman, J.A.; Frankel, W.L.; de La Chapelle, A. The Frequency of Muir-Torre Syndrome among Lynch Syndrome Families. J. Natl. Cancer Inst. 2008, 100, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Berends, M.J.W.; Mensink, R.G.J.; Kempinga, C.; Sijmons, R.H.; van der Zee, A.G.J.; Hollema, H.; Kleibeuker, J.H.; Buys, C.H.C.M.; Hofstra, R.M.W. Association of Hereditary Nonpolyposis Colorectal Cancer-Related Tumors Displaying Low Microsatellite Instability with MSH6 Germline Mutations. Am. J. Hum. Genet. 1999, 65, 1291–1298. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, Y.; Hughes, K.S.; Parmigiani, G.; Braun, D. Penetrance of Colorectal Cancer Among Mismatch Repair Gene Mutation Carriers: A Meta-Analysis. JNCI Cancer Spectr. 2020, 4, pkaa027. [Google Scholar] [CrossRef]
- Senter, L.; Clendenning, M.; Sotamaa, K.; Hampel, H.; Green, J.; Potter, J.D.; Lindblom, A.; Lagerstedt, K.; Thibodeau, S.N.; Lindor, N.M.; et al. The Clinical Phenotype of Lynch Syndrome Due to Germ-Line PMS2 Mutations. Gastroenterology 2008, 135, 419–428.e1. [Google Scholar] [CrossRef]
- Broeke, S.W.T.; Klift, H.M.V.; Tops, C.M.J.; Aretz, S.; Bernstein, I.; Buchanan, D.D.; dela Chapelle, A.; Capella, G.; Clendenning, M.; Engel, C.; et al. Cancer Risks for PMS2-Associated Lynch Syndrome. J. Clin. Oncol. 2018, 36, 2961–2968. [Google Scholar] [CrossRef]
- Suerink, M.; Rodríguez-Girondo, M.; van der Klift, H.M.; Colas, C.; Brugieres, L.; Lavoine, N.; Jongmans, M.; Munar, G.C.; Evans, D.G.; Farrell, M.P.; et al. An Alternative Approach to Establishing Unbiased Colorectal Cancer Risk Estimation in Lynch Syndrome. Genet. Med. 2019, 21, 2706–2712. [Google Scholar] [CrossRef]
- NCCN Guidelines Version 2.2022 Genetic/Familial High-Risk Assessment: Colorectal. Available online: https://www.nccn.org/professionals/physician_gls/pdf/genetics_colon.pdf (accessed on 19 January 2023).
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2022. CA Cancer J. Clin 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Dominguez-Valentin, M.; Sampson, J.R.; Seppälä, T.T.; ten Broeke, S.W.; Plazzer, J.P.; Nakken, S.; Engel, C.; Aretz, S.; Jenkins, M.A.; Sunde, L.; et al. Cancer Risks by Gene, Age, and Gender in 6350 Carriers of Pathogenic Mismatch Repair Variants: Findings from the Prospective Lynch Syndrome Database. Genet. Med. 2020, 22, 15–25. [Google Scholar] [CrossRef]
- Baglietto, L.; Lindor, N.M.; Dowty, J.G.; White, D.M.; Wagner, A.; Gomez Garcia, E.B.; Vriends, A.H.J.T.; Cartwright, N.R.; Barnetson, R.A.; Farrington, S.M.; et al. Risks of Lynch Syndrome Cancers for MSH6 Mutation Carriers. J. Natl. Cancer Inst. 2010, 102, 193–201. [Google Scholar] [CrossRef]
- Bonneville, R.; Krook, M.A.; Kautto, E.A.; Miya, J.; Wing, M.R.; Chen, H.-Z.; Reeser, J.W.; Yu, L.; Roychowdhury, S. Landscape of Microsatellite Instability Across 39 Cancer Types. JCO Precis. Oncol. 2017, 1, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.J.; O’Haire, S.; Franchini, F.; Ijzerman, M.; Zalcberg, J.; Macrae, F.; Canfell, K.; Steinberg, J. A Scoping Review and Meta-Analysis on the Prevalence of Pan-Tumour Biomarkers (DMMR, MSI, High TMB) in Different Solid Tumours. Sci. Rep. 2022, 12, 20495. [Google Scholar] [CrossRef] [PubMed]
- Carr, P.R.; Alwers, E.; Bienert, S.; Weberpals, J.; Kloor, M.; Brenner, H.; Hoffmeister, M. Lifestyle Factors and Risk of Sporadic Colorectal Cancer by Microsatellite Instability Status: A Systematic Review and Meta-Analyses. Ann. Oncol. 2018, 29, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Ashktorab, H.; Ahuja, S.; Kannan, L.; Llor, X.; Nathan, E.; Xicola, R.M.; Laiyemo, A.O.; Carethers, J.M.; Brim, H.; Nouraie, M. A Meta-Analysis of MSI Frequency and Race in Colorectal Cancer. Oncotarget 2016, 7, 34546–34557. [Google Scholar] [CrossRef]
- Gkekas, I.; Novotny, J.; Pecen, L.; Strigård, K.; Palmqvist, R.; Gunnarsson, U. Microsatellite Instability as a Prognostic Factor in Stage II Colon Cancer Patients, a Meta-Analysis of Published Literature. Anticancer Res. 2017, 37, 6563–6574. [Google Scholar] [CrossRef]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The Consensus Molecular Subtypes of Colorectal Cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Petrelli, F.; Ghidini, M.; Cabiddu, M.; Pezzica, E.; Corti, D.; Turati, L.; Costanzo, A.; Varricchio, A.; Ghidini, A.; Barni, S.; et al. Microsatellite Instability and Survival in Stage II Colorectal Cancer: A Systematic Review and Meta-Analysis. Anticancer Res. 2019, 39, 6431–6441. [Google Scholar] [CrossRef]
- Aaltonen, L.A.; Peltomäki, P.; Leach, F.S.; Sistonen, P.; Pylkkänen, L.; Mecklin, J.P.; Järvinen, H.; Powell, S.M.; Jen, J.; Hamilton, S.R.; et al. Clues to the Pathogenesis of Familial Colorectal Cancer. Science 1993, 260, 812–816. [Google Scholar] [CrossRef]
- Mohamed, A.; Jiang, R.; Philip, P.A.; Diab, M.; Behera, M.; Wu, C.; Alese, O.; Shaib, W.L.; Gaines, T.M.; Balch, G.G.; et al. High-Risk Features Are Prognostic in DMMR/MSI-H Stage II Colon Cancer. Front. Oncol. 2021, 11, 755113. [Google Scholar] [CrossRef]
- Hecht, J.L.; Mutter, G.L. Molecular and Pathologic Aspects of Endometrial Carcinogenesis. J. Clin. Oncol. 2006, 24, 4783–4791. [Google Scholar] [CrossRef]
- Zighelboim, I.; Goodfellow, P.J.; Gao, F.; Gibb, R.K.; Powell, M.A.; Rader, J.S.; Mutch, D.G. Microsatellite Instability and Epigenetic Inactivation of MLH1 and Outcome of Patients with Endometrial Carcinomas of the Endometrioid Type. J. Clin. Oncol. 2007, 25, 2042–2048. [Google Scholar] [CrossRef] [PubMed]
- Getz, G.; Gabriel, S.B.; Cibulskis, K.; Lander, E.; Sivachenko, A.; Sougnez, C.; Lawrence, M.; Kandoth, C.; Dooling, D.; Fulton, R.; et al. Integrated Genomic Characterization of Endometrial Carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [CrossRef]
- Kwon, M.; An, M.; Klempner, S.J.; Lee, H.; Kim, K.M.; Sa, J.K.; Cho, H.J.; Hong, J.Y.; Lee, T.; Min, Y.W.; et al. Determinants of Response and Intrinsic Resistance to PD-1 Blockade in Microsatellite Instability-High Gastric Cancer. Cancer Discov. 2021, 11, 2168–2185. [Google Scholar] [CrossRef] [PubMed]
- Puliga, E.; Corso, S.; Pietrantonio, F.; Giordano, S. Microsatellite Instability in Gastric Cancer: Between Lights and Shadows. Cancer Treat. Rev. 2021, 95, 102175. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.A.; Wentzensen, N. Frequency of Mismatch Repair Deficiency in Ovarian Cancer: A Systematic Review This Article Is a US Government Work and, as Such, Is in the Public Domain of the United States of America. Int. J. Cancer 2011, 129, 1914–1922. [Google Scholar] [CrossRef]
- Pal, T.; Permuth-Wey, J.; Kumar, A.; Sellers, T.A. Systematic Review and Meta-Analysis of Ovarian Cancers: Estimation of Microsatellite-High Frequency and Characterization of Mismatch Repair Deficient Tumor Histology. Clin. Cancer Res. 2008, 14, 6847–6854. [Google Scholar] [CrossRef]
- Raphael, B.J.; Hruban, R.H.; Aguirre, A.J.; Moffitt, R.A.; Yeh, J.J.; Stewart, C.; Robertson, A.G.; Cherniack, A.D.; Gupta, M.; Getz, G.; et al. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203.e13. [Google Scholar] [CrossRef]
- Luchini, C.; Brosens, L.A.A.; Wood, L.D.; Chatterjee, D.; Shin, J.I.; Sciammarella, C.; Fiadone, G.; Malleo, G.; Salvia, R.; Kryklyva, V.; et al. Comprehensive Characterisation of Pancreatic Ductal Adenocarcinoma with Microsatellite Instability: Histology, Molecular Pathology and Clinical Implications. Gut 2021, 70, 148–156. [Google Scholar] [CrossRef]
- Luchini, C.; Scarpa, A. Microsatellite Instability in Pancreatic and Ampullary Carcinomas: Histology, Molecular Pathology, and Clinical Implications. Hum. Pathol. 2022, 132, 176–182. [Google Scholar] [CrossRef]
- Kullmann, F.; Strissel, P.L.; Strick, R.; Stoehr, R.; Eckstein, M.; Bertz, S.; Wullich, B.; Sikic, D.; Wach, S.; Taubert, H.; et al. Frequency of Microsatellite Instability (MSI) in Upper Tract Urothelial Carcinoma: Comparison of the Bethesda Panel and the Idylla MSI Assay in a Consecutively Collected, Multi-Institutional Cohort. J. Clin. Pathol. 2023, 76, 126–132. [Google Scholar] [CrossRef]
- Sobrino-Reig, E.; Meizoso, T.; García, J.; Varillas-Delgado, D.; Martin, Y.B. Morphological Predictors for Microsatellite Instability in Urothelial Carcinoma. Diagn. Pathol. 2021, 16, 106. [Google Scholar] [CrossRef] [PubMed]
- Schneider, B.; Glass, Ä.; Jagdmann, S.; Hühns, M.; Claus, J.; Zettl, H.; Dräger, D.L.; Maruschke, M.; Hakenberg, O.W.; Erbersdobler, A.; et al. Loss of Mismatch-Repair Protein Expression and Microsatellite Instability in Upper Tract Urothelial Carcinoma and Clinicopathologic Implications. Clin. Genitourin. Cancer 2020, 18, e563–e572. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch Repair Deficiency Predicts Response of Solid Tumors to PD-1 Blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Latham, A.; Srinivasan, P.; Kemel, Y.; Shia, J.; Bandlamudi, C.; Mandelker, D.; Middha, S.; Hechtman, J.; Zehir, A.; Dubard-Gault, M.; et al. Microsatellite Instability Is Associated with the Presence of Lynch Syndrome Pan-Cancer. J. Clin. Oncol. 2019, 37, 286. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Frankel, W.; Panescu, J.; Lockman, J.; Sotamaa, K.; Fix, D.; Comeras, I.; la Jeunesse, J.; Nakagawa, H.; Westman, J.A.; et al. Screening for Lynch Syndrome (Hereditary Nonpolyposis Colorectal Cancer) among Endometrial Cancer Patients. Cancer Res. 2006, 66, 7810–7817. [Google Scholar] [CrossRef]
- Stelloo, E.; Jansen, A.M.L.; Osse, E.M.; Nout, R.A.; Creutzberg, C.L.; Ruano, D.; Church, D.N.; Morreau, H.; Smit, V.T.H.B.M.; van Wezel, T.; et al. Practical Guidance for Mismatch Repair-Deficiency Testing in Endometrial Cancer. Ann. Oncol. 2017, 28, 96–102. [Google Scholar] [CrossRef]
- Goodfellow, P.J.; Billingsley, C.C.; Lankes, H.A.; Ali, S.; Cohn, D.E.; Broaddus, R.J.; Ramirez, N.; Pritchard, C.C.; Hampel, H.; Chassen, A.S.; et al. Combined Microsatellite Instability, MLH1 Methylation Analysis, and Immunohistochemistry for Lynch Syndrome Screening in Endometrial Cancers from GOG210: An NRG Oncology and Gynecologic Oncology Group Study. J. Clin. Oncol. 2015, 33, 4301–4308. [Google Scholar] [CrossRef]
- Simpkins, S.B.; Bocker, T.; Swisher, E.M.; Mutch, D.G.; Gersell, D.J.; Kovatich, A.J.; Palazzo, J.P.; Fishel, R.; Goodfellow, P.J. MLH1 Promoter Methylation and Gene Silencing Is the Primary Cause of Microsatellite Instability in Sporadic Endometrial Cancers. Hum. Mol. Genet. 1999, 8, 661–666. [Google Scholar] [CrossRef]
- Deshpande, M.; Romanski, P.A.; Rosenwaks, Z.; Gerhardt, J. Gynecological Cancers Caused by Deficient Mismatch Repair and Microsatellite Instability. Cancers 2020, 12, 3319. [Google Scholar] [CrossRef]
- Leite, M.; Corso, G.; Sousa, S.; Milanezi, F.; Afonso, L.P.; Henrique, R.; Soares, J.M.; Castedo, S.; Carneiro, F.; Roviello, F.; et al. MSI Phenotype and MMR Alterations in Familial and Sporadic Gastric Cancer. Int. J. Cancer 2011, 128, 1606–1613. [Google Scholar] [CrossRef]
- Gay, L.J.; Arends, M.J.; Mitrou, P.N.; Bowman, R.; Ibrahim, A.E.; Happerfield, L.; Luben, R.; McTaggart, A.; Ball, R.Y.; Rodwell, S.A. MLH1 Promoter Methylation, Diet, and Lifestyle Factors in Mismatch Repair Deficient Colorectal Cancer Patients from EPIC-Norfolk. Nutr. Cancer 2011, 63, 1000–1010. [Google Scholar] [CrossRef]
- Meyer, L.A.; Broaddus, R.R.; Lu, K.H. Endometrial Cancer and Lynch Syndrome: Clinical and Pathologic Considerations. Cancer Control 2009, 16, 14–22. [Google Scholar] [CrossRef]
- Ibrahim, A.E.K.; Arends, M.J. Molecular Typing of Colorectal Cancer: Applications in Diagnosis and Treatment. Diagn. Histopathol. 2012, 18, 70–80. [Google Scholar] [CrossRef]
- Jin, J.; Shi, Y.; Zhang, S.; Yang, S. PIK3CA Mutation and Clinicopathological Features of Colorectal Cancer: A Systematic Review and Meta-Analysis. Acta Oncol. 2020, 59, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Brennetot, C.; Duval, A.; Hamelin, R.; Pinto, M.; Oliveira, C.; Seruca, R.; Schwartz, S. Frequent Ki-Ras Mutations in Gastric Tumors of the MSI Phenotype. Gastroenterology 2003, 125, 1282–1283. [Google Scholar] [CrossRef]
- Polom, K.; Das, K.; Marrelli, D.; Roviello, G.; Pascale, V.; Voglino, C.; Rho, H.; Tan, P.; Roviello, F. KRAS Mutation in Gastric Cancer and Prognostication Associated with Microsatellite Instability Status. Pathol. Oncol. Res. 2019, 25, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.I.; Tseng, L.H.; Gocke, C.D.; Reil, S.; Le, D.T.; Azad, N.S.; Eshleman, J.R. Mutational Profiling of Colorectal Cancers with Microsatellite Instability. Oncotarget 2015, 6, 42334–42344. [Google Scholar] [CrossRef]
- Andreyev, H.J.N.; Norman, A.R.; Cunningham, D.; Oates, J.; Dix, B.R.; Iacopetta, B.J.; Young, J.; Walsh, T.; Ward, R.; Hawkins, N.; et al. Kirsten Ras Mutations in Patients with Colorectal Cancer: The “RASCAL II” Study. Br. J. Cancer 2001, 85, 692–696. [Google Scholar] [CrossRef]
- Venderbosch, S.; Nagtegaal, I.D.; Maughan, T.S.; Smith, C.G.; Cheadle, J.P.; Fisher, D.; Kaplan, R.; Quirke, P.; Seymour, M.T.; Richman, S.D.; et al. Mismatch Repair Status and BRAF Mutation Status in Metastatic Colorectal Cancer Patients: A Pooled Analysis of the CAIRO, CAIRO2, COIN, and FOCUS Studies. Clin. Cancer Res. 2014, 20, 5322–5330. [Google Scholar] [CrossRef]
- Salem, M.; Kopetz, S.; El-Refai, S.; Tabernero, J.; Sinicrope, F.; Tie, J.; George, T.; van Cutsem, E.; Mauer, E.; Lonardi, S.; et al. LBA SO-34 Impact of BRAF-V600E Mutation on Immunologic Characteristics of the Tumor Microenvironment (TME) and Associated Genomic Alterations in Patients with Microsatellite Instability-High (MSI-H) or Mismatch-Repair–Deficient (DMMR) Colorectal Cancer (CRC). Ann. Oncol. 2022, 33, S378. [Google Scholar] [CrossRef]
- McGivern, A.; Wynter, C.V.A.; Whitehall, V.L.J.; Kambara, T.; Spring, K.J.; Walsh, M.D.; Barker, M.A.; Arnold, S.; Simms, L.A.; Leggett, B.A.; et al. Promoter Hypermethylation Frequency and BRAF Mutations Distinguish Hereditary Non-Polyposis Colon Cancer from Sporadic MSI-H Colon Cancer. Fam. Cancer 2004, 3, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Bessa, X.; Ballesté, B.; Andreu, M.; Castells, A.; Bellosillo, B.; Balaguer, F.; Castellví-bel, S.; Paya, A.; Jover, R.; Alenda, C.; et al. A Prospective, Multicenter, Population-Based Study of BRAF Mutational Analysis for Lynch Syndrome Screening. Clin. Gastroenterol. Hepatol. 2008, 6, 206–214. [Google Scholar] [CrossRef]
- Porkka, N.; Lahtinen, L.; Ahtiainen, M.; Böhm, J.P.; Kuopio, T.; Eldfors, S.; Mecklin, J.P.; Seppälä, T.T.; Peltomäki, P. Epidemiological, Clinical and Molecular Characterization of Lynch-like Syndrome: A Population-Based Study. Int. J. Cancer 2019, 145, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Parsons, M.T.; Buchanan, D.D.; Thompson, B.; Young, J.P.; Spurdle, A.B. Correlation of Tumour BRAF Mutations and MLH1 Methylation with Germline Mismatch Repair (MMR) Gene Mutation Status: A Literature Review Assessing Utility of Tumour Features for MMR Variant Classification. J. Med. Genet. 2012, 49, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Kambara, T.; Simms, L.A.; Whitehall, V.L.J.; Spring, K.J.; Wynter, C.V.A.; Walsh, M.D.; Barker, M.A.; Arnold, S.; McGivern, A.; Matsubara, N.; et al. BRAF Mutation Is Associated with DNA Methylation in Serrated Polyps and Cancers of the Colorectum. Gut 2004, 53, 1137–1144. [Google Scholar] [CrossRef] [PubMed]
- Weisenberger, D.J.; Siegmund, K.D.; Campan, M.; Young, J.; Long, T.I.; Faasse, M.A.; Kang, G.H.; Widschwendter, M.; Weener, D.; Buchanan, D.; et al. CpG Island Methylator Phenotype Underlies Sporadic Microsatellite Instability and Is Tightly Associated with BRAF Mutation in Colorectal Cancer. Nat. Genet. 2006, 38, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Alvi, M.A.; Loughrey, M.B.; Dunne, P.; McQuaid, S.; Turkington, R.; Fuchs, M.A.; McGready, C.; Bingham, V.; Pang, B.; Moore, W.; et al. Molecular Profiling of Signet Ring Cell Colorectal Cancer Provides a Strong Rationale for Genomic Targeted and Immune Checkpoint Inhibitor Therapies. Br. J. Cancer 2017, 117, 203–209. [Google Scholar] [CrossRef]
- Fennell, L.J.; Jamieson, S.; McKeone, D.; Corish, T.; Rohdmann, M.; Furner, T.; Bettington, M.; Liu, C.; Kawamata, F.; Bond, C.; et al. MLH1-93 G/a Polymorphism Is Associated with MLH1 Promoter Methylation and Protein Loss in Dysplastic Sessile Serrated Adenomas with BRAFV600E Mutation. BMC Cancer 2018, 18, 35. [Google Scholar] [CrossRef]
- Tabernero, J.; Ros, J.; Élez, E. The Evolving Treatment Landscape in BRAF-V600E-Mutated Metastatic Colorectal Cancer. Am. Soc. Clin. Oncol. Educ. Book 2022, 42, 254–263. [Google Scholar] [CrossRef]
- Bettington, M.; Walker, N.; Rosty, C.; Brown, I.; Clouston, A.; McKeone, D.; Pearson, S.A.; Leggett, B.; Whitehall, V. Clinicopathological and Molecular Features of Sessile Serrated Adenomas with Dysplasia or Carcinoma. Gut 2017, 66, 97–106. [Google Scholar] [CrossRef]
- Bettington, M.L.; Walker, N.I.; Rosty, C.; Brown, I.S.; Clouston, A.D.; McKeone, D.M.; Pearson, S.A.; Klein, K.; Leggett, B.A.; Whitehall, V.L.J. A Clinicopathological and Molecular Analysis of 200 Traditional Serrated Adenomas. Mod. Pathol. 2015, 28, 414–427. [Google Scholar] [CrossRef] [PubMed]
- Bell, D.; Berchuck, A.; Birrer, M.; Chien, J.; Cramer, D.W.; Dao, F.; Dhir, R.; Disaia, P.; Gabra, H.; Glenn, P.; et al. Integrated Genomic Analyses of Ovarian Carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Weinstein, J.N.; Akbani, R.; Broom, B.M.; Wang, W.; Verhaak, R.G.W.; McConkey, D.; Lerner, S.; Morgan, M.; Creighton, C.J.; Smith, C.; et al. Comprehensive Molecular Characterization of Urothelial Bladder Carcinoma. Nature 2014, 507, 315–322. [Google Scholar] [CrossRef]
- Muzny, D.M.; Bainbridge, M.N.; Chang, K.; Dinh, H.H.; Drummond, J.A.; Fowler, G.; Kovar, C.L.; Lewis, L.R.; Morgan, M.B.; Newsham, I.F.; et al. Comprehensive Molecular Characterization of Human Colon and Rectal Cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- Wang, Y.K.; Bashashati, A.; Anglesio, M.S.; Cochrane, D.R.; Grewal, D.S.; Ha, G.; McPherson, A.; Horlings, H.M.; Senz, J.; Prentice, L.M.; et al. Genomic Consequences of Aberrant DNA Repair Mechanisms Stratify Ovarian Cancer Histotypes. Nat. Genet. 2017, 49, 856–864. [Google Scholar] [CrossRef]
- Wang, J.; Li, R.; He, Y.; Yi, Y.; Wu, H.; Liang, Z. Next-Generation Sequencing Reveals Heterogeneous Genetic Alterations in Key Signaling Pathways of Mismatch Repair Deficient Colorectal Carcinomas. Mod. Pathol. 2020, 33, 2591–2601. [Google Scholar] [CrossRef] [PubMed]
- Qiu, M.Z.; He, C.Y.; Yang, X.H.; Yang, L.Q.; Lin, J.Z.; Zhou, D.L.; Long, Y.K.; Guan, W.L.; Jin, Y.; Li, Y.H.; et al. Relationship of HER2 Alteration and Microsatellite Instability Status in Colorectal Adenocarcinoma. Oncologist 2021, 26, e1161–e1170. [Google Scholar] [CrossRef]
- Saygin, I.; Cakir, E. The Status of HER2 in Colorectal Carcinoma and the Relation of HER2 with Prognostic Parameters and MSI. Indian J. Pathol. Microbiol. 2022, 65, 336–342. [Google Scholar] [CrossRef]
- Engel, C.; Ahadova, A.; Seppälä, T.T.; Aretz, S.; Bigirwamungu-Bargeman, M.; Bläker, H.; Bucksch, K.; Büttner, R.; de Vos tot Nederveen Cappel, W.T.; Endris, V.; et al. Associations of Pathogenic Variants in MLH1, MSH2, and MSH6 With Risk of Colorectal Adenomas and Tumors and With Somatic Mutations in Patients with Lynch Syndrome. Gastroenterology 2020, 158, 1326–1333. [Google Scholar] [CrossRef]
- Mäki-Nevala, S.; Valo, S.; Ristimäki, A.; Sarhadi, V.; Knuutila, S.; Nyström, M.; Renkonen-Sinisalo, L.; Lepistö, A.; Mecklin, J.P.; Peltomäki, P. DNA Methylation Changes and Somatic Mutations as Tumorigenic Events in Lynch Syndrome-Associated Adenomas Retaining Mismatch Repair Protein Expression. EBioMedicine 2019, 39, 280–291. [Google Scholar] [CrossRef]
- Yurgelun, M.B.; Goel, A.; Hornick, J.L.; Sen, A.; Turgeon, D.K.; Ruffin IV, M.T.; Marcon, N.E.; Baron, J.A.; Bresalier, R.S.; Syngal, S.; et al. Microsatellite Instability and DNA Mismatch Repair Protein Deficiency in Lynch Syndrome Colorectal Polyps. Cancer Prev. Res. 2012, 5, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.K.; Ricciardiello, L.; Goel, A.; Chang, C.L.; Boland, C.R. Steady-State Regulation of the Human DNA Mismatch Repair System. J. Biol. Chem. 2000, 275, 18424–18431. [Google Scholar] [CrossRef] [PubMed]
- Ahadova, A.; Gallon, R.; Gebert, J.; Ballhausen, A.; Endris, V.; Kirchner, M.; Stenzinger, A.; Burn, J.; von Knebel Doeberitz, M.; Bläker, H.; et al. Three Molecular Pathways Model Colorectal Carcinogenesis in Lynch Syndrome. Int. J. Cancer 2018, 143, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Stormorken, A.T.; Clark, N.; Grindedal, E.; Mæhle, L.; Møller, P. Prevention of Colorectal Cancer by Colonoscopic Surveillance in Families with Hereditary Colorectal Cancer. Scand. J. Gastroenterol. 2007, 42, 611–617. [Google Scholar] [CrossRef]
- Møller, P. The Prospective Lynch Syndrome Database Reports Enable Evidence-Based Personal Precision Health Care. Hered. Cancer Clin. Pract. 2020, 18, 6. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Program—NCI. Available online: https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga (accessed on 10 January 2023).
- Salem, M.E.; Bodor, J.N.; Puccini, A.; Xiu, J.; Goldberg, R.M.; Grothey, A.; Korn, W.M.; Shields, A.F.; Worrilow, W.M.; Kim, E.S.; et al. Relationship between MLH1, PMS2, MSH2 and MSH6 Gene-Specific Alterations and Tumor Mutational Burden in 1057 Microsatellite Instability-High Solid Tumors. Int. J. Cancer 2020, 147, 2948–2956. [Google Scholar] [CrossRef]
- Goodman, A.M.; Sokol, E.S.; Frampton, G.M.; Lippman, S.M.; Kurzrock, R. Microsatellite-Stable Tumors with High Mutational Burden Benefit from Immunotherapy. Cancer Immunol. Res. 2019, 7, 1570–1573. [Google Scholar] [CrossRef]
- Fabrizio, D.A.; George, T.J.; Dunne, R.F.; Frampton, G.; Sun, J.; Gowen, K.; Kennedy, M.; Greenbowe, J.; Schrock, A.B.; Hezel, A.F.; et al. Beyond Microsatellite Testing: Assessment of Tumor Mutational Burden Identifies Subsets of Colorectal Cancer Who May Respond to Immune Checkpoint Inhibition. J. Gastrointest. Oncol. 2018, 9, 610–617. [Google Scholar] [CrossRef]
- Lee, S.; Lara, O.; Karpel, H.; Pothuri, B. The Association of Tumor Mutational Burden, Microsatellite Stability, and Mismatch Repair Deficiency in an Endometrial Cancer Patient Cohort (194). Gynecol. Oncol. 2022, 166, S111. [Google Scholar] [CrossRef]
- Oaknin, A.; Gilbert, L.; Tinker, A.V.; Brown, J.; Mathews, C.; Press, J.; Sabatier, R.; O’Malley, D.M.; Samouelian, V.; Boni, V.; et al. Safety and Antitumor Activity of Dostarlimab in Patients with Advanced or Recurrent DNA Mismatch Repair Deficient/Microsatellite Instability-High (DMMR/MSI-H) or Proficient/Stable (MMRp/MSS) Endometrial Cancer: Interim Results from GARNET-a Phase I, Single-Arm Study. J. Immunother. Cancer 2022, 10, e003777. [Google Scholar] [CrossRef]
- Jones, N.L.; Xiu, J.; Rocconi, R.P.; Herzog, T.J.; Winer, I.S. Immune Checkpoint Expression, Microsatellite Instability, and Mutational Burden: Identifying Immune Biomarker Phenotypes in Uterine Cancer. Gynecol. Oncol. 2020, 156, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Ahn, S.; Son, D.S.; Kim, N.K.D.; Lee, K.W.; Kim, S.; Lee, J.; Park, S.H.; Park, J.O.; Kang, W.K.; et al. Bridging Genomics and Phenomics of Gastric Carcinoma. Int. J. Cancer 2019, 145, 2407–2417. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.F.; Ibrahim, A.E.K.; Arends, M.J. Molecular Pathological Classification of Colorectal Cancer. Virchows Arch. 2016, 469, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Dienstmann, R.; Vermeulen, L.; Guinney, J.; Kopetz, S.; Tejpar, S.; Tabernero, J. Consensus Molecular Subtypes and the Evolution of Precision Medicine in Colorectal Cancer. Nat. Rev. Cancer 2017, 17, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Włodarczyk, M.; Włodarczyk, J.; Siwiński, P.; Sobolewska-Włodarczyk, A.; Fichna, J. Genetic Molecular Subtypes in Optimizing Personalized Therapy for Metastatic Colorectal Cancer. Curr. Drug Targets 2018, 19, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.H.; Goel, A.; Chung, D.C. Pathways of Colorectal Carcinogenesis. Gastroenterology 2020, 158, 291–302. [Google Scholar] [CrossRef]
- Salem, M.E.; Xiu, J.; Lenz, H.-J.; Atkins, M.B.; Philip, P.A.; Hwang, J.J.; Gatalica, Z.; Xiao, N.; Gibney, G.T.; El-Deiry, W.S.; et al. Characterization of Tumor Mutation Load (TML) in Solid Tumors. J. Clin. Oncol. 2017, 35, 11517. [Google Scholar] [CrossRef]
- Salem, M.E.; Puccini, A.; Grothey, A.; Raghavan, D.; Goldberg, R.M.; Xiu, J.; Michael Korn, W.; Weinberg, B.A.; Hwang, J.J.; Shields, A.F.; et al. Landscape of Tumor Mutation Load, Mismatch Repair Deficiency, and PD-L1 Expression in a Large Patient Cohort of Gastrointestinal Cancers. Mol. Cancer Res. 2018, 16, 805–812. [Google Scholar] [CrossRef]
- Kumar, S. A Perfect Biomarker for Immune Checkpoint Inhibition—An Elusive Goal? Cancer Res. Stat. Treat. 2021, 4, 594–595. [Google Scholar]
- Grossman, J.E.; Vasudevan, D.; Joyce, C.E.; Hildago, M. Is PD-L1 a Consistent Biomarker for Anti-PD-1 Therapy? The Model of Balstilimab in a Virally-Driven Tumor. Oncogene 2021, 40, 1393–1395. [Google Scholar] [CrossRef]
- Marabelle, A.; Fakih, M.; Lopez, J.; Shah, M.; Shapira-Frommer, R.; Nakagawa, K.; Chung, H.C.; Kindler, H.L.; Lopez-Martin, J.A.; Miller, W.H.; et al. Association of Tumour Mutational Burden with Outcomes in Patients with Advanced Solid Tumours Treated with Pembrolizumab: Prospective Biomarker Analysis of the Multicohort, Open-Label, Phase 2 KEYNOTE-158 Study. Lancet Oncol. 2020, 21, 1353–1365. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K.; et al. Tumor-Associated B7-H1 Promotes T-Cell Apoptosis: A Potential Mechanism of Immune Evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.K.; Vandsemb, E.N.; Herbst, R.S.; Chen, L. Adaptive Immune Resistance at the Tumour Site: Mechanisms and Therapeutic Opportunities. Nat. Rev. Drug Discov. 2022, 21, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.S.; Kähler, K.C.; Hauschild, A. Management of Immune-Related Adverse Events and Kinetics of Response with Ipilimumab. J. Clin. Oncol. 2012, 30, 2691–2697. [Google Scholar] [CrossRef] [PubMed]
- Tivol, E.A.; Borriello, F.; Schweitzer, A.N.; Lynch, W.P.; Bluestone, J.A.; Sharpe, A.H. Loss of CTLA-4 Leads to Massive Lymphoproliferation and Fatal Multiorgan Tissue Destruction, Revealing a Critical Negative Regulatory Role of CTLA-4. Immunity 1995, 3, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 Human Cancer Genomes Reveals the Landscape of Tumor Mutational Burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef]
- Mandal, R.; Samstein, R.M.; Lee, K.W.; Havel, J.J.; Wang, H.; Krishna, C.; Sabio, E.Y.; Makarov, V.; Kuo, F.; Blecua, P.; et al. Genetic Diversity of Tumors with Mismatch Repair Deficiency Influences Anti-PD-1 Immunotherapy Response. Science 2019, 364, 485–491. [Google Scholar] [CrossRef]
- Turajlic, S.; Litchfield, K.; Xu, H.; Rosenthal, R.; McGranahan, N.; Reading, J.L.; Wong, Y.N.S.; Rowan, A.; Kanu, N.; al Bakir, M.; et al. Insertion-and-Deletion-Derived Tumour-Specific Neoantigens and the Immunogenic Phenotype: A Pan-Cancer Analysis. Lancet Oncol. 2017, 18, 1009–1021. [Google Scholar] [CrossRef]
- Sena, L.A.; Fountain, J.; Isaacsson Velho, P.; Lim, S.J.; Wang, H.; Nizialek, E.; Rathi, N.; Nussenzveig, R.; Maughan, B.L.; Velez, M.G.; et al. Tumor Frameshift Mutation Proportion Predicts Response to Immunotherapy in Mismatch Repair-Deficient Prostate Cancer. Oncologist 2021, 26, e270–e278. [Google Scholar] [CrossRef]
- Schiappacasse Cocio, G.V.; Schiappacasse, E.D. Is Adjuvant Chemotherapy Efficient in Colon Cancer with High Microsatellite Instability? A Look Towards the Future. Cancer Res. 2019, 79, 441–444. [Google Scholar] [CrossRef]
- des Guetz, G.; des Guetz, G.; Lecaille, C.; Mariani, P.; Bennamoun, M.; Uzzan, B.; Nicolas, P.; Boisseau, A.; Sastre, X.; Cucherousset, J.; et al. Prognostic Impact of Microsatellite Instability in Colorectal Cancer Patients Treated with Adjuvant FOLFOX. Anticancer Res. 2010, 30, 4297–4302. [Google Scholar] [PubMed]
- Vasen, H.F.A.; Blanco, I.; Aktan-Collan, K.; Gopie, J.P.; Alonso, A.; Aretz, S.; Bernstein, I.; Bertario, L.; Burn, J.; Capella, G.; et al. Revised Guidelines for the Clinical Management of Lynch Syndrome (HNPCC): Recommendations by a Group of European Experts. Gut 2013, 62, 812–823. [Google Scholar] [CrossRef]
- Popat, S.; Hubner, R.; Houlston, R.S. Systematic Review of Microsatellite Instability and Colorectal Cancer Prognosis. J. Clin. Oncol. 2005, 23, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Cohen, R.; Taieb, J.; Fiskum, J.; Yothers, G.; Goldberg, R.; Yoshino, T.; Alberts, S.; Allegra, C.; de Gramont, A.; Seitz, J.F.; et al. Microsatellite Instability in Patients with Stage III Colon Cancer Receiving Fluoropyrimidine with or without Oxaliplatin: An ACCENT Pooled Analysis of 12 Adjuvant Trials. J. Clin. Oncol. 2021, 39, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Sinicrope, F.A.; Chakrabarti, S.; Laurent-Puig, P.; Huebner, L.; Smyrk, T.C.; Tabernero, J.; Mini, E.; Goldberg, R.M.; Zaanan, A.; Folprecht, G.; et al. Prognostic Variables in Low and High Risk Stage III Colon Cancers Treated in Two Adjuvant Chemotherapy Trials. Eur. J. Cancer 2021, 144, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Bertagnolli, M.M.; Niedzwiecki, D.; Compton, C.C.; Hahn, H.P.; Hall, M.; Damas, B.; Jewell, S.D.; Mayer, R.J.; Goldberg, R.M.; Saltz, L.B.; et al. Microsatellite Instability Predicts Improved Response to Adjuvant Therapy with Irinotecan, Fluorouracil, and Leucovorin in Stage III Colon Cancer: Cancer and Leukemia Group B Protocol 89803. J. Clin. Oncol 2009, 27, 1814–1821. [Google Scholar] [CrossRef] [PubMed]
- Fallik, D.; Borrini, F.; Boige, V.; Viguier, J.; Jacob, S.; Miquel, C.; Sabourin, J.-C.; Ducreux, M.; Praz, F. Microsatellite Instability Is a Predictive Factor of the Tumor Response to Irinotecan in Patients with Advanced Colorectal Cancer. Cancer Res. 2003, 63, 5738–5744. [Google Scholar] [PubMed]
- Tajima, A.; Hess, M.T.; Cabrera, B.L.; Kolodner, R.D.; Carethers, J.M. The Mismatch Repair Complex HMutSα Recognizes 5-Fluorouracil-Modified DNA: Implications for Chemosensitivity and Resistance. Gastroenterology 2004, 127, 1678–1684. [Google Scholar] [CrossRef]
- Jover, R.; Zapater, P.; Castells, A.; Llor, X.; Andreu, M.; Cubiella, J.; Piñol, V.; Xicola, R.M.; Bujanda, L.; Reñé, J.M.; et al. Mismatch Repair Status in the Prediction of Benefit from Adjuvant Fluorouracil Chemotherapy in Colorectal Cancer. Gut 2006, 55, 848–855. [Google Scholar] [CrossRef]
- Hutchins, G.; Southward, K.; Handley, K.; Magill, L.; Beaumont, C.; Stahlschmidt, J.; Richman, S.; Chambers, P.; Seymour, M.; Kerr, D.; et al. Value of Mismatch Repair, KRAS, and BRAF Mutations in Predicting Recurrence and Benefits from Chemotherapy in Colorectal Cancer. J. Clin. Oncol. 2011, 29, 1261–1270. [Google Scholar] [CrossRef]
- Klingbiel, D.; Saridaki, Z.; Roth, A.D.; Bosman, F.T.; Delorenzi, M.; Tejpar, S. Prognosis of Stage II and III Colon Cancer Treated with Adjuvant 5-Fluorouracil or FOLFIRI in Relation to Microsatellite Status: Results of the PETACC-3 Trial. Ann. Oncol. 2015, 26, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Ribic, C.M.; Sargent, D.J.; Moore, M.J.; Thibodeau, S.N.; French, A.J.; Goldberg, R.M.; Hamilton, S.R.; Laurent-Puig, P.; Gryfe, R.; Shepherd, L.E.; et al. Tumor Microsatellite-Instability Status as a Predictor of Benefit from Fluorouracil-Based Adjuvant Chemotherapy for Colon Cancer. N. Engl. J. Med. 2003, 349, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Gavin, P.G.; Colangelo, L.H.; Fumagalli, D.; Tanaka, N.; Remillard, M.Y.; Yothers, G.; Kim, C.; Taniyama, Y.; Kim, S.I.; Choi, H.J.; et al. Mutation Profiling and Microsatellite Instability in Stage II and III Colon Cancer: An Assessment of Their Prognostic and Oxaliplatin Predictive Value. Clin. Cancer Res. 2012, 18, 6531–6541. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Hong, Y.S.; Kim, H.J.; Kim, K.P.; Kim, S.Y.; Lim, S.B.; Park, I.J.; Kim, C.W.; Yoon, Y.S.; Yu, C.S.; et al. Microsatellite Instability Was Not Associated with Survival in Stage III Colon Cancer Treated with Adjuvant Chemotherapy of Oxaliplatin and Infusional 5-Fluorouracil and Leucovorin (FOLFOX). Ann. Surg. Oncol. 2017, 24, 1289–1294. [Google Scholar] [CrossRef]
- Jover, R.; Zapater, P.; Castells, A.; Llor, X.; Andreu, M.; Cubiella, J.; Balaguer, F.; Sempere, L.; Xicola, R.M.; Bujanda, L.; et al. The Efficacy of Adjuvant Chemotherapy with 5-Fluorouracil in Colorectal Cancer Depends on the Mismatch Repair Status. Eur. J. Cancer 2009, 45, 365–373. [Google Scholar] [CrossRef]
- Kim, G.P.; Colangelo, L.H.; Wieand, H.S.; Paik, S.; Kirsch, I.R.; Wolmark, N.; Allegra, C.J. Prognostic and Predictive Roles of High-Degree Microsatellite Instability in Colon Cancer: A National Cancer Institute-National Surgical Adjuvant Breast and Bowel Project Collaborative Study. J. Clin. Oncol. 2007, 25, 767–772. [Google Scholar] [CrossRef]
- Sargent, D.J.; Marsoni, S.; Monges, G.; Thibodeau, S.N.; Labianca, R.; Hamilton, S.R.; French, A.J.; Kabat, B.; Foster, N.R.; Torri, V.; et al. Defective Mismatch Repair as a Predictive Marker for Lack of Efficacy of Fluorouracil-Based Adjuvant Therapy in Colon Cancer. J. Clin. Oncol. 2010, 28, 3219–3226. [Google Scholar] [CrossRef]
- Innocenti, F.; Ou, F.S.; Qu, X.; Zemla, T.J.; Niedzwiecki, D.; Tam, R.; Mahajan, S.; Goldberg, R.M.; Bertagnolli, M.M.; Blanke, C.D.; et al. Mutational Analysis of Patients with Colorectal Cancer in CALGB/SWOG 80405 Identifies New Roles of Microsatellite Instability and Tumor Mutational Burden for Patient Outcome. J. Clin. Oncol. 2019, 37, 1217–1227. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration Website. JEMPERLI (Dostarlimab-Gxly). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/761174s002lbl.pdf (accessed on 7 February 2023).
- Andre, T.; Berton, D.; Curigliano, G.; Jimenez-Rodriguez, B.; Ellard, S.; Gravina, A.; Miller, R.; Tinker, A.; Jewell, A.; Pikiel, J.; et al. Efficacy and Safety of Dostarlimab in Patients (Pts) with Mismatch Repair Deficient (DMMR) Solid Tumors: Analysis of 2 Cohorts in the GARNET Study. J. Clin. Oncol. 2022, 40, 2587. [Google Scholar] [CrossRef]
- Lipson, E.J.; Sharfman, W.H.; Drake, C.G.; Wollner, I.; Taube, J.M.; Anders, R.A.; Xu, H.; Yao, S.; Pons, A.; Chen, L.; et al. Durable Cancer Regression Off-Treatment and Effective Reinduction Therapy with an Anti-PD-1 Antibody. Clin. Cancer Res. 2013, 19, 462–468. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Diaz, L.A.; Shiu, K.K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab versus Chemotherapy for Microsatellite Instability-High or Mismatch Repair-Deficient Metastatic Colorectal Cancer (KEYNOTE-177): Final Analysis of a Randomised, Open-Label, Phase 3 Study. Lancet Oncol. 2022, 23, 659–670. [Google Scholar] [CrossRef] [PubMed]
- André, T.; Shiu, K.-K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in Microsatellite-Instability-High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef] [PubMed]
- Casak, S.J.; Marcus, L.; Fashoyin-Aje, L.; Mushti, S.L.; Cheng, J.; Shen, Y.L.; Pierce, W.F.; Her, L.; Goldberg, K.B.; Theoret, M.R.; et al. FDA Approval Summary: Pembrolizumab for the First-Line Treatment of Patients with MSI-H/DMMR Advanced Unresectable or Metastatic Colorectal Carcinoma. Clin. Cancer Res. 2021, 27, 4680–4684. [Google Scholar] [CrossRef] [PubMed]
- Ludford, K.; Ho, W.J.; Thomas, J.V.; Raghav, K.P.S.; Murphy, M.B.; Fleming, N.D.; Lee, M.S.; Smaglo, B.G.; You, Y.N.; Tillman, M.M.; et al. Neoadjuvant Pembrolizumab in Localized Microsatellite Instability High/Deficient Mismatch Repair Solid Tumors. J. Clin. Oncol. 2023, JCO2201351. [Google Scholar] [CrossRef] [PubMed]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable Clinical Benefit with Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in Patients with Metastatic DNA Mismatch Repair-Deficient or Microsatellite Instability-High Colorectal Cancer (CheckMate 142): An Open-Label, Multicentre, Phase 2 Study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- Overman, M.J.; Bergamo, F.; McDermott, R.S.; Aglietta, M.; Chen, F.; Gelsomino, F.; Wong, M.; Morse, M.; van Cutsem, E.; Hendlisz, A.; et al. Nivolumab in Patients with DNA Mismatch Repair-Deficient/Microsatellite Instability-High (DMMR/MSI-H) Metastatic Colorectal Cancer (MCRC): Long-Term Survival According to Prior Line of Treatment from CheckMate-142. J. Clin. Oncol. 2018, 36, 554. [Google Scholar] [CrossRef]
- André, T.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; van Cutsem, E.; McDermott, R.; Hill, A.; et al. Nivolumab plus Low-Dose Ipilimumab in Previously Treated Patients with Microsatellite Instability-High/Mismatch Repair-Deficient Metastatic Colorectal Cancer: 4-Year Follow-up from CheckMate 142. Ann. Oncol. 2022, 33, 1052–1060. [Google Scholar] [CrossRef]
- Cohen, R.; Meurisse, A.; Pudlarz, T.; Bennouna, J.; Tournigand, C.; la Fouchardiere, C.D.; Tougeron, D.; Borg, C.; Mazard, T.; Chibaudel, B.; et al. One-Year Duration of Nivolumab plus Ipilimumab in Patients (Pts) with Microsatellite Instability-High/Mismatch Repair-Deficient (MSI/DMMR) Metastatic Colorectal Cancer (MCRC): Long-Term Follow-up of the GERCOR NIPICOL Phase II Study. J. Clin. Oncol. 2022, 40, 13. [Google Scholar] [CrossRef]
- Chalabi, M.; Fanchi, L.F.; Dijkstra, K.K.; van den Berg, J.G.; Aalbers, A.G.; Sikorska, K.; Lopez-Yurda, M.; Grootscholten, C.; Beets, G.L.; Snaebjornsson, P.; et al. Neoadjuvant Immunotherapy Leads to Pathological Responses in MMR-Proficient and MMR-Deficient Early-Stage Colon Cancers. Nat. Med. 2020, 26, 566. [Google Scholar] [CrossRef] [PubMed]
- Verschoor, Y.L.; van den Berg, J.; Beets, G.; Sikorska, K.; Aalbers, A.; van Lent, A.; Grootscholten, C.; Huibregtse, I.; Marsman, H.; Oosterling, S.; et al. Neoadjuvant Nivolumab, Ipilimumab, and Celecoxib in MMR-Proficient and MMR-Deficient Colon Cancers: Final Clinical Analysis of the NICHE Study. J. Clin. Oncol. 2022, 40, 3511. [Google Scholar] [CrossRef]
- Chalabi, M.; Verschoor, Y.L.; van den Berg, J.; Sikorska, K.; Beets, G.; Lent, A.V.; Grootscholten, M.C.; Aalbers, A.; Buller, N.; Marsman, H.; et al. LBA7 Neoadjuvant Immune Checkpoint Inhibition in Locally Advanced MMR-Deficient Colon Cancer: The NICHE-2 Study. Ann. Oncol. 2022, 33, S1389. [Google Scholar] [CrossRef]
- Andre, T.; Berton, D.; Curigliano, G.; Ellard, S.; Pérez, J.M.T.; Arkenau, H.-T.; Abdeddaim, C.; Moreno, V.; Guo, W.; Im, E.; et al. Safety and Efficacy of Anti–PD-1 Antibody Dostarlimab in Patients (Pts) with Mismatch Repair-Deficient (DMMR) Solid Cancers: Results from GARNET Study. J. Clin. Oncol. 2021, 39, 9. [Google Scholar] [CrossRef]
- Andre, T.; de Braud, F.G.; Jimenez-Rodriguez, B.; Berton, D.; Curigliano, G.; Arkenau, T.; Torres, A.A.; Paez, D.; Ellard, S.; Abdeddaim, C.; et al. Antitumor Activity and Safety of Dostarlimab Monotherapy in Patients with Mismatch Repair Deficient Non-Endometrial Solid Tumors: A Post-Hoc Subgroup Analysis of Patients with Colorectal Cancer. J. Clin. Oncol. 2022, 40, 201. [Google Scholar] [CrossRef]
- Taieb, J.; Bouche, O.; André, T.; Barbier, E.; Laurent-Puig, P.; Bez, J.; Toullec, C.; Borg, C.; Randrian, V.; Evesque, L.; et al. Avelumab versus Standard Second-Line Treatment Chemotherapy in Metastatic Colorectal Cancer (MCRC) Patients with Microsatellite Instability (MSI): The SAMCO-PRODIGE 54 Randomised Phase II Trial. Ann. Oncol. 2022, 22, S808–S869. [Google Scholar] [CrossRef]
- Tintelnot, J.; Ristow, I.; Sauer, M.; Simnica, D.; Schultheiß, C.; Scholz, R.; Goekkurt, E.; von Wenserski, L.; Willscher, E.; Paschold, L.; et al. Translational Analysis and Final Efficacy of the AVETUX Trial—Avelumab, Cetuximab and FOLFOX in Metastatic Colorectal Cancer. Front. Oncol. 2022, 12, 993611. [Google Scholar] [CrossRef]
- Stein, A.; Binder, M.; Goekkurt, E.; Lorenzen, S.; Riera-Knorrenschild, J.; Depenbusch, R.; Ettrich, T.J.; Doerfel, S.; Al-Batran, S.-E.; Karthaus, M.; et al. Avelumab and Cetuximab in Combination with FOLFOX in Patients with Previously Untreated Metastatic Colorectal Cancer (MCRC): Final Results of the Phase II AVETUX Trial (AIO-KRK-0216). J. Clin. Oncol. 2020, 38, 96. [Google Scholar] [CrossRef]
- Segal, N.H.; Wainberg, Z.A.; Overman, M.J.; Ascierto, P.A.; Arkenau, H.-T.; Butler, M.O.; Eder, J.P.; Keilholz, U.; Kim, D.-W.; Cunningham, D.; et al. Safety and Clinical Activity of Durvalumab Monotherapy in Patients with Microsatellite Instability–High (MSI-H) Tumors. J. Clin. Oncol. 2019, 37, 670. [Google Scholar] [CrossRef]
- Oh, C.R.; Kim, J.E.; Hong, Y.S.; Kim, S.Y.; Ahn, J.B.; Baek, J.Y.; Lee, M.A.; Kang, M.J.; Cho, S.H.; Beom, S.H.; et al. Phase II Study of Durvalumab Monotherapy in Patients with Previously Treated Microsatellite Instability-High/Mismatch Repair-Deficient or POLE-Mutated Metastatic or Unresectable Colorectal Cancer. Int. J. Cancer 2022, 150, 2038–2045. [Google Scholar] [CrossRef]
- McGregor, M.; Price, T.J. IMblaze 370: Lessons Learned and Future Strategies in Colorectal Cancer Treatment. Ann. Transl. Med. 2019, 7, 602. [Google Scholar] [CrossRef] [PubMed]
- Friedman, C.F.; Hainsworth, J.D.; Kurzrock, R.; Spigel, D.R.; Burris, H.A.; Sweeney, C.J.; Meric-Bernstam, F.; Wang, Y.; Levy, J.; Grindheim, J.; et al. Atezolizumab Treatment of Tumors with High Tumor Mutational Burden from MyPathway, a Multicenter, Open-Label, Phase IIa Multiple Basket Study. Cancer Discov. 2022, 12, 654–669. [Google Scholar] [CrossRef] [PubMed]
- Bever, K.; Wang, H.; Durham, J.; Apostol, C.; Azad, N.; Browner, I.; Gaillard, S.; Laheru, D.; Lee, V.; Sharfman, W.; et al. 711 Interim Results of a Phase 2 Study of Nivolumab and Relatlimab in Advanced Mismatch Repair Deficient (DMMR) Cancers Resistant to Prior PD-(L)1 Inhibition. J. Immunother. Cancer 2022, 10, A743. [Google Scholar] [CrossRef]
- Hervieu, A.; Guigay, J.; Borcoman, E.; Lavaud, P.; Coquan, E.; Frenel, J.-S.; de La Motte Rouge, T.; Augereau, P.; Cropet, C.; Legrand, F.; et al. 478P Metronomic Oral Vinorelbine (MOV) Combined with Tremelimumab (T) + Durvalumab (D): Results of the Tumor Mutational Burden-High (TMB-h) and/or Microsatellite Instability-High (MSI-h) Cohort of the MOVIE Study. Ann. Oncol. 2022, 33, S758–S759. [Google Scholar] [CrossRef]
- Therkildsen, C.; Jensen, L.H.; Rasmussen, M.; Bernstein, I. An Update on Immune Checkpoint Therapy for the Treatment of Lynch Syndrome. Clin. Exp. Gastroenterol. 2021, 14, 181–197. [Google Scholar] [CrossRef]
- O’Malley, D.M.; Bariani, G.M.; Cassier, P.A.; Marabelle, A.; Hansen, A.R.; de Jesus Acosta, A.; Miller, W.H.; Safra, T.; Italiano, A.; Mileshkin, L.; et al. Pembrolizumab in Patients with Microsatellite Instability–High Advanced Endometrial Cancer: Results from the KEYNOTE-158 Study. J. Clin. Oncol. 2022, 40, 752–761. [Google Scholar] [CrossRef]
- Makker, V.; Colombo, N.; Casado Herráez, A.; Santin, A.D.; Colomba, E.; Miller, D.S.; Fujiwara, K.; Pignata, S.; Baron-Hay, S.; Ray-Coquard, I.; et al. Lenvatinib plus Pembrolizumab for Advanced Endometrial Cancer. New Engl. J. Med. 2022, 386, 437–448. [Google Scholar] [CrossRef]
- Oaknin, A.; Pothuri, B.; Gilbert, L.; Sabatier, R.; Ghamande, S.A.; Gravina, A.; Calvo, E.; Banerjee, S.N.; Miller, R.; Pikiel, J.; et al. Dostarlimab in Advanced/Recurrent (AR) Mismatch Repair Deficient/Microsatellite Instability–High or Proficient/Stable (DMMR/MSI-H or MMRp/MSS) Endometrial Cancer (EC): The GARNET Study. J. Clin. Oncol. 2022, 40, 5509. [Google Scholar] [CrossRef]
- Chao, J.; Fuchs, C.S.; Shitara, K.; Tabernero, J.; Muro, K.; van Cutsem, E.; Bang, Y.J.; de Vita, F.; Landers, G.; Yen, C.J.; et al. Assessment of Pembrolizumab Therapy for the Treatment of Microsatellite Instability-High Gastric or Gastroesophageal Junction Cancer Among Patients in the KEYNOTE-059, KEYNOTE-061, and KEYNOTE-062 Clinical Trials. JAMA Oncol. 2021, 7, 895–902. [Google Scholar] [CrossRef]
- André, T.; Tougeron, D.; Piessen, G.; de La Fouchardière, C.; Louvet, C.; Adenis, A.; Jary, M.; Tournigand, C.; Aparicio, T.; Desrame, J.; et al. Neoadjuvant Nivolumab Plus Ipilimumab and Adjuvant Nivolumab in Localized Deficient Mismatch Repair/Microsatellite Instability-High Gastric or Esophagogastric Junction Adenocarcinoma: The GERCOR NEONIPIGA Phase II Study. J. Clin. Oncol. 2023, 41, 255–265. [Google Scholar] [CrossRef]
- Versluis, J.M.; Long, G.V.; Blank, C.U. Learning from Clinical Trials of Neoadjuvant Checkpoint Blockade. Nat. Med. 2020, 26, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Blake, S.J.; Yong, M.C.R.; Harjunpää, H.; Ngiow, S.F.; Takeda, K.; Young, A.; O’Donnell, J.S.; Allen, S.; Smyth, M.J.; et al. Improved Efficacy of Neoadjuvant Compared to Adjuvant Immunotherapy to Eradicate Metastatic Disease. Cancer Discov. 2016, 6, 1382–1399. [Google Scholar] [CrossRef] [PubMed]
- Blank, C.U.; Rozeman, E.A.; Fanchi, L.F.; Sikorska, K.; van de Wiel, B.; Kvistborg, P.; Krijgsman, O.; van den Braber, M.; Philips, D.; Broeks, A.; et al. Neoadjuvant versus Adjuvant Ipilimumab plus Nivolumab in Macroscopic Stage III Melanoma. Nat. Med. 2018, 24, 1655–1661. [Google Scholar] [CrossRef] [PubMed]
- Versluis, J.M.; Reijers, I.L.M.; Rozeman, E.A.; Sikorska, K.; van Houdt, W.J.; van Thienen, J.V.; Adriaansz, S.A.; Mallo, H.; van Tinteren, H.; van de Wiel, B.A.; et al. 4-Year Relapse-Free Survival (RFS), Overall Survival (OS) and Long-Term Toxicity of (Neo)Adjuvant Ipilimumab (IPI) + Nivolumab (NIVO) in Macroscopic Stage III Melanoma: OpACIN Trial. Ann. Oncol. 2020, 31, S742–S743. [Google Scholar] [CrossRef]
- Patel, S.P.; Othus, M.; Chen, Y.; Wright, G.P.; Yost, K.J.; Hyngstrom, J.R.; Hu-Lieskovan, S.; Lao, C.D.; Fecher, L.A.; Truong, T.-G.; et al. Neoadjuvant-Adjuvant or Adjuvant-Only Pembrolizumab in Advanced Melanoma. N. Engl. J. Med. 2023, 388, 813–823. [Google Scholar] [CrossRef]
- Lau, D.; Kalaitzaki, E.; Church, D.N.; Pandha, H.; Tomlinson, I.; Annels, N.; Gerlinger, M.; Sclafani, F.; Smith, G.; Begum, R.; et al. Rationale and Design of the POLEM Trial: Avelumab plus Fluoropyrimidine-Based Chemotherapy as Adjuvant Treatment for Stage III Mismatch Repair Deficient or POLE Exonuclease Domain Mutant Colon Cancer: A Phase III Randomised Study. ESMO Open 2020, 5, e000638. [Google Scholar] [CrossRef]
- Sinicrope, F.A.; Ou, F.-S.; Zemla, T.; Nixon, A.B.; Mody, K.; Levasseur, A.; Dueck, A.C.; Dhanarajan, A.R.; Lieu, C.H.; Cohen, D.J.; et al. Randomized Trial of Standard Chemotherapy Alone or Combined with Atezolizumab as Adjuvant Therapy for Patients with Stage III Colon Cancer and Deficient Mismatch Repair (ATOMIC, Alliance A021502). J. Clin. Oncol. 2019, 37, e15169. [Google Scholar] [CrossRef]
- ClinicalTrials.Gov. Avelumab Plus 5-FU Based Chemotherapy as Adjuvant Treatment for Stage 3 MSI-High or POLE Mutant Colon Cancer—Full Text View. Available online: https://www.clinicaltrials.gov/ct2/show/NCT03827044 (accessed on 3 April 2023).
- Le, D.T.; Kim, T.W.; van Cutsem, E.; Geva, R.; Jäger, D.; Hara, H.; Burge, M.; O’Neil, B.; Kavan, P.; Yoshino, T.; et al. Phase II Open-Label Study of Pembrolizumab in Treatment-Refractory, Microsatellite Instability-High/Mismatch Repair-Deficient Metastatic Colorectal Cancer: KEYNOTE-164. J. Clin. Oncol. 2020, 38, 11–19. [Google Scholar] [CrossRef]
- Cohen, R.; Bennouna, J.; Meurisse, A.; Tournigand, C.; de La Fouchardière, C.; Tougeron, D.; Borg, C.; Mazard, T.; Chibaudel, B.; Garcia-Larnicol, M.L.; et al. RECIST and IRECIST Criteria for the Evaluation of Nivolumab plus Ipilimumab in Patients with Microsatellite Instability-High/Mismatch Repair-Deficient Metastatic Colorectal Cancer: The GERCOR NIPICOL Phase II Study. J. Immunother. Cancer 2020, 8, e001499. [Google Scholar] [CrossRef]
- Sahin, I.H.; Goyal, S.; Pumpalova, Y.; Sonbol, M.B.; Das, S.; Haraldsdottir, S.; Ahn, D.; Ciombor, K.K.; Chen, Z.; Draper, A.; et al. Mismatch Repair (MMR) Gene Alteration and BRAF V600E Mutation Are Potential Predictive Biomarkers of Immune Checkpoint Inhibitors in MMR-Deficient Colorectal Cancer. Oncologist 2021, 26, 668. [Google Scholar] [CrossRef]
- Park, R.; da Silva, L.L.; Lee, S.; Saeed, A. Impact of BRAF Mutations on Prognosis and Immunotherapy Response in Microsatellite Instability/Mismatch Repair Deficient Metastatic Colorectal Cancer: A Systematic Review and Meta-Analysis. J. Clin. Oncol. 2021, 39, 3557. [Google Scholar] [CrossRef]
- Schrock, A.B.; Ouyang, C.; Sandhu, J.; Sokol, E.; Jin, D.; Ross, J.S.; Miller, V.A.; Lim, D.; Amanam, I.; Chao, J.; et al. Tumor Mutational Burden Is Predictive of Response to Immune Checkpoint Inhibitors in MSI-High Metastatic Colorectal Cancer. Ann. Oncol. 2019, 30, 1096–1103. [Google Scholar] [CrossRef] [PubMed]
- Chida, K.; Kawazoe, A.; Kawazu, M.; Suzuki, T.; Nakamura, Y.; Nakatsura, T.; Kuwata, T.; Ueno, T.; Kuboki, Y.; Kotani, D.; et al. A Low Tumor Mutational Burden and PTEN Mutations Are Predictors of a Negative Response to PD-1 Blockade in MSI-H/DMMR Gastrointestinal Tumors. Clin. Cancer Res. 2021, 27, 3714–3724. [Google Scholar] [CrossRef]
- Loupakis, F.; Depetris, I.; Biason, P.; Intini, R.; Prete, A.A.; Leone, F.; Lombardi, P.; Filippi, R.; Spallanzani, A.; Cascinu, S.; et al. Prediction of Benefit from Checkpoint Inhibitors in Mismatch Repair Deficient Metastatic Colorectal Cancer: Role of Tumor Infiltrating Lymphocytes. Oncologist 2020, 25, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Li, W.; Huang, Y.; Huang, M.; Li, S.; Zhai, X.; Zhao, J.; Gao, C.; Xie, W.; Qin, H.; et al. A Next-Generation Sequencing-Based Strategy Combining Microsatellite Instability and Tumor Mutation Burden for Comprehensive Molecular Diagnosis of Advanced Colorectal Cancer. BMC Cancer 2021, 21, 282. [Google Scholar] [CrossRef]
- Lu, C.; Guan, J.; Lu, S.; Jin, Q.; Rousseau, B.; Lu, T.; Stephens, D.; Zhang, H.; Zhu, J.; Yang, M.; et al. DNA Sensing in Mismatch Repair-Deficient Tumor Cells Is Essential for Anti-Tumor Immunity. Cancer Cell 2021, 39, 96–108. [Google Scholar] [CrossRef] [PubMed]
- Smyrk, T.C.; Watson, P.; Kaul, K.; Lynch, H.T. Tumor-Infiltrating Lymphocytes Are a Marker for Microsatellite Instability in Colorectal Carcinoma. Cancer 2001, 91, 2417–2422. [Google Scholar] [CrossRef]
- Bruni, D.; Angell, H.K.; Galon, J. The Immune Contexture and Immunoscore in Cancer Prognosis and Therapeutic Efficacy. Nat. Rev. Cancer 2020, 20, 662–680. [Google Scholar] [CrossRef]
- Noepel-Duennebacke, S.; Juette, H.; Schulmann, K.; Graeven, U.; Porschen, R.; Stoehlmacher, J.; Hegewisch-Becker, S.; Raulf, A.; Arnold, D.; Reinacher-Schick, A.; et al. Microsatellite Instability (MSI-H) Is Associated with a High Immunoscore but Not with PD-L1 Expression or Increased Survival in Patients (Pts.) with Metastatic Colorectal Cancer (MCRC) Treated with Oxaliplatin (Ox) and Fluoropyrimidine (FP) with and without Bevacizumab (Bev): A Pooled Analysis of the AIO KRK 0207 and RO91 Trials. J. Cancer Res. Clin. Oncol. 2021, 147, 3063–3072. [Google Scholar] [CrossRef]
- Sui, Q.; Zhang, X.; Chen, C.; Tang, J.; Yu, J.; Li, W.; Han, K.; Jiang, W.; Liao, L.; Kong, L.; et al. Inflammation Promotes Resistance to Immune Checkpoint Inhibitors in High Microsatellite Instability Colorectal Cancer. Nat. Commun. 2022, 13, 7316. [Google Scholar] [CrossRef]
- Vétizou, M.; Pitt, J.M.; Daillère, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.M.; et al. Anticancer Immunotherapy by CTLA-4 Blockade Relies on the Gut Microbiota. Science 2015, 350, 1079–1084. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut Microbiome Modulates Response to Anti-PD-1 Immunotherapy in Melanoma Patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Wu, J.; Shi, L.; Zhou, B.; Shang, F.; Chang, X.; Dong, X.; Deng, S.; Liu, L.; Cai, K.; et al. Gut Microbiota Distinct between Colorectal Cancers with Deficient and Proficient Mismatch Repair: A Study of 230 CRC Patients. Front. Microbiol. 2022, 13, 993285. [Google Scholar] [CrossRef] [PubMed]
- Serpas Higbie, V.; Rogers, J.; Hwang, H.; Qiao, W.; Xiao, L.; Dasari, A.; Mola-Rudd, K.; Morris, V.K.; Wolff, R.A.; Raghav, K.; et al. Antibiotic Exposure Does Not Impact Immune Checkpoint Blockade Response in MSI-H/DMMR Metastatic Colorectal Cancer: A Single-Center Experience. Oncologist 2022, 27, 952–957. [Google Scholar] [CrossRef] [PubMed]
- Desai, R.; Coxon, A.T.; Dunn, G.P. Therapeutic Applications of the Cancer Immunoediting Hypothesis. Semin. Cancer Biol. 2022, 78, 63–77. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Checkpoint | Drug | Indications for MSI and/or dMMR Tumors |
|---|---|---|
| CTLA4 | Ipilimumab (in combination with nivolumab) | MSI/dMMR colorectal cancer |
| Tremelimumab | Indications do not include MSI/dMMR tumors | |
| PD1 | Nivolumab | MSI/dMMR colorectal cancer |
| Pembrolizumab | All types of MSI/dMMR tumors (site-agnostic) MSI/dMMR colorectal cancer MSI/dMMR endometrial cancer | |
| Cemiplimab | Indications do not include MSI/dMMR tumors | |
| Dostarlimab | All types of dMMR tumors (site agnostic) dMMR endometrial cancer | |
| PD-L1 | Atezolizumab | Indications do not include MSI/dMMR tumors |
| Avelumab | Indications do not include MSI/dMMR tumors | |
| Durvalumab | Indications do not include MSI/dMMR tumors | |
| LAG3 | Relatlimab | Indications do not include MSI/dMMR tumors |
| Study | Total Number of Patients | Drug | ORR or pCR (for Neoadjuvant Settings) | Lynch Syndrome Accounted | BRAF/RAS Status Evaluated | PD-L1 Expression Status Evaluated by IHC |
|---|---|---|---|---|---|---|
| Tissue/Site-Agnostic Treatment in Pretreated Metastatic Patients | ||||||
| KEYNOTE-016, KEYNOTE-164, KEYNOTE-012, KEYNOTE-028, KEYNOTE-158 [39] | 149 patients: 89 CRC 14 EC 45 other | Pembrolizumab | 40% | No | Yes | Yes |
| KEYNOTE-158 [40] | 233 non-CRC | Pembrolizumab | 34% | No | No | Yes |
| GARNET [193] (including POLE mutant) | 209 non-EC | Dostarlimab | 43% | No | No | Yes |
| Colorectal cancer | ||||||
| Late lines | ||||||
| KEYNOTE-016 [105] | 40 | Pembrolizumab | 52% | Yes | No | Yes |
| CheckMate-142 [200] | 119 | Nivolumab plus Ipilimumab | 55% | Yes | Yes | Yes |
| NCT01693562 [213] | 36 | Durvalumab | 22% | No information | No information | No information |
| NCT03435107 [214] | 33 | Durvalumab | 42% | No | Yes | No |
| SAMCO-PRODIGE [210] | 61 | Avelumab | 28% | No | Yes | Yes |
| First line | ||||||
| KEYNOTE-177 [196] | 153 | Pembrolizumab | 45% | No | Yes | No |
| CheckMate-142 [41] | 45 | Nivolumab plus Ipilimumab | 69% | No | Yes | Yes |
| Neoadjuvant settings | ||||||
| NICHE [205] | 20 | Nivolumab plus Ipilimumab | 60% | Yes | Yes | Yes |
| NICHE-2 [207] | 107 | Nivolumab plus Ipilimumab | 67% | No information | No information | No information |
| NCT04165772 [42] | 12 | Nivolumab plus Ipilimumab | 100% | Yes | Yes | Yes |
| Endometrial cancer | ||||||
| KEYNOTE-158 [220] | 79 | Pembrolizumab | 48% | No | No | Yes |
| GARNET [153,222] | 143 | Dostarlimab | 46% | No | No | Yes |
| Gastric cancer | ||||||
| KEYNOTE-059, KEYNOTE-061, KEYNOTE-062 [223] | 7 27 50 | Pembrolizumab | 57% 47% 57% | No | No | Yes |
| KEYNOTE-158 [44] | 42 | Pembrolizumab | 31% | No | No | Yes |
| Small intestine cancer | ||||||
| KEYNOTE-158 [44] | 25 | Pembrolizumab | 48% | No | No | Yes |
| Ovarian cancer | ||||||
| KEYNOTE-158 [44] | 24 | Pembrolizumab | 33% | No | No | Yes |
| Cholangiocarcinoma/biliary tract cancer | ||||||
| KEYNOTE-158 [44] | 22 | Pembrolizumab | 41% | No | No | Yes |
| Pancreatic cancer | ||||||
| KEYNOTE-158 [44] | 22 | Pembrolizumab | 18% | No | No | Yes |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kavun, A.; Veselovsky, E.; Lebedeva, A.; Belova, E.; Kuznetsova, O.; Yakushina, V.; Grigoreva, T.; Mileyko, V.; Fedyanin, M.; Ivanov, M. Microsatellite Instability: A Review of Molecular Epidemiology and Implications for Immune Checkpoint Inhibitor Therapy. Cancers 2023, 15, 2288. https://doi.org/10.3390/cancers15082288
Kavun A, Veselovsky E, Lebedeva A, Belova E, Kuznetsova O, Yakushina V, Grigoreva T, Mileyko V, Fedyanin M, Ivanov M. Microsatellite Instability: A Review of Molecular Epidemiology and Implications for Immune Checkpoint Inhibitor Therapy. Cancers. 2023; 15(8):2288. https://doi.org/10.3390/cancers15082288
Chicago/Turabian StyleKavun, Alexandra, Egor Veselovsky, Alexandra Lebedeva, Ekaterina Belova, Olesya Kuznetsova, Valentina Yakushina, Tatiana Grigoreva, Vladislav Mileyko, Mikhail Fedyanin, and Maxim Ivanov. 2023. "Microsatellite Instability: A Review of Molecular Epidemiology and Implications for Immune Checkpoint Inhibitor Therapy" Cancers 15, no. 8: 2288. https://doi.org/10.3390/cancers15082288
APA StyleKavun, A., Veselovsky, E., Lebedeva, A., Belova, E., Kuznetsova, O., Yakushina, V., Grigoreva, T., Mileyko, V., Fedyanin, M., & Ivanov, M. (2023). Microsatellite Instability: A Review of Molecular Epidemiology and Implications for Immune Checkpoint Inhibitor Therapy. Cancers, 15(8), 2288. https://doi.org/10.3390/cancers15082288