

Cerivastatin Synergizes with Trametinib and Enhances Its Efficacy in the Therapy of Uveal Melanoma

 , , , , ,
, , , , ,  and
and
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Method
2.1. Cancer Cell Lines
2.2. Reagents
2.3. Drug Screening Assay
2.4. Cell Cycle Analysis
2.5. Annexin V-FITC/PI Apoptosis Analysis
2.6. Western Blotting Analysis
2.7. Xenograft Mouse Model
2.8. Microarray
2.9. Statistical Analysis
3. Results
3.1. Anti-Proliferative Activity of MEK and YAP/TAZ Inhibitors in UM Cell Lines
3.2. In Vitro Anti-Proliferative Activity of Trametinib-Based Combinations, in UM Cell Lines
3.3. The Combination of Trametinib and Cerivastatin Induces Apoptosis and Cell Cycle Arrest in UM Cell Lines
3.4. Trametinib and Cerivastatin Synergistic Concentrations Increase Apoptosis and Decrease Survival/Proliferation Pathways
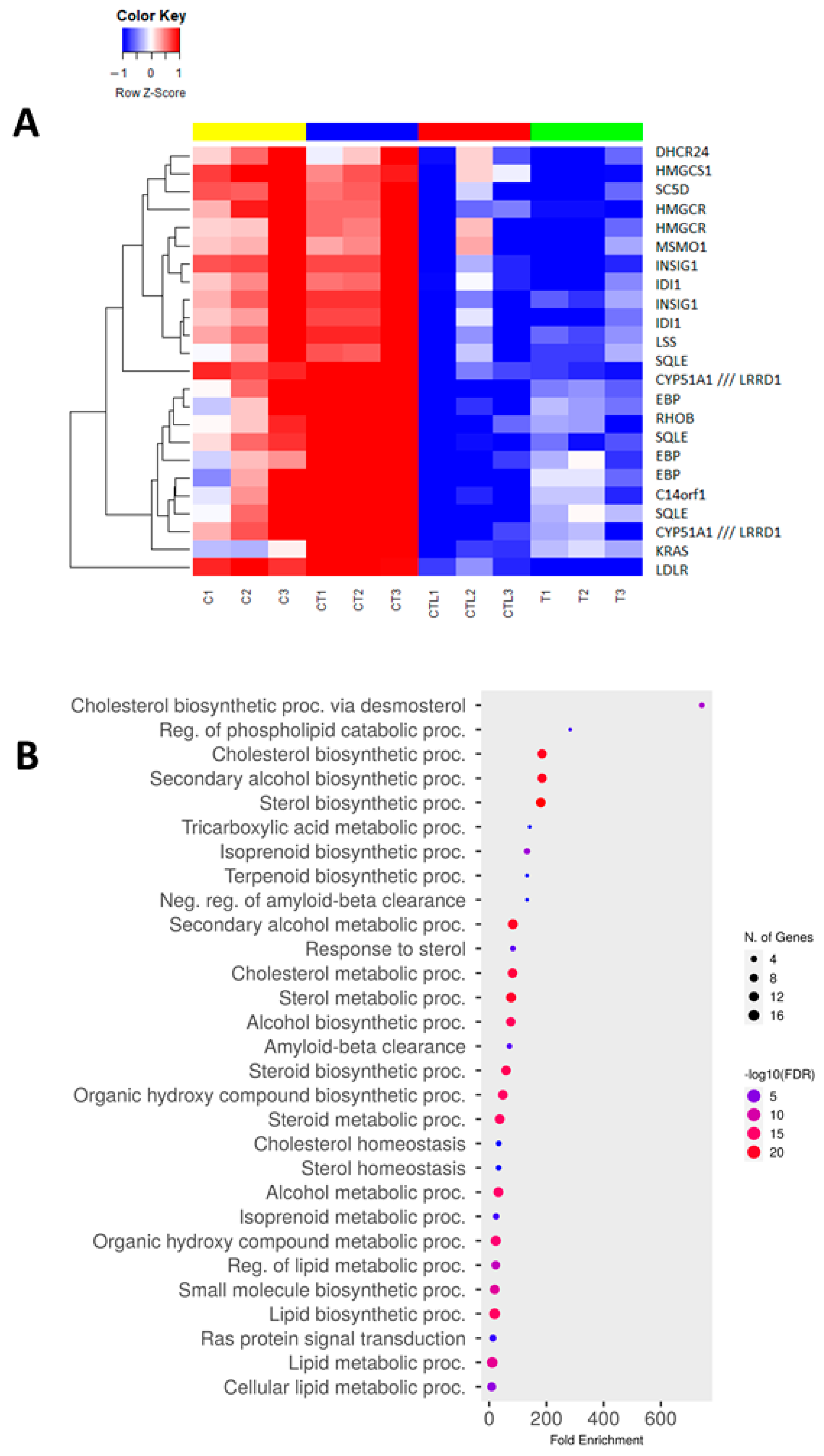
3.5. Trametinib and Cerivastatin Synergistic Concentrations Down-Modulate Genes Involved in Cell Cycle Regulation and DNA Replication
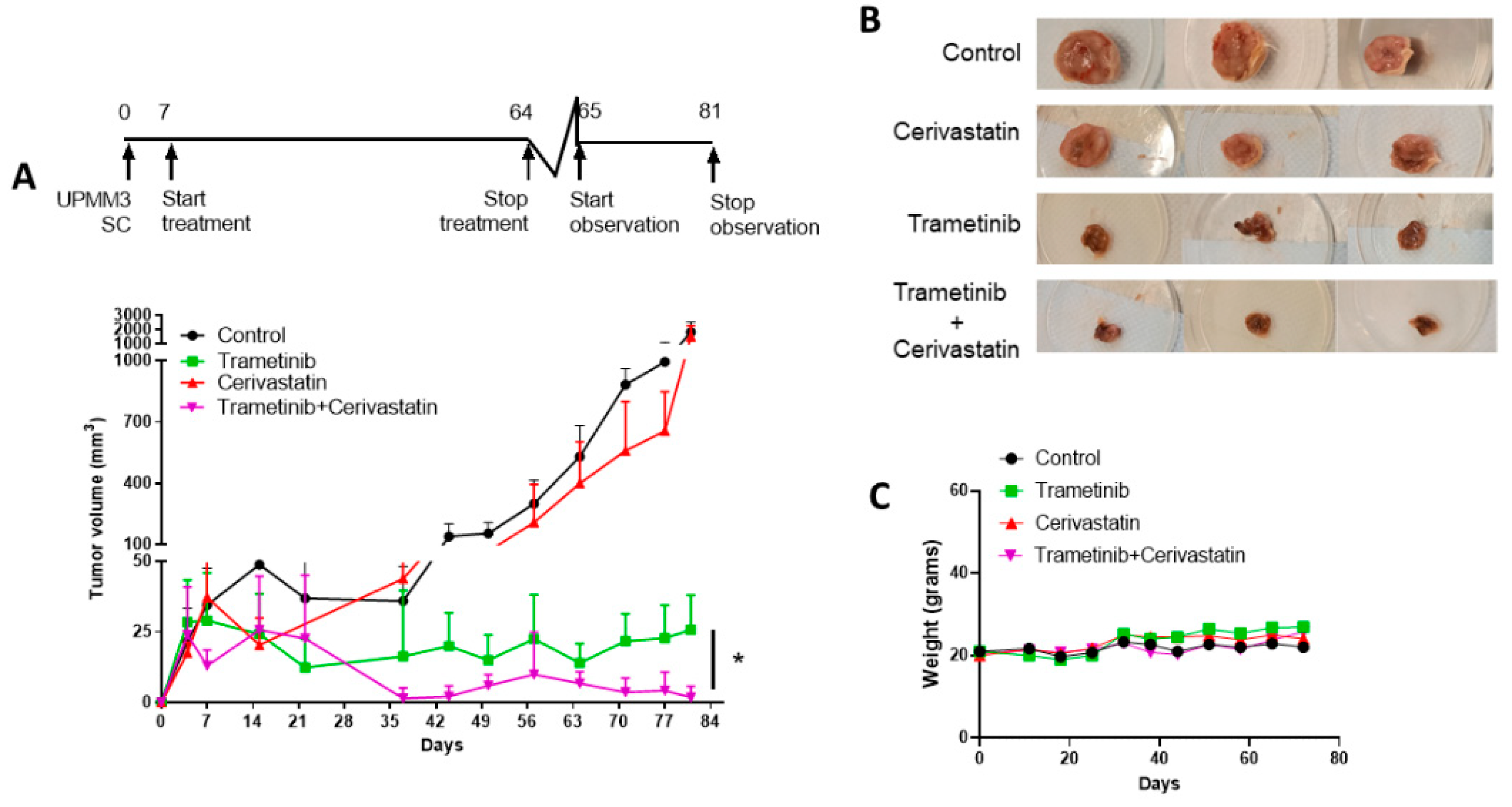
3.6. Trametinib and Cerivastatin Synergistic Concentrations Inhibit the Growth of the Monosomic BAP1 Mutated UM Cell Line In Vivo
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jager, M.J.; Shields, C.L.; Cebulla, C.M.; Abdel-Rahman, M.H.; Grossniklaus, H.E.; Stern, M.-H.; Carvajal, R.D.; Belfort, R.N.; Jia, R.; Shields, J.A.; et al. Uveal Melanoma. Nat. Rev. Dis. Prim. 2020, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Robertson, A.G.; Shih, J.; Yau, C.; Gibb, E.A.; Oba, J.; Mungall, K.L.; Hess, J.M.; Uzunangelov, V.; Walter, V.; Danilova, L.; et al. Integrative Analysis Identifies Four Molecular and Clinical Subsets in Uveal Melanoma. Cancer Cell 2017, 32, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Field, M.G.; Durante, M.A.; Anbunathan, H.; Cai, L.Z.; Decatur, C.L.; Bowcock, A.M.; Kurtenbach, S.; Harbour, J.W. Punctuated Evolution of Canonical Genomic Aberrations in Uveal Melanoma. Nat. Commun. 2018, 9, 116. [Google Scholar] [CrossRef]
- Johansson, P.; Aoude, L.G.; Wadt, K.; Glasson, W.J.; Warrier, S.K.; Hewitt, A.W.; Kiilgaard, J.F.; Heegaard, S.; Isaacs, T.; Franchina, M.; et al. Deep Sequencing of Uveal Melanoma Identifies a Recurrent Mutation in PLCB4. Oncotarget 2016, 7, 4624–4631. [Google Scholar] [CrossRef]
- Zhang, T.; Dutton-Regester, K.; Brown, K.M.; Hayward, N.K. The Genomic Landscape of Cutaneous Melanoma. Pigment. Cell Melanoma Res. 2016, 29, 266–283. [Google Scholar] [CrossRef] [PubMed]
- Van Raamsdonk, C.D.; Griewank, K.G.; Crosby, M.B.; Garrido, M.C.; Vemula, S.; Wiesner, T.; Obenauf, A.C.; Wackernagel, W.; Green, G.; Bouvier, N.; et al. Mutations in GNA11 in Uveal Melanoma. N. Engl. J. Med. 2010, 363, 2191–2199. [Google Scholar] [CrossRef] [PubMed]
- Van Raamsdonk, C.D.; Bezrookove, V.; Green, G.; Bauer, J.; Gaugler, L.; O’Brien, J.M.; Simpson, E.M.; Barsh, G.S.; Bastian, B.C. Frequent Somatic Mutations of GNAQ in Uveal Melanoma and Blue Naevi. Nature 2009, 457, 599–602. [Google Scholar] [CrossRef] [PubMed]
- Harbour, J.W.; Onken, M.D.; Roberson, E.D.O.; Duan, S.; Cao, L.; Worley, L.A.; Council, M.L.; Matatall, K.A.; Helms, C.; Bowcock, A.M. Frequent Mutation of BAP1 in Metastasizing Uveal Melanomas. Science 2010, 330, 1410–1413. [Google Scholar] [CrossRef]
- Piaggio, F.; Tozzo, V.; Bernardi, C.; Croce, M.; Puzone, R.; Viaggi, S.; Patrone, S.; Barla, A.; Coviello, D.; Jager, M.J.; et al. Secondary Somatic Mutations in G-Protein-Related Pathways and Mutation Signatures in Uveal Melanoma. Cancers 2019, 11, 1688. [Google Scholar] [CrossRef] [PubMed]
- Piaggio, F.; Croce, M.; Reggiani, F.; Monti, P.; Bernardi, C.; Ambrosio, M.; Banelli, B.; Dogrusöz, M.; Jockers, R.; Bordo, D.; et al. In Uveal Melanoma Gα-Protein GNA11 Mutations Convey a Shorter Disease-Specific Survival and Are More Strongly Associated with Loss of BAP1 and Chromosomal Alterations than Gα-Protein GNAQ Mutations. Eur. J. Cancer 2022, 170, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Croce, M.; Ferrini, S.; Pfeffer, U.; Gangemi, R. Targeted Therapy of Uveal Melanoma: Recent Failures and New Perspectives. Cancers 2019, 11, 846. [Google Scholar] [CrossRef] [PubMed]
- Conway, E.; Rossi, F.; Fernandez-Perez, D.; Ponzo, E.; Ferrari, K.J.; Zanotti, M.; Manganaro, D.; Rodighiero, S.; Tamburri, S.; Pasini, D. BAP1 Enhances Polycomb Repression by Counteracting Widespread H2AK119ub1 Deposition and Chromatin Condensation. Mol. Cell 2021, 81, 3526–3541. [Google Scholar] [CrossRef]
- Bakhoum, M.F.; Francis, J.H.; Agustinus, A.; Earlie, E.M.; Di Bona, M.; Abramson, D.H.; Duran, M.; Masilionis, I.; Molina, E.; Shoushtari, A.N.; et al. Loss of Polycomb Repressive Complex 1 Activity and Chromosomal Instability Drive Uveal Melanoma Progression. Nat. Commun. 2021, 12, 5402. [Google Scholar] [CrossRef] [PubMed]
- O’Hayre, M.; Degese, M.S.; Gutkind, J.S. Novel Insights into G Protein and G Protein-Coupled Receptor Signaling in Cancer. Curr. Opin. Cell Biol. 2014, 27, 126–135. [Google Scholar] [CrossRef]
- Rossi, E.; Croce, M.; Reggiani, F.; Schinzari, G.; Ambrosio, M.; Gangemi, R.; Tortora, G.; Pfeffer, U.; Amaro, A. Uveal Melanoma Metastasis. Cancers 2021, 13, 5684. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Chen, Q.; Gutkind, J.S. Oncotargeting G Proteins: The Hippo in the Room. Oncotarget 2014, 5, 10997–10999. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.-X.; Zhang, K.; Guan, K.-L. YAP as Oncotarget in Uveal Melanoma. Oncoscience 2014, 1, 480–481. [Google Scholar] [CrossRef]
- Moroishi, T.; Park, H.W.; Qin, B.; Chen, Q.; Meng, Z.; Plouffe, S.W.; Taniguchi, K.; Yu, F.-X.; Karin, M.; Pan, D.; et al. A YAP/TAZ-Induced Feedback Mechanism Regulates Hippo Pathway Homeostasis. Genes Dev. 2015, 29, 1271–1284. [Google Scholar] [CrossRef]
- Gujral, T.S.; Kirschner, M.W. Hippo Pathway Mediates Resistance to Cytotoxic Drugs. Proc. Natl. Acad. Sci. USA 2017, 114, E3729–E3738. [Google Scholar] [CrossRef]
- Lin, L.; Sabnis, A.J.; Chan, E.; Olivas, V.; Cade, L.; Pazarentzos, E.; Asthana, S.; Neel, D.; Yan, J.J.; Lu, X.; et al. The Hippo Effector YAP Promotes Resistance to RAF- and MEK-Targeted Cancer Therapies. Nat. Genet. 2015, 47, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.-F.; Tseng, Y.-C.; Nguyen, P.A.; Li, Y.-C.; Ho, C.-C.; Wu, C.-W. Enhanced YAP Expression Leads to EGFR TKI Resistance in Lung Adenocarcinomas. Sci. Rep. 2018, 8, 271. [Google Scholar] [CrossRef] [PubMed]
- Totaro, A.; Zhuang, Q.; Panciera, T.; Battilana, G.; Azzolin, L.; Brumana, G.; Gandin, A.; Brusatin, G.; Cordenonsi, M.; Piccolo, S. Cell Phenotypic Plasticity Requires Autophagic Flux Driven by YAP/TAZ Mechanotransduction. Proc. Natl. Acad. Sci. USA 2019, 116, 17848–17857. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Tumaneng, K.; Guan, K.-L. The Hippo Pathway in Organ Size Control, Tissue Regeneration and Stem Cell Self-Renewal. Nat. Cell Biol. 2011, 13, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Zhang, L.; Wang, L.; Ji, C.; Xia, R.; Yang, Y. A Systematic Review and Meta-Analysis on the Efficacy of Statins in the Treatment of Atherosclerosis. Ann. Palliat. Med. 2021, 10, 6793–6803. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Longo, J.; van Leeuwen, J.E.; Mullen, P.J.; Ba-Alawi, W.; Haibe-Kains, B.; Penn, L.Z. Statin-Induced Cancer Cell Death Can Be Mechanistically Uncoupled from Prenylation of RAS Family Proteins. Cancer Res. 2018, 78, 1347–1357. [Google Scholar] [CrossRef] [PubMed]
- Guerra, B.; Recio, C.; Aranda-Tavío, H.; Guerra-Rodríguez, M.; García-Castellano, J.M.; Fernández-Pérez, L. The Mevalonate Pathway, a Metabolic Target in Cancer Therapy. Front. Oncol. 2021, 11, 626971. [Google Scholar] [CrossRef]
- Sorrentino, G.; Ruggeri, N.; Specchia, V.; Cordenonsi, M.; Mano, M.; Dupont, S.; Manfrin, A.; Ingallina, E.; Sommaggio, R.; Piazza, S.; et al. Metabolic Control of YAP and TAZ by the Mevalonate Pathway. Nat. Cell Biol. 2014, 16, 357–366. [Google Scholar] [CrossRef]
- Yu, W.Y.; Hill, S.T.; Chan, E.R.; Pink, J.J.; Cooper, K.; Leachman, S.; Lund, A.W.; Kulkarni, R.; Bordeaux, J.S. Computational Drug Repositioning Identifies Statins as Modifiers of Prognostic Genetic Expression Signatures and Metastatic Behavior in Melanoma. J. Invest. Dermatol. 2021, 141, 1802–1809. [Google Scholar] [CrossRef]
- Brouwer, N.J.; Konstantinou, E.K.; Gragoudas, E.S.; Marinkovic, M.; Luyten, G.P.M.; Kim, I.K.; Jager, M.J.; Vavvas, D.G. Targeting the YAP/TAZ Pathway in Uveal and Conjunctival Melanoma With Verteporfin. Invest. Ophthalmol. Vis. Sci. 2021, 62, 3. [Google Scholar] [CrossRef]
- Palamaris, K.; Moutafi, M.; Gakiopoulou, H.; Theocharis, S. Histone Deacetylase (HDAC) Inhibitors: A Promising Weapon to Tackle Therapy Resistance in Melanoma. Int. J. Mol. Sci. 2022, 23, 3660. [Google Scholar] [CrossRef]
- Goutas, D.; Theocharis, S.; Tsourouflis, G. Unraveling the Epigenetic Role and Clinical Impact of Histone Deacetylases in Neoplasia. Diagnostics 2021, 11, 1346. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.Z.; Maniar, A.B.; Carvajal, R.D. New Targeted and Epigenetic Therapeutic Strategies for the Treatment of Uveal Melanoma. Cancer Gene Ther. 2022, 29, 1819–1826. [Google Scholar] [CrossRef] [PubMed]
- Landreville, S.; Agapova, O.A.; Matatall, K.A.; Kneass, Z.T.; Onken, M.D.; Lee, R.S.; Bowcock, A.M.; Harbour, J.W. Histone Deacetylase Inhibitors Induce Growth Arrest and Differentiation in Uveal Melanoma. Clin. Cancer Res. 2012, 18, 408–416. [Google Scholar] [CrossRef] [PubMed]
- De Waard-Siebinga, I.; Blom, D.J.; Griffioen, M.; Schrier, P.I.; Hoogendoorn, E.; Beverstock, G.; Danen, E.H.; Jager, M.J. Establishment and Characterization of an Uveal-Melanoma Cell Line. Int. J. Cancer 1995, 62, 155–161. [Google Scholar] [CrossRef]
- Chen, P.W.; Murray, T.G.; Salgaller, M.L.; Ksander, B.R. Expression of MAGE Genes in Ocular Melanoma Cell Lines. J. Immunother. 1997, 20, 265–275. [Google Scholar] [CrossRef]
- Luyten, G.P.M.; Naus, N.C.; Mooy, C.M.; Hagemeijer, A.; Kan-Mitchell, J.; Van Drunen, E.; Vuzevski, V.; De Jong, P.T.V.M.; Luider, T.M. Establishment and Characterization of Primary and Metastatic Uveal Melanoma Cell Lines. Int. J. Cancer 1996, 66, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Verbik, D.J.; Murray, T.G.; Tran, J.M.; Ksander, B.R. Melanomas That Develop within the Eye Inhibit Lymphocyte Proliferation. Int. J. Cancer 1997, 73, 470–478. [Google Scholar] [CrossRef]
- Nareyeck, G.; Zeschnigk, M.; Bornfeld, N.; Anastassiou, G. Novel Cell Lines Derived by Long-Term Culture of Primary Uveal Melanomas. Ophthalmologica 2009, 223, 196–201. [Google Scholar] [CrossRef]
- Chou, T.C.; Talalay, P. Quantitative Analysis of Dose-Effect Relationships: The Combined Effects of Multiple Drugs or Enzyme Inhibitors. Adv. Enzym. Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef] [PubMed]
- Gangemi, R.; Amaro, A.; Gino, A.; Barisione, G.; Fabbi, M.; Pfeffer, U.; Brizzolara, A.; Queirolo, P.; Salvi, S.; Boccardo, S.; et al. ADAM10 Correlates with Uveal Melanoma Metastasis and Promotes in Vitro Invasion. Pigment. Cell Melanoma Res. 2014, 27, 1138–1148. [Google Scholar] [CrossRef]
- Irizarry, R.A.; Hobbs, B.; Collin, F.; Beazer-Barclay, Y.D.; Antonellis, K.J.; Scherf, U.; Speed, T.P. Exploration, Normalization, and Summaries of High Density Oligonucleotide Array Probe Level Data. Biostatistics 2003, 4, 249–264. [Google Scholar] [CrossRef] [PubMed]
- Tusher, V.G.; Tibshirani, R.; Chu, G. Significance Analysis of Microarrays Applied to the Ionizing Radiation Response. Proc. Natl. Acad. Sci. USA 2001, 98, 5116–5121. [Google Scholar] [CrossRef] [PubMed]
- Amaro, A.; Mirisola, V.; Angelini, G.; Musso, A.; Tosetti, F.; Esposito, A.I.; Perri, P.; Lanza, F.; Nasciuti, F.; Mosci, C.; et al. Evidence of Epidermal Growth Factor Receptor Expression in Uveal Melanoma: Inhibition of Epidermal Growth Factor-Mediated Signalling by Gefitinib and Cetuximab Triggered Antibody-Dependent Cellular Cytotoxicity. Eur. J. Cancer 2013, 49, 3353–3365. [Google Scholar] [CrossRef] [PubMed]
- Liu-Chittenden, Y.; Huang, B.; Shim, J.S.; Chen, Q.; Lee, S.-J.; Anders, R.A.; Liu, J.O.; Pan, D. Genetic and Pharmacological Disruption of the TEAD-YAP Complex Suppresses the Oncogenic Activity of YAP. Genes Dev. 2012, 26, 1300–1305. [Google Scholar] [CrossRef]
- Faião-Flores, F.; Emmons, M.F.; Durante, M.A.; Kinose, F.; Saha, B.; Fang, B.; Koomen, J.M.; Chellappan, S.P.; Maria-Engler, S.S.; Rix, U.; et al. HDAC Inhibition Enhances the In Vivo Efficacy of MEK Inhibitor Therapy in Uveal Melanoma. Clin. Cancer Res. 2019, 25, 5686–5701. [Google Scholar] [CrossRef]
- Mergener, S.; Siveke, J.T.; Peña-Llopis, S. Monosomy 3 Is Linked to Resistance to MEK Inhibitors in Uveal Melanoma. Int. J. Mol. Sci. 2021, 22, 6727. [Google Scholar] [CrossRef]
- Gbelcová, H.; Rimpelová, S.; Ruml, T.; Fenclová, M.; Kosek, V.; Hajšlová, J.; Strnad, H.; Kolář, M.; Vítek, L. Variability in Statin-Induced Changes in Gene Expression Profiles of Pancreatic Cancer. Sci. Rep. 2017, 7, 44219. [Google Scholar] [CrossRef]
- Calvert, A.E.; Chalastanis, A.; Wu, Y.; Hurley, L.A.; Kouri, F.M.; Bi, Y.; Kachman, M.; May, J.L.; Bartom, E.; Hua, Y.; et al. Cancer-Associated IDH1 Promotes Growth and Resistance to Targeted Therapies in the Absence of Mutation. Cell Rep. 2017, 19, 1858–1873. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Li, Z.; Wu, Q.; Liu, H.; Sun, Z.; Wu, Y.; Luo, J. B4GALT5 High Expression Associated with Poor Prognosis of Hepatocellular Carcinoma. BMC Cancer 2022, 22, 392. [Google Scholar] [CrossRef]
- Kudo, T.; Ikeda, M.; Nishikawa, M.; Yang, Z.; Ohno, K.; Nakagawa, K.; Hata, Y. The RASSF3 Candidate Tumor Suppressor Induces Apoptosis and G1–S Cell-Cycle Arrest via P53. Cancer Res. 2012, 72, 2901–2911. [Google Scholar] [CrossRef]
- Bin, B.-H.; Bhin, J.; Yang, S.H.; Shin, M.; Nam, Y.-J.; Choi, D.-H.; Shin, D.W.; Lee, A.-Y.; Hwang, D.; Cho, E.-G.; et al. Membrane-Associated Transporter Protein (MATP) Regulates Melanosomal PH and Influences Tyrosinase Activity. PLoS ONE 2015, 10, e0129273. [Google Scholar] [CrossRef]
- Phillips, S.; Lizee, G.; Brown, C.; Lara, J.M.; Bassett, R.L.; Beal, L.G.; Murthy, R.; Talukder, A.; Hawke, D.H.; Lai, I.; et al. A Phase Ib Study of Endogenous SLC45A2-Specific Cytotoxic T Cells for the Treatment of Patients with Metastatic Uveal Melanoma. J. Clin. Oncol. 2020, 38, TPS10086. [Google Scholar] [CrossRef]
- Rimpelová, S.; Kolář, M.; Strnad, H.; Ruml, T.; Vítek, L.; Gbelcová, H. Comparison of Transcriptomic Profiles of MiaPaCa-2 Pancreatic Cancer Cells Treated with Different Statins. Molecules 2021, 26, 3528. [Google Scholar] [CrossRef] [PubMed]
- Dongoran, R.A.; Wang, K.-H.; Lin, T.-J.; Yuan, T.-C.; Liu, C.-H. Anti-Proliferative Effect of Statins Is Mediated by DNMT1 Inhibition and P21 Expression in OSCC Cells. Cancers 2020, 12, 2084. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A Graphical Gene-Set Enrichment Tool for Animals and Plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef]
- Marseglia, M.; Amaro, A.; Solari, N.; Gangemi, R.; Croce, E.; Tanda, E.T.; Spagnolo, F.; Filaci, G.; Pfeffer, U.; Croce, M. How to Make Immunotherapy an Effective Therapeutic Choice for Uveal Melanoma. Cancers 2021, 13, 2043. [Google Scholar] [CrossRef]
- Yu, F.-X.; Luo, J.; Mo, J.-S.; Liu, G.; Kim, Y.C.; Meng, Z.; Zhao, L.; Peyman, G.; Ouyang, H.; Jiang, W.; et al. Mutant Gq/11 Promote Uveal Melanoma Tumorigenesis by Activating YAP. Cancer Cell 2014, 25, 822–830. [Google Scholar] [CrossRef]
- Neelature Sriramareddy, S.; Smalley, K.S.M. MEK-Ing the Most of It: Strategies to Co-Target Gαq and MAPK in Uveal Melanoma. Clin. Cancer Res. 2021, 27, 1217–1219. [Google Scholar] [CrossRef]
- Scott, L.J.; Goa, K.L. Verteporfin. Drugs Aging 2000, 16, 139–146; discussion 147–148. [Google Scholar] [CrossRef]
- Ju, Y.; Dai, X.; Tang, Z.; Ming, Z.; Ni, N.; Zhu, D.; Zhang, J.; Ma, B.; Wang, J.; Huang, R.; et al. Verteporfin-Mediated on/off Photoswitching Functions Synergistically to Treat Choroidal Vascular Diseases. Bioact. Mater. 2022, 14, 402–415. [Google Scholar] [CrossRef]
- Souri, Z.; Jochemsen, A.G.; Versluis, M.; Wierenga, A.P.A.; Nemati, F.; van der Velden, P.A.; Kroes, W.G.M.; Verdijk, R.M.; Luyten, G.P.M.; Jager, M.J. HDAC Inhibition Increases HLA Class I Expression in Uveal Melanoma. Cancers 2020, 12, 3690. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsoff, J.N.; Owens, D.A.; Lopez, A.; Rodriguez, D.A.; Chee, N.T.; Kurtenbach, S.; Bilbao, D.; Roberts, E.R.; Volmar, C.-H.; Wahlestedt, C.; et al. Dual Screen for Efficacy and Toxicity Identifies HDAC Inhibitor with Distinctive Activity Spectrum for BAP1-Mutant Uveal Melanoma. Mol. Cancer Res. 2021, 19, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Orloff, M.M.; Valsecchi, M.E.; Shimada, A.; Chervoneva, I.; Sharpe-Mills, E.; Klose, H.; Norcini, J.; Belinsky, J.; Sato, S.; et al. A Randomized Phase II Study of Adjuvant Sunitinib or Valproic Acid in High-Risk Patients with Uveal Melanoma. J. Clin. Oncol. 2020, 38, e22059. [Google Scholar] [CrossRef]
- Jiang, W.; Hu, J.-W.; He, X.-R.; Jin, W.-L.; He, X.-Y. Statins: A Repurposed Drug to Fight Cancer. J. Exp. Clin. Cancer Res. 2021, 40, 241. [Google Scholar] [CrossRef] [PubMed]
- Yun, M.R.; Choi, H.M.; Lee, Y.W.; Joo, H.S.; Park, C.W.; Choi, J.W.; Kim, D.H.; Kang, H.N.; Pyo, K.-H.; Shin, E.J.; et al. Targeting YAP to Overcome Acquired Resistance to ALK Inhibitors in ALK-Rearranged Lung Cancer. EMBO Mol. Med. 2019, 11, e10581. [Google Scholar] [CrossRef]
- Long, S.A.; Huang, S.; Kambala, A.; Ren, L.; Wilson, J.; Goetz, M.; Hao, X.; Yang, X.; Goncharova, E.I.; Jia, L.; et al. Identification of Potential Modulators of Osteosarcoma Metastasis by High-Throughput Cellular Screening of Natural Products. Chem. Biol. Drug Des. 2021, 97, 77–86. [Google Scholar] [CrossRef]
- Davidson, M.H. Reducing Residual Risk for Patients on Statin Therapy: The Potential Role of Combination Therapy. Am. J. Cardiol. 2005, 96, 3–13; discussion 34–35. [Google Scholar] [CrossRef]
- Gonçalves, J.; Emmons, M.F.; Faião-Flores, F.; Aplin, A.E.; Harbour, J.W.; Licht, J.D.; Wink, M.R.; Smalley, K.S.M. Decitabine Limits Escape from MEK Inhibition in Uveal Melanoma. Pigment. Cell Melanoma Res. 2020, 33, 507–514. [Google Scholar] [CrossRef]
- Calipel, A.; Abonnet, V.; Nicole, O.; Mascarelli, F.; Coupland, S.E.; Damato, B.; Mouriaux, F. Status of RASSF1A in Uveal Melanocytes and Melanoma Cells. Mol. Cancer Res. 2011, 9, 1187–1198. [Google Scholar] [CrossRef]
- Dittfeld, C.; Richter, A.M.; Steinmann, K.; Klagge-Ulonska, A.; Dammann, R.H. The SARAH Domain of RASSF1A and Its Tumor Suppressor Function. Mol. Biol. Int. 2012, 2012, 196715. [Google Scholar] [CrossRef]
- Qin, F.; Tian, J.; Zhou, D.; Chen, L. Mst1 and Mst2 Kinases: Regulations and Diseases. Cell Biosci. 2013, 3, 31. [Google Scholar] [CrossRef] [PubMed]
- Zinatizadeh, M.R.; Miri, S.R.; Zarandi, P.K.; Chalbatani, G.M.; Rapôso, C.; Mirzaei, H.R.; Akbari, M.E.; Mahmoodzadeh, H. The Hippo Tumor Suppressor Pathway (YAP/TAZ/TEAD/MST/LATS) and EGFR-RAS-RAF-MEK in Cancer Metastasis. Genes Dis. 2021, 8, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Furberg, C.D.; Pitt, B. Withdrawal of Cerivastatin from the World Market. Curr. Control. Trials Cardiovasc. Med. 2001, 2, 205–207. [Google Scholar] [CrossRef] [PubMed]
- Staffa, J.A.; Chang, J.; Green, L. Cerivastatin and Reports of Fatal Rhabdomyolysis. N. Engl. J. Med. 2002, 346, 539–540. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Kimura, J. Mechanism of Statin-Induced Rhabdomyolysis. J. Pharmacol. Sci. 2013, 123, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Osaki, Y.; Nakagawa, Y.; Miyahara, S.; Iwasaki, H.; Ishii, A.; Matsuzaka, T.; Kobayashi, K.; Yatoh, S.; Takahashi, A.; Yahagi, N.; et al. Skeletal Muscle-Specific HMG-CoA Reductase Knockout Mice Exhibit Rhabdomyolysis: A Model for Statin-Induced Myopathy. Biochem. Biophys. Res. Commun. 2015, 466, 536–540. [Google Scholar] [CrossRef] [PubMed]
- Adams, S.P.; Tiellet, N.; Alaeiilkhchi, N.; Wright, J.M. Cerivastatin for Lowering Lipids. Cochrane Database Syst. Rev. 2020, 1, CD012501. [Google Scholar] [CrossRef] [PubMed]
- Cooper-DeHoff, R.M.; Niemi, M.; Ramsey, L.B.; Luzum, J.A.; Tarkiainen, E.K.; Straka, R.J.; Gong, L.; Tuteja, S.; Wilke, R.A.; Wadelius, M.; et al. The Clinical Pharmacogenetics Implementation Consortium Guideline for SLCO1B1, ABCG2, and CYP2C9 Genotypes and Statin-Associated Musculoskeletal Symptoms. Clin. Pharmacol. Ther. 2022, 111, 1007–1021. [Google Scholar] [CrossRef]
- Antons, K.A.; Williams, C.D.; Baker, S.K.; Phillips, P.S. Clinical Perspectives of Statin-Induced Rhabdomyolysis. Am. J. Med. 2006, 119, 400–409. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Origin | GNAQ | GNA11 | BAP1 | chr3 Status | Reference |
|---|---|---|---|---|---|---|
| 92.1 | primary | Q209L | WT | WT | trisomic | [34] |
| MEL270 | primary $ | Q209P | WT | WT | disomic | [35] |
| OMM1 | metastatic | Q209L | Q209L | WT | disomic | [36] |
| OMM2.5 | metastatic $ | Q209P | WT | WT | disomic | [37] |
| UPMM2 | primary | Q209L | WT | I586H fs * 57 | monosomic | [38] |
| UPMM3 | primary | Q209P | WT | G45F48del | monosomic | [38] |
| OMM2.5 | UPMM3 | MEL270 | 92.1 | OMM1 | UPMM2 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Drug | Class | Unit | IC50 | SEM | IC50 | SEM | IC50 | SEM | IC50 | SEM | IC50 | SEM | IC50 | SEM |
| trametinib | MEK-inhibitor | nM | 171.60 | ±44 | 16.39 | ±6 | 5.00 | ±1.4 | 9.90 | ±4.8 | 6500.00 | ±3983 | 8.10 | ±3 |
| valproic acid | HDAC-inhibitor | mM | 40.90 | ±26.5 | 2.77 | ±93 | 10.88 | ±2.56 | 3.39 | ±0.98 | 9.56 | ±2.73 | 23.24 | ±2.34 |
| verteporfin | Photosensitizer | µM | 8.22 | ±3.88 | 30.24 | ±1.44 | 18.67 | ±0.9 | 16.55 | ±0.788 | 13.15 | ±4.75 | 720.60 | ±703.5 |
| cerivastatin | Statin | µM | 0.99 | ±0.16 | 0.35 | ±0.1 | 0.40 | ±0.09 | 1.56 | ±0.26 | 0.06 | ±0.02 | 0.38 | ±0.12 |
| atorvastatin | Statin | µM | 62.11 | ±46.45 | 116.50 | ±96.26 | 137.30 | ±55.17 | na | na | 5.63 | ±0.62 | 50.75 | ±20.84 |
| simvastatin | Statin | µM | 8.29 | ±0.8 | 8.32 | ±1.12 | 15.57 | ±5.9 | 115.40 | ±32.6 | 2.83 | ±0.34 | 36.17 | ±14.6 |
| A: Trametinib and Valproic Acid (VPA) | |||||
| Cell Line | Trametinib | VPA | Trametinib | VPA | Combination Index |
| IC50 | Synergistic Concentration | ||||
| nM | mM | nM | mM | ||
| 92.1 | 14.60 | 5.56 | 3.64 | 1.00 | 0.43 |
| Mel270 | 7.13 | 14.65 | 2.24 | 1.00 | 0.38 |
| Mel270 | 7.13 | 14.65 | 1.77 | 2.00 | 0.38 |
| UPMM3 | 20.38 | 1.84 | 13.02 | 0.25 | 0.77 |
| UPMM3 | 20.38 | 1.84 | 9.61 | 0.50 | 0.74 |
| UPMM3 | 20.38 | 1.84 | 2.39 | 1.00 | 0.65 |
| B: Trametinib and Verteporfin | |||||
| Cell Line | Trametinib | Verteporfin | Trametinib | Verteporfin | Combination Index |
| IC50 | Synergistic Concentration | ||||
| nM | µM | nM | µM | ||
| 92.1 | 26.74 | 15.76 | 0.15 | 10.00 | 0.63 |
| Mel270 | 1.20 | 17.78 | 0.00 | 10.00 | 0.56 |
| UPMM3 | 11.08 | 28.84 | 0.43 | 10.00 | 0.39 |
| UPMM3 | 11.08 | 28.84 | 0.55 | 14.00 | 0.53 |
| C: Trametinib and Cerivastatin | |||||
| Cell Line | Trametinib | Cerivastatin | Trametinib | Cerivastatin | Combination Index |
| IC50 | Synergistic Concentration | ||||
| nM | µM | nM | µM | ||
| UPMM2 | 19.53 | 2.66 | 10.20 | 0.50 | 0.70 |
| UPMM2 | 19.53 | 2.66 | 6.15 | 1.00 | 0.68 |
| UPMM3 | 26.31 | 0.77 | 6.00 | 0.25 | 0.54 |
| UPMM3 | 26.31 | 0.77 | 0.27 | 0.50 | 0.66 |
| OMM2.5 * | 2286.00 | 1.11 | 71.00 | 0.25 | 0.25 |
| OMM2.5 * | 2286.00 | 1.11 | 21.00 | 0.50 | 0.44 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amaro, A.A.; Gangemi, R.; Emionite, L.; Castagnola, P.; Filaci, G.; Jager, M.J.; Tanda, E.T.; Spagnolo, F.; Mascherini, M.; Pfeffer, U.; et al. Cerivastatin Synergizes with Trametinib and Enhances Its Efficacy in the Therapy of Uveal Melanoma. Cancers 2023, 15, 886. https://doi.org/10.3390/cancers15030886
Amaro AA, Gangemi R, Emionite L, Castagnola P, Filaci G, Jager MJ, Tanda ET, Spagnolo F, Mascherini M, Pfeffer U, et al. Cerivastatin Synergizes with Trametinib and Enhances Its Efficacy in the Therapy of Uveal Melanoma. Cancers. 2023; 15(3):886. https://doi.org/10.3390/cancers15030886
Chicago/Turabian StyleAmaro, Adriana Agnese, Rosaria Gangemi, Laura Emionite, Patrizio Castagnola, Gilberto Filaci, Martine J. Jager, Enrica Teresa Tanda, Francesco Spagnolo, Matteo Mascherini, Ulrich Pfeffer, and et al. 2023. "Cerivastatin Synergizes with Trametinib and Enhances Its Efficacy in the Therapy of Uveal Melanoma" Cancers 15, no. 3: 886. https://doi.org/10.3390/cancers15030886
APA StyleAmaro, A. A., Gangemi, R., Emionite, L., Castagnola, P., Filaci, G., Jager, M. J., Tanda, E. T., Spagnolo, F., Mascherini, M., Pfeffer, U., & Croce, M. (2023). Cerivastatin Synergizes with Trametinib and Enhances Its Efficacy in the Therapy of Uveal Melanoma. Cancers, 15(3), 886. https://doi.org/10.3390/cancers15030886