
FAK Inhibitor-Based Combinations with MEK or PKC Inhibitors Trigger Synergistic Antitumor Effects in Uveal Melanoma
 , ,
, ,  , ,
, ,  ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Compounds
2.3. Drug Combination Cell Viability Screen
2.4. Caspase-3/7 Activity Assay
2.5. Immunoblotting Analyses
2.6. UM PDX Models
2.7. In Vivo Tumor Growth and Antitumor Efficacy
3. Results
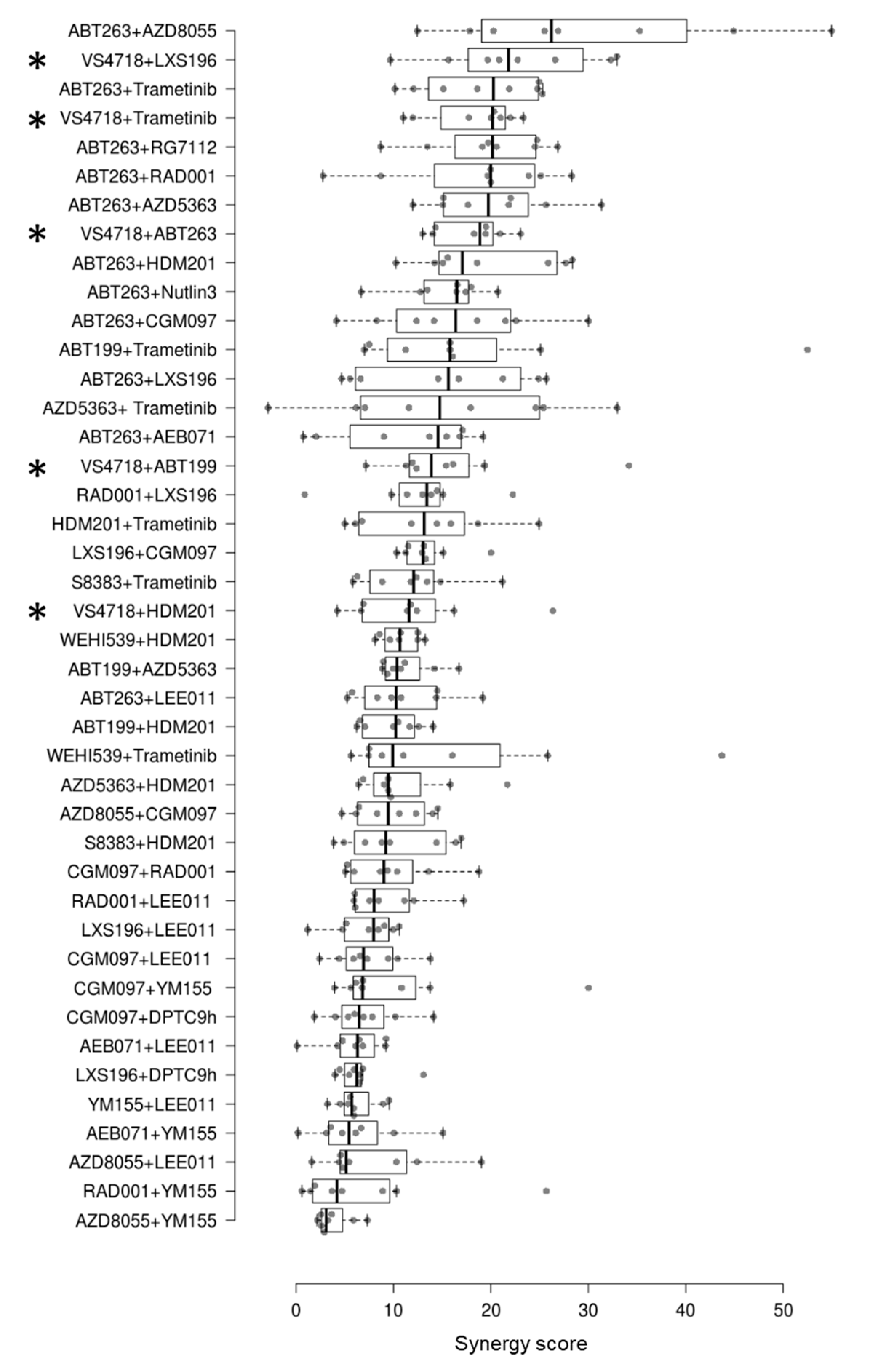
3.1. Identification of Synergistic FAK Inhibitor-Based Combinations in Uveal Melanoma Cell Lines
3.2. Apoptosis Induction after Treatment with VS4718 Combined with LXS196 or with Trametinib
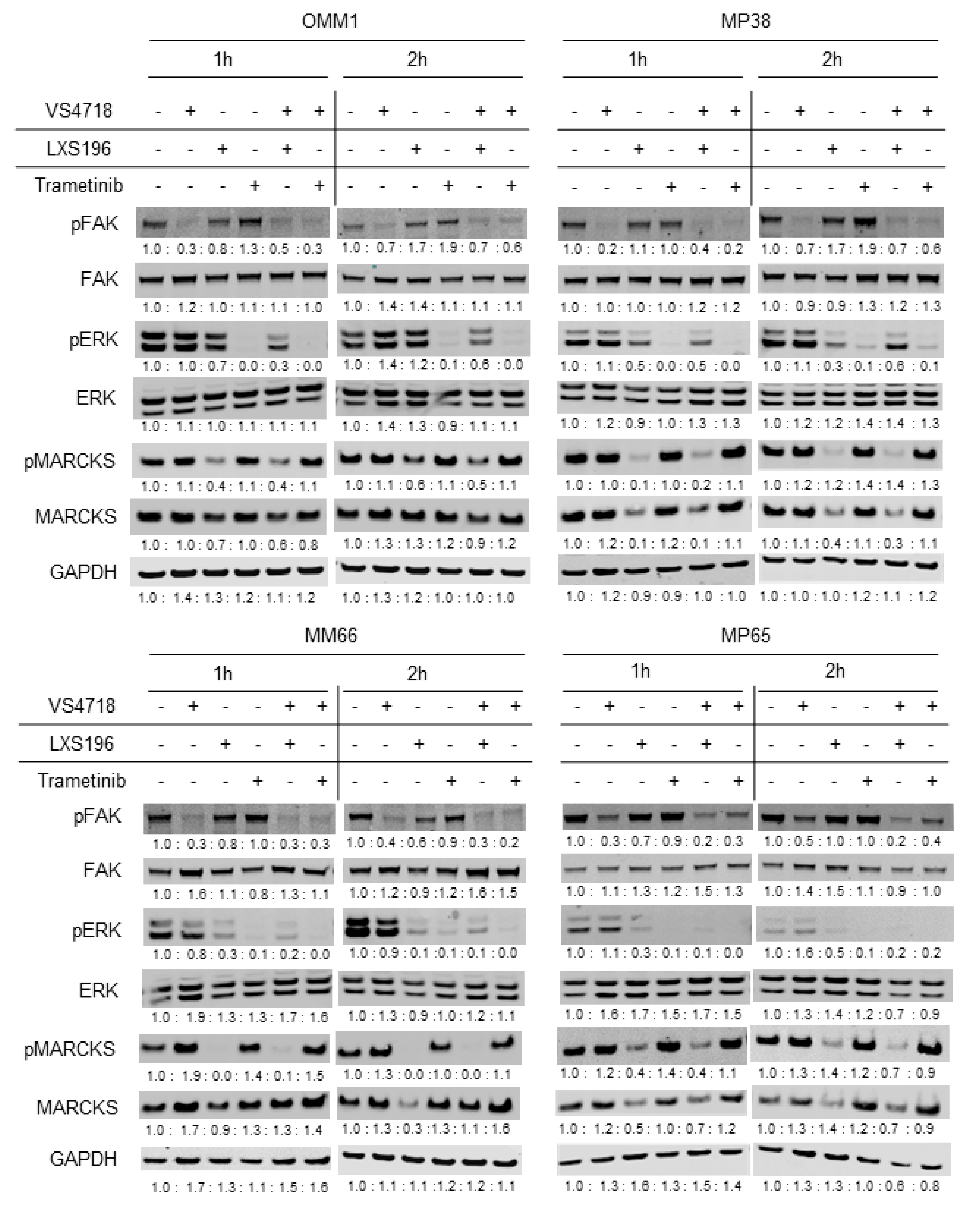
3.3. Validation of Target Engagement under Treatment with FAK Inhibitor Drug Combinations
3.4. In Vivo Evaluation of FAK Inhibitor-Based Combinations in UM PDXs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mahendraraj, K.; Lau, C.S.M.; Lee, I.; Chamberlain, R.S. Trends in incidence, survival, and management of uveal melanoma: A population-based study of 7516 patients from the Surveillance, Epidemiology, and End Results database (1973–2012). Clin. Ophthalmol. 2016, 10, 2113–2119. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Bergman, L.; Seregard, S. Uveal Melanoma: Epidemiologic Aspects. Ophthalmol. Clin. N. Am. 2005, 18, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Durairaj, P.; Yeung, J. Uveal melanoma: A review of the literature. Oncol. Ther. 2018, 6, 87–104. [Google Scholar] [CrossRef] [PubMed]
- Vivet-Noguer, R.; Tarin, M.; Roman-Roman, S.; Alsafadi, S. Emerging Therapeutic Opportunities Based on Current Knowledge of Uveal Melanoma Biology. Cancers 2019, 11, 1019. [Google Scholar] [CrossRef] [PubMed]
- Hoiom, V.; Helgadottir, H. The genetics of uveal melanoma: Current insights. Appl. Clin. Genet. 2016, 9, 147–155. [Google Scholar] [CrossRef]
- Van Raamsdonk, C.D.; Bezrookove, V.; Green, G.; Bauer, J.; Gaugler, L.; O’Brien, J.M.; Simpson, E.M.; Barsh, G.S.; Bastian, B.C. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature 2009, 457, 599–602. [Google Scholar] [CrossRef]
- Robertson, A.G.; Shih, J.; Yau, C.; Gibb, E.A.; Oba, J.; Mungall, K.L.; Hess, J.M.; Uzunangelov, V.; Walter, V.; Danilova, L.; et al. Integrative Analysis Identifies Four Molecular and Clinical Subsets in Uveal Melanoma. Cancer Cell 2017, 32, 204–220.e215. [Google Scholar] [CrossRef]
- Van Raamsdonk, C.D.; Griewank, K.G.; Crosby, M.B.; Garrido, M.C.; Vemula, S.; Wiesner, T.; Obenauf, A.C.; Wackernagel, W.; Green, G.; Bouvier, N.; et al. Mutations in GNA11 in Uveal Melanoma. N. Engl. J. Med. 2010, 363, 2191–2199. [Google Scholar] [CrossRef]
- Moore, A.R.; Ceraudo, E.; Sher, J.J.; Guan, Y.; Shoushtari, A.N.; Chang, M.T.; Zhang, J.Q.; Walczak, E.G.; Kazmi, M.A.; Taylor, B.S.; et al. Recurrent activating mutations of G-protein-coupled receptor CYSLTR2 in uveal melanoma. Nat. Genet. 2016, 48, 675–680. [Google Scholar] [CrossRef]
- Johansson, P.; Aoude, L.G.; Wadt, K.; Glasson, W.J.; Warrier, S.K.; Hewitt, A.W.; Kiilgaard, J.F.; Heegaard, S.; Isaacs, T.; Franchina, M.; et al. Deep sequencing of uveal melanoma identifies a recurrent mutation in PLCB4. Oncotarget 2016, 7, 4624–4631. [Google Scholar] [CrossRef]
- Shain, A.H.; Bagger, M.M.; Yu, R.; Chang, D.; Liu, S.; Vemula, S.; Weier, J.F.; Wadt, K.; Heegaard, S.; Bastian, B.C.; et al. The genetic evolution of metastatic uveal melanoma. Nat. Genet. 2019, 51, 1123–1130. [Google Scholar] [CrossRef]
- Harbour, J.W.; Onken, M.D.; Roberson, E.D.O.; Duan, S.; Cao, L.; Worley, L.A.; Council, M.L.; Matatall, K.A.; Helms, C.; Bowcock, A.M. Frequent Mutation of BAP1 in Metastasizing Uveal Melanomas. Science 2010, 330, 1410–1413. [Google Scholar] [CrossRef]
- Harbour, J.W.; Roberson, E.D.O.; Anbunathan, H.; Onken, M.D.; Worley, L.A.; Bowcock, A.M. Recurrent mutations at codon 625 of the splicing factor SF3B1 in uveal melanoma. Nat. Genet. 2013, 45, 133–135. [Google Scholar] [CrossRef]
- Furney, S.J.; Pedersen, M.; Gentien, D.; Dumont, A.G.; Rapinat, A.; Desjardins, L.; Turajlic, S.; Piperno-Neumann, S.; de la Grange, P.; Roman-Roman, S.; et al. SF3B1 Mutations Are Associated with Alternative Splicing in Uveal Melanoma. Cancer Discov. 2013, 3, 1122–1129. [Google Scholar] [CrossRef]
- Martin, M.; Maßhöfer, L.; Temming, P.; Rahmann, S.; Metz, C.; Bornfeld, N.; Van De Nes, J.; Klein-Hitpass, L.; Hinnebusch, A.G.; Horsthemke, B.; et al. Exome sequencing identifies recurrent somatic mutations in EIF1AX and SF3B1 in uveal melanoma with disomy 3. Nat. Genet. 2013, 45, 933–936. [Google Scholar] [CrossRef]
- Feng, X.; Degese, M.S.; Iglesias-Bartolome, R.; Vaque, J.P.; Molinolo, A.A.; Rodrigues, M.; Zaidi, M.R.; Ksander, B.R.; Merlino, G.; Sodhi, A.; et al. Hippo-Independent Activation of YAP by the GNAQ Uveal Melanoma Oncogene through a Trio-Regulated Rho GTPase Signaling Circuitry. Cancer Cell 2014, 25, 831–845. [Google Scholar] [CrossRef]
- Paradis, J.S.; Acosta, M.; Saddawi-Konefka, R.; Kishore, A.; Lubrano, S.; Gomes, F.G.; Arang, N.; Tiago, M.; Coma, S.; Wu, X.; et al. Synthetic Lethal Screens Reveal Cotargeting FAK and MEK as a Multimodal Precision Therapy for GNAQ-Driven Uveal Melanoma. Clin. Cancer Res. 2021, 27, 3190–3200. [Google Scholar] [CrossRef]
- Feng, X.; Arang, N.; Rigiracciolo, D.C.; Lee, J.S.; Yeerna, H.; Wang, Z.; Lubrano, S.; Kishore, A.; Pachter, J.A.; König, G.M.; et al. A Platform of Synthetic Lethal Gene Interaction Networks Reveals that the GNAQ Uveal Melanoma Oncogene Controls the Hippo Pathway through FAK. Cancer Cell 2019, 35, 457–472.e5. [Google Scholar] [CrossRef]
- Carvajal, R.D.; Sosman, J.A.; Quevedo, J.F.; Milhem, M.M.; Joshua, A.M.; Kudchadkar, R.R.; Linette, G.P.; Gajewski, T.F.; Lutzky, J.; Lawson, D.H.; et al. Effect of Selumetinib vs Chemotherapy on Progression-Free Survival in Uveal Melanoma: A randomized clinical trial. JAMA 2014, 311, 2397–2405. [Google Scholar] [CrossRef]
- Carvajal, R.D.; Piperno-Neumann, S.; Kapiteijn, E.; Chapman, P.B.; Frank, S.; Joshua, A.M.; Piulats, J.M.; Wolter, P.; Cocquyt, V.; Chmielowski, B.; et al. Selumetinib in Combination With Dacarbazine in Patients With Metastatic Uveal Melanoma: A Phase III, Multicenter, Randomized Trial (SUMIT). J. Clin. Oncol. 2018, 36, 1232–1239. [Google Scholar] [CrossRef]
- Piperno-Neumann, S.; Larkin, J.; Carvajal, R.D.; Luke, J.J.; Schwartz, G.K.; Hodi, F.S.; Sablin, M.-P.; Shoushtari, A.N.; Szpakowski, S.; Chowdhury, N.R.; et al. Genomic Profiling of Metastatic Uveal Melanoma and Clinical Results of a Phase I Study of the Protein Kinase C Inhibitor AEB071. Mol. Cancer Ther. 2020, 19, 1031–1039. [Google Scholar] [CrossRef] [PubMed]
- Piperno-Neumann, S.; Carlino, M.S.; Boni, V.; Loirat, D.; Speetjens, F.M.; Park, J.J.; Calvo, E.; Carvajal, R.D.; Nyakas, M.; Gonzalez-Maffe, J.; et al. A phase I trial of LXS196, a protein kinase C (PKC) inhibitor, for metastatic uveal melanoma. Br. J. Cancer 2023, 128, 1040–1051. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, R.D.; Sacco, J.J.; Jager, M.J.; Eschelman, D.J.; Bagge, R.O.; Harbour, J.W.; Chieng, N.D.; Patel, S.P.; Joshua, A.M.; Piperno-Neumann, S. Advances in the clinical management of uveal melanoma. Nat. Rev. Clin. Oncol. 2023, 20, 99–115. [Google Scholar] [CrossRef]
- Nathan, P.; Hassel, J.C.; Rutkowski, P.; Baurain, J.-F.; Butler, M.O.; Schlaak, M.; Sullivan, R.J.; Ochsenreither, S.; Dummer, R.; Kirkwood, J.M.; et al. Overall Survival Benefit with Tebentafusp in Metastatic Uveal Melanoma. N. Engl. J. Med. 2021, 385, 1196–1206. [Google Scholar] [CrossRef] [PubMed]
- Seedor, R.S.; Orloff, M.; Gutkind, J.S.; Aplin, A.E.; Terai, M.; Sharpe-Mills, E.; Klose, H.; Mastrangelo, M.J.; Sato, T. Clinical trial in progress: Phase II trial of defactinib (VS-6063) combined with VS-6766 (CH5126766) in patients with metastatic uveal melanoma. J. Clin. Oncol. 2021, 39 (Suppl. S15), TPS9588. [Google Scholar] [CrossRef]
- Shinde, R.; Terbuch, A.; Little, M.; Caldwell, R.; Kurup, R.; Riisnaes, R.; Crespo, M.; Ruddle, R.; Gurel, B.; Stewart, A.; et al. Abstract CT143: Phase I study of the combination of a RAF-MEK inhibitor CH5126766 and FAK inhibitor defactinib in an intermittent dosing schedule with expansions in KRAS mutant cancers. Cancer Res 2020, 80 (Suppl. S15), CT143. [Google Scholar] [CrossRef]
- Decaudin, D.; Leitz, E.F.D.; Nemati, F.; Tarin, M.; Naguez, A.; Zerara, M.; Marande, B.; Vivet-Noguer, R.; Halilovic, E.; Fabre, C.; et al. Preclinical evaluation of drug combinations identifies co-inhibition of Bcl-2/XL/W and MDM2 as a potential therapy in uveal melanoma. Eur. J. Cancer 2020, 126, 93–103. [Google Scholar] [CrossRef]
- Amirouchene-Angelozzi, N.; Némati, F.; Gentien, D.; Nicolas, A.; Dumont, A.; Carita, G.; Camonis, J.; Desjardins, L.; Cassoux, N.; Piperno-Neumann, S.; et al. Establishment of novel cell lines recapitulating the genetic landscape of uveal melanoma and preclinical validation of mTOR as a therapeutic target. Mol. Oncol. 2014, 8, 1508–1520. [Google Scholar] [CrossRef]
- Amirouchene-Angelozzi, N.; Frisch-Dit-Leitz, E.; Carita, G.; Dahmani, A.; Raymondie, C.; Liot, G.; Gentien, D.; Némati, F.; Decaudin, D.; Roman-Roman, S.; et al. The mTOR inhibitor Everolimus synergizes with the PI3K inhibitor GDC0941 to enhance anti-tumor efficacy in uveal melanoma. Oncotarget 2016, 7, 23633–23646. [Google Scholar] [CrossRef]
- Némati, F.; Sastre-Garau, X.; Laurent, C.; Couturier, J.; Mariani, P.; Desjardins, L.; Piperno-Neumann, S.; Lantz, O.; Asselain, B.; Plancher, C.; et al. Establishment and Characterization of a Panel of Human Uveal Melanoma Xenografts Derived from Primary and/or Metastatic Tumors. Clin. Cancer Res. 2010, 16, 2352–2362. [Google Scholar] [CrossRef]
- Decaudin, D.; El Botty, R.; Diallo, B.; Massonnet, G.; Fleury, J.; Naguez, A.; Raymondie, C.; Davies, E.; Smith, A.; Wilson, J.; et al. Selumetinib-based therapy in uveal melanoma patient-derived xenografts. Oncotarget 2018, 9, 21674–21686. [Google Scholar] [CrossRef]
- Gao, H.; Korn, J.M.; Ferretti, S.; Monahan, J.E.; Wang, Y.; Singh, M.; Zhang, C.; Schnell, C.; Yang, G.; Zhang, Y.; et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat. Med. 2015, 21, 1318–1325. [Google Scholar] [CrossRef]
- de Koning, L.; Decaudin, D.; El Botty, R.; Nicolas, A.; Carita, G.; Schuller, M.; Ouine, B.; Cartier, A.; Naguez, A.; Fleury, J.; et al. PARP Inhibition Increases the Response to Chemotherapy in Uveal Melanoma. Cancers 2019, 11, 751. [Google Scholar] [CrossRef]
- Marty, B.; Maire, V.; Gravier, E.; Rigaill, G.; Vincent-Salomon, A.; Kappler, M.; Lebigot, I.; Djelti, F.; Tourdès, A.; Gestraud, P.; et al. Frequent PTEN genomic alterations and activated phosphatidylinositol 3-kinase pathway in basal-like breast cancer cells. Breast Cancer Res. 2008, 10, R101. [Google Scholar] [CrossRef]
- Ma, J.; Weng, L.; Bastian, B.C.; Chen, X. Functional characterization of uveal melanoma oncogenes. Oncogene 2021, 40, 806–820. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tarin, M.; Némati, F.; Decaudin, D.; Canbezdi, C.; Marande, B.; Silva, L.; Derrien, H.; Jochemsen, A.G.; Gardrat, S.; Piperno-Neumann, S.; et al. FAK Inhibitor-Based Combinations with MEK or PKC Inhibitors Trigger Synergistic Antitumor Effects in Uveal Melanoma. Cancers 2023, 15, 2280. https://doi.org/10.3390/cancers15082280
Tarin M, Némati F, Decaudin D, Canbezdi C, Marande B, Silva L, Derrien H, Jochemsen AG, Gardrat S, Piperno-Neumann S, et al. FAK Inhibitor-Based Combinations with MEK or PKC Inhibitors Trigger Synergistic Antitumor Effects in Uveal Melanoma. Cancers. 2023; 15(8):2280. https://doi.org/10.3390/cancers15082280
Chicago/Turabian StyleTarin, Malcy, Fariba Némati, Didier Decaudin, Christine Canbezdi, Benjamin Marande, Lisseth Silva, Héloïse Derrien, Aart G. Jochemsen, Sophie Gardrat, Sophie Piperno-Neumann, and et al. 2023. "FAK Inhibitor-Based Combinations with MEK or PKC Inhibitors Trigger Synergistic Antitumor Effects in Uveal Melanoma" Cancers 15, no. 8: 2280. https://doi.org/10.3390/cancers15082280
APA StyleTarin, M., Némati, F., Decaudin, D., Canbezdi, C., Marande, B., Silva, L., Derrien, H., Jochemsen, A. G., Gardrat, S., Piperno-Neumann, S., Rodrigues, M., Mariani, P., Cassoux, N., Stern, M.-H., Roman-Roman, S., & Alsafadi, S. (2023). FAK Inhibitor-Based Combinations with MEK or PKC Inhibitors Trigger Synergistic Antitumor Effects in Uveal Melanoma. Cancers, 15(8), 2280. https://doi.org/10.3390/cancers15082280