

Lipidomic Profiling Reveals Biological Differences between Tumors of Self-Identified African Americans and Non-Hispanic Whites with Cancer



Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Specimen Procurement
2.2. Patient Characteristics
2.3. Specimen Processing
2.4. Lipid Extraction
2.5. Internal Standards
2.6. Retention-Time Standards
2.7. Mass Spectrometry Analysis
2.8. Statistical Analysis
3. Results
3.1. Self-Identified Race-Independent and Disease-Specific Analysis of Sphingolipid Metabolism Alterations
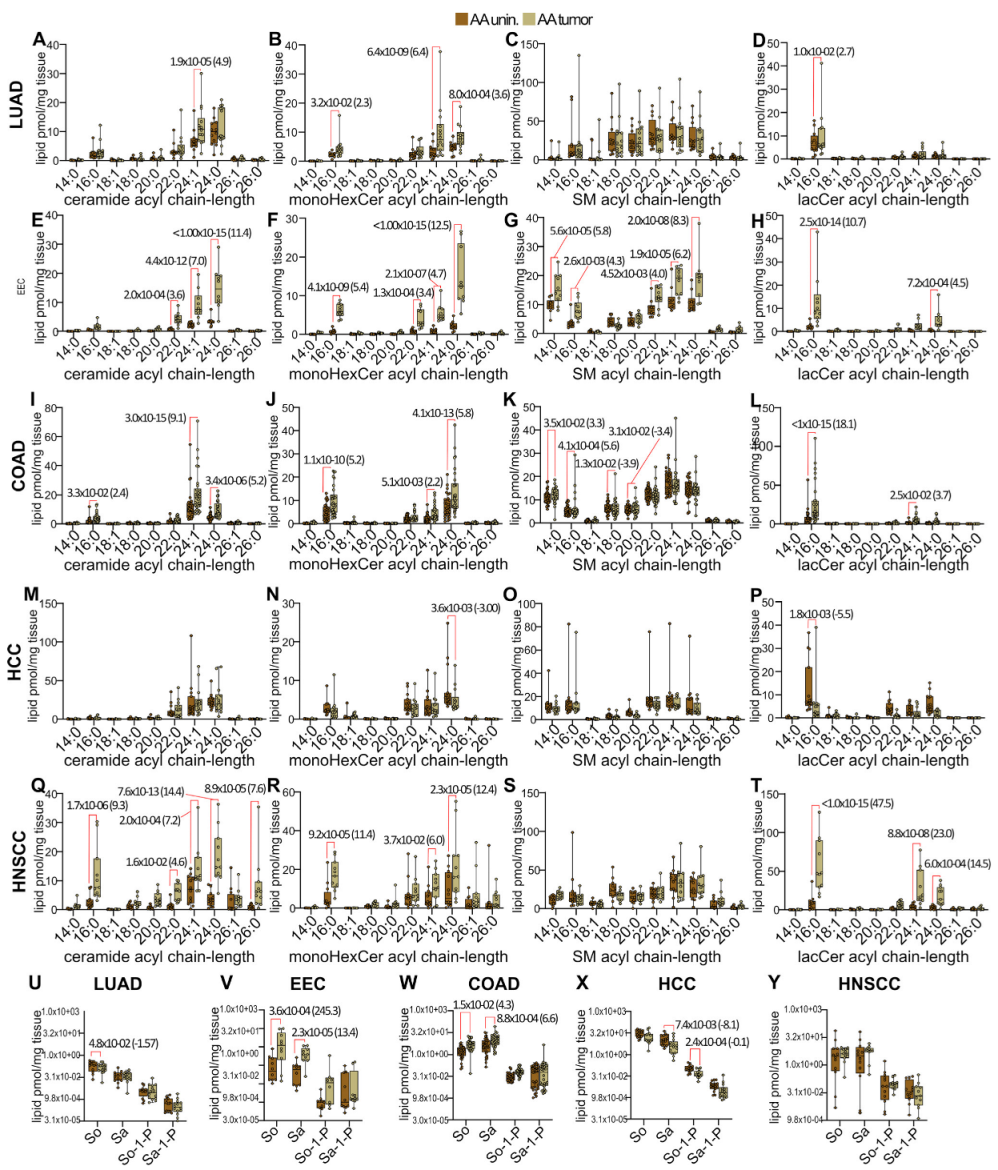
3.2. Disease-Specific Analysis of Sphingolipid Metabolism Alterations in Self-Identified AA Males and Females
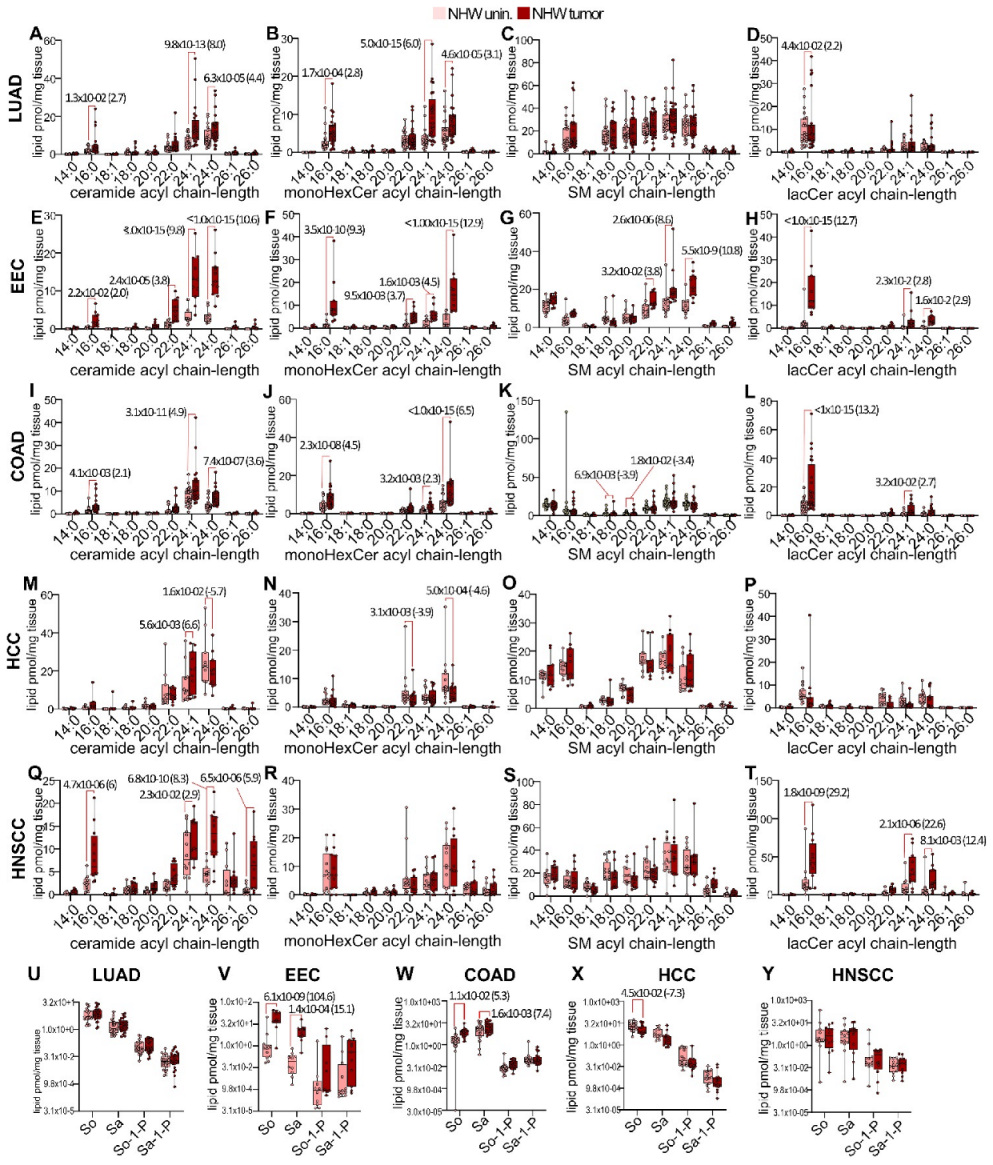
3.3. Disease-Specific Analysis of Sphingolipid Metabolism Alterations in Self-Identified NHW Males and Females
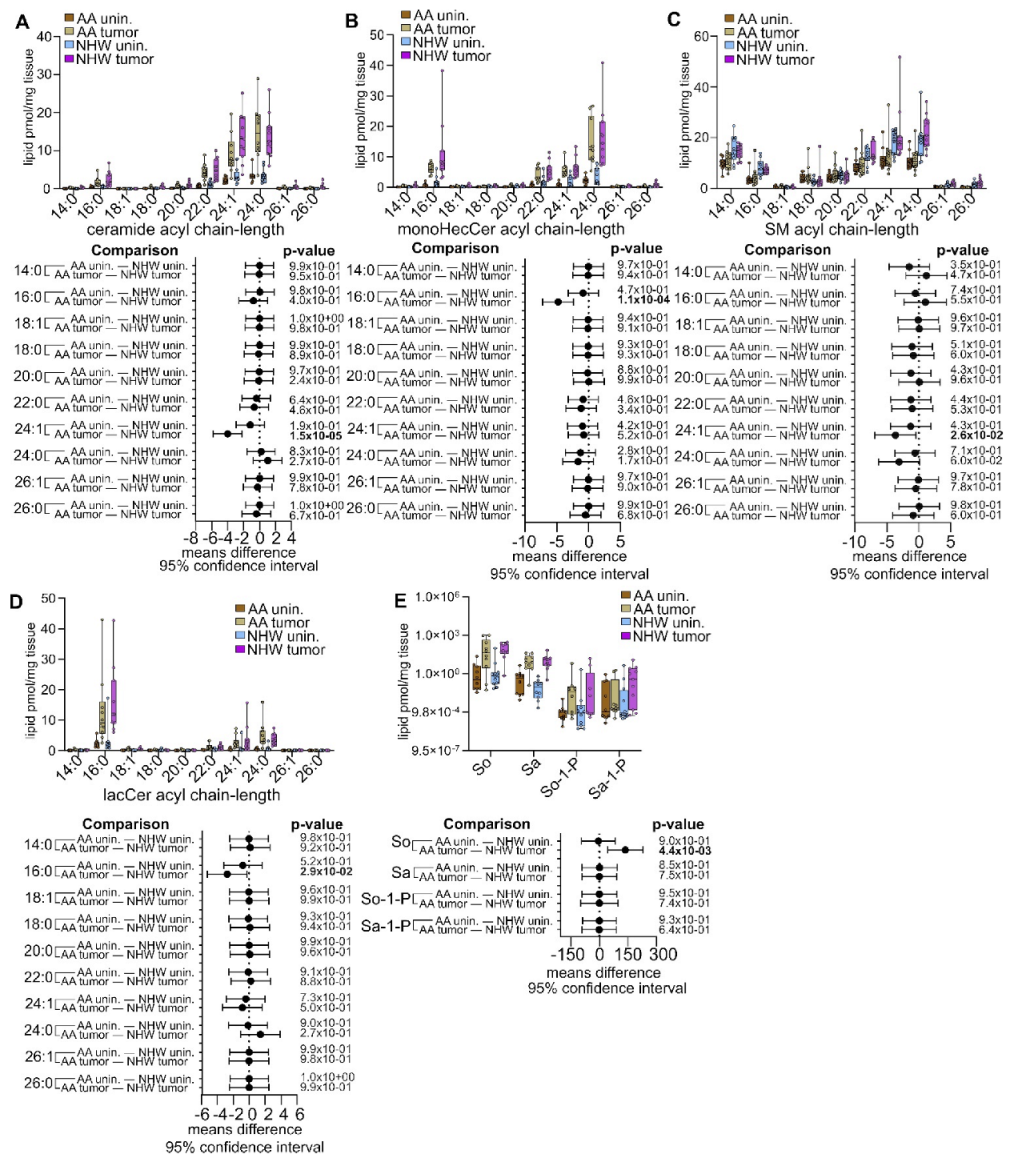
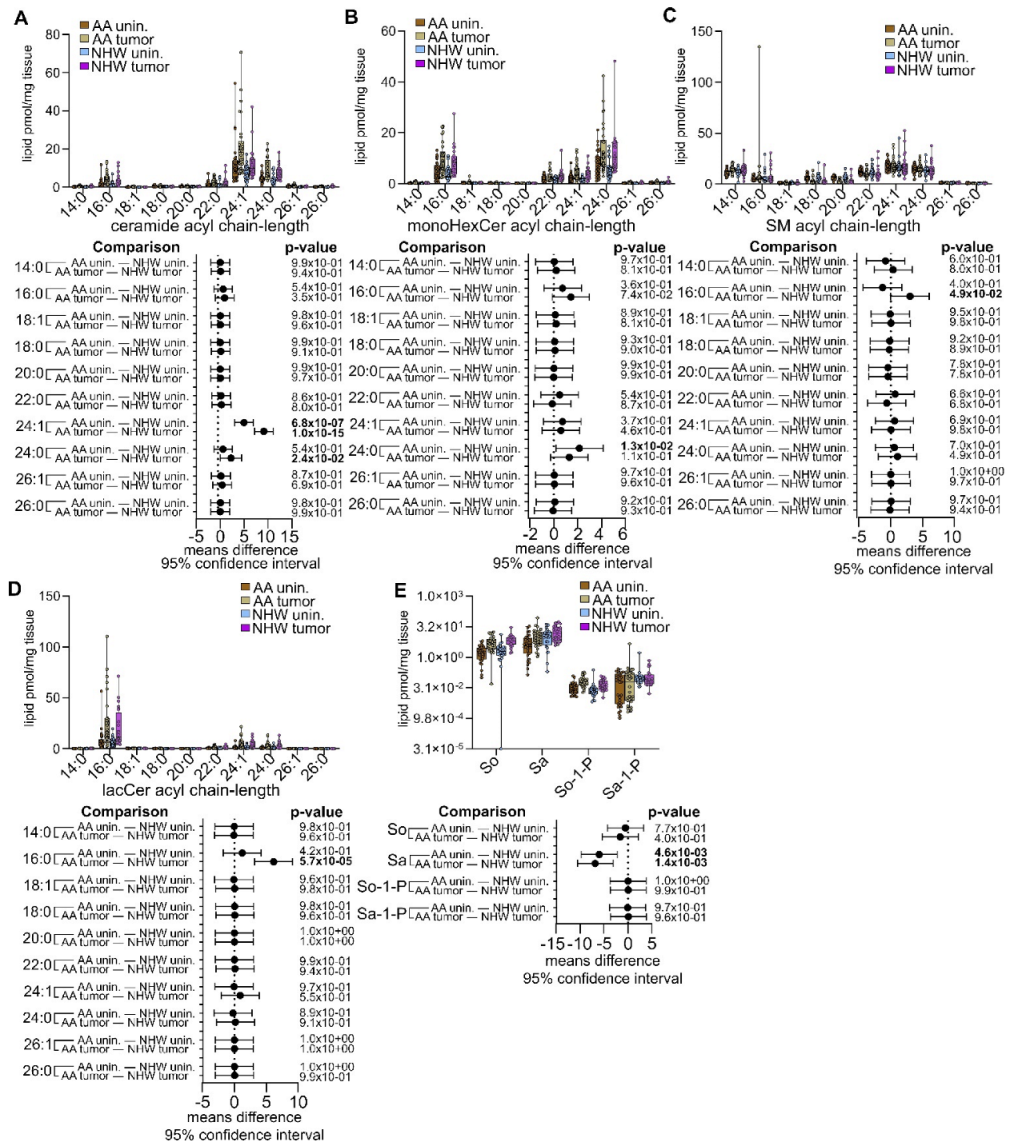
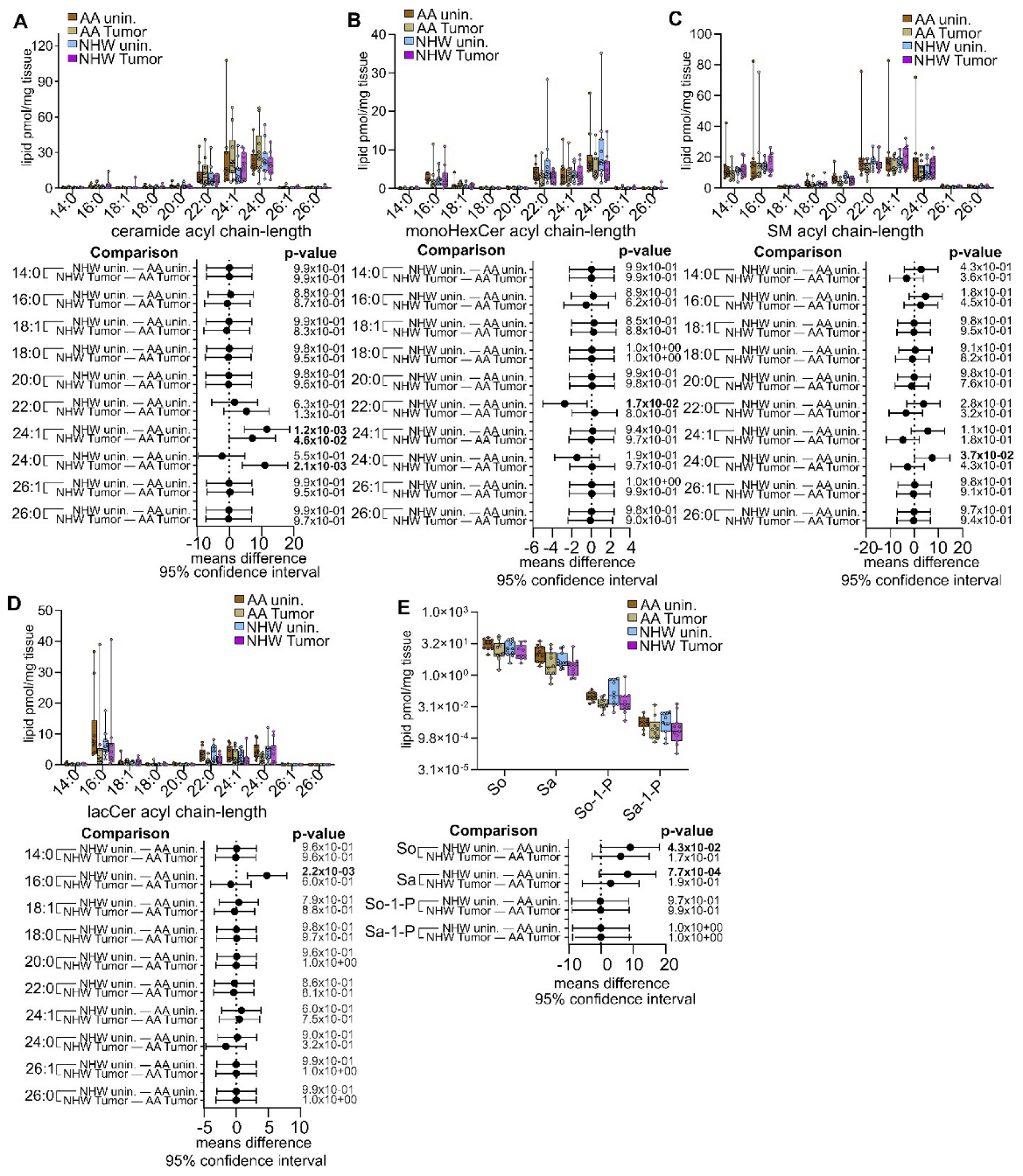
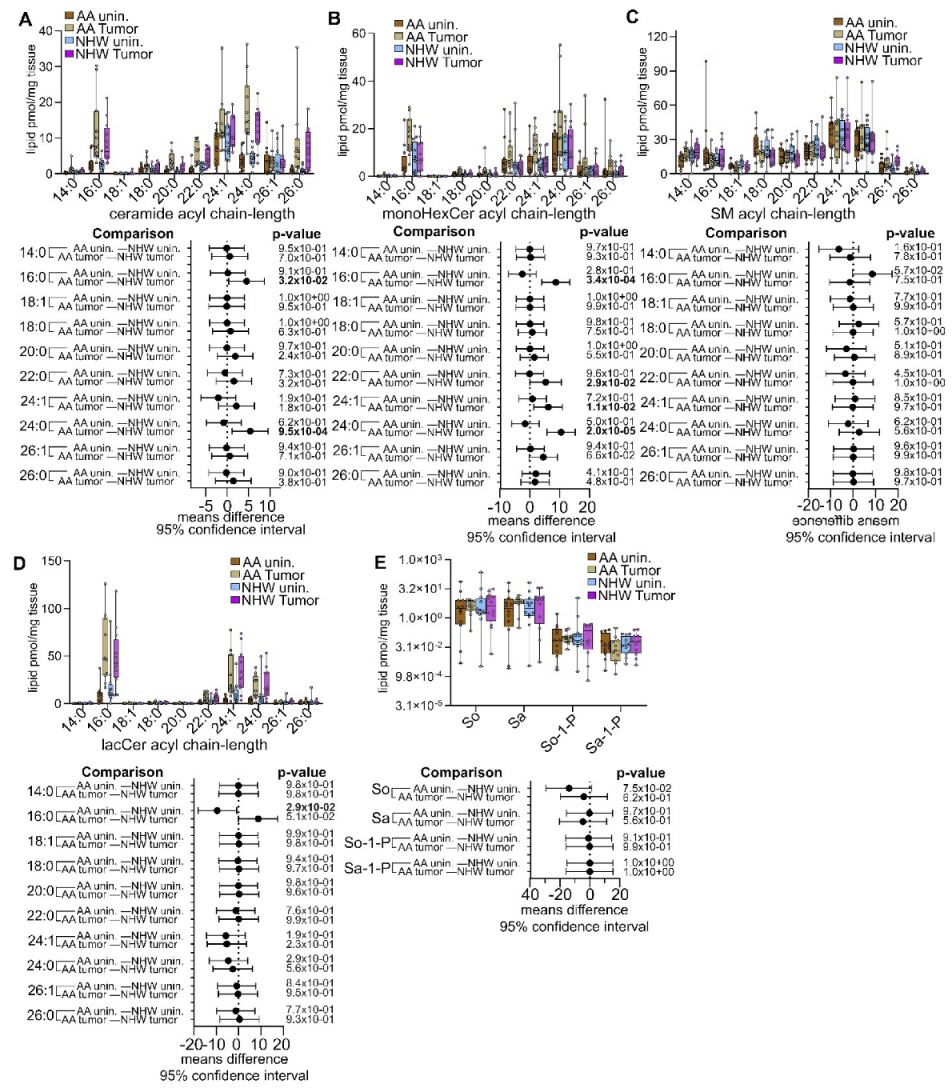
3.4. Disease-Specific Comparisons between Self-Identified AA and NHW with Cancer
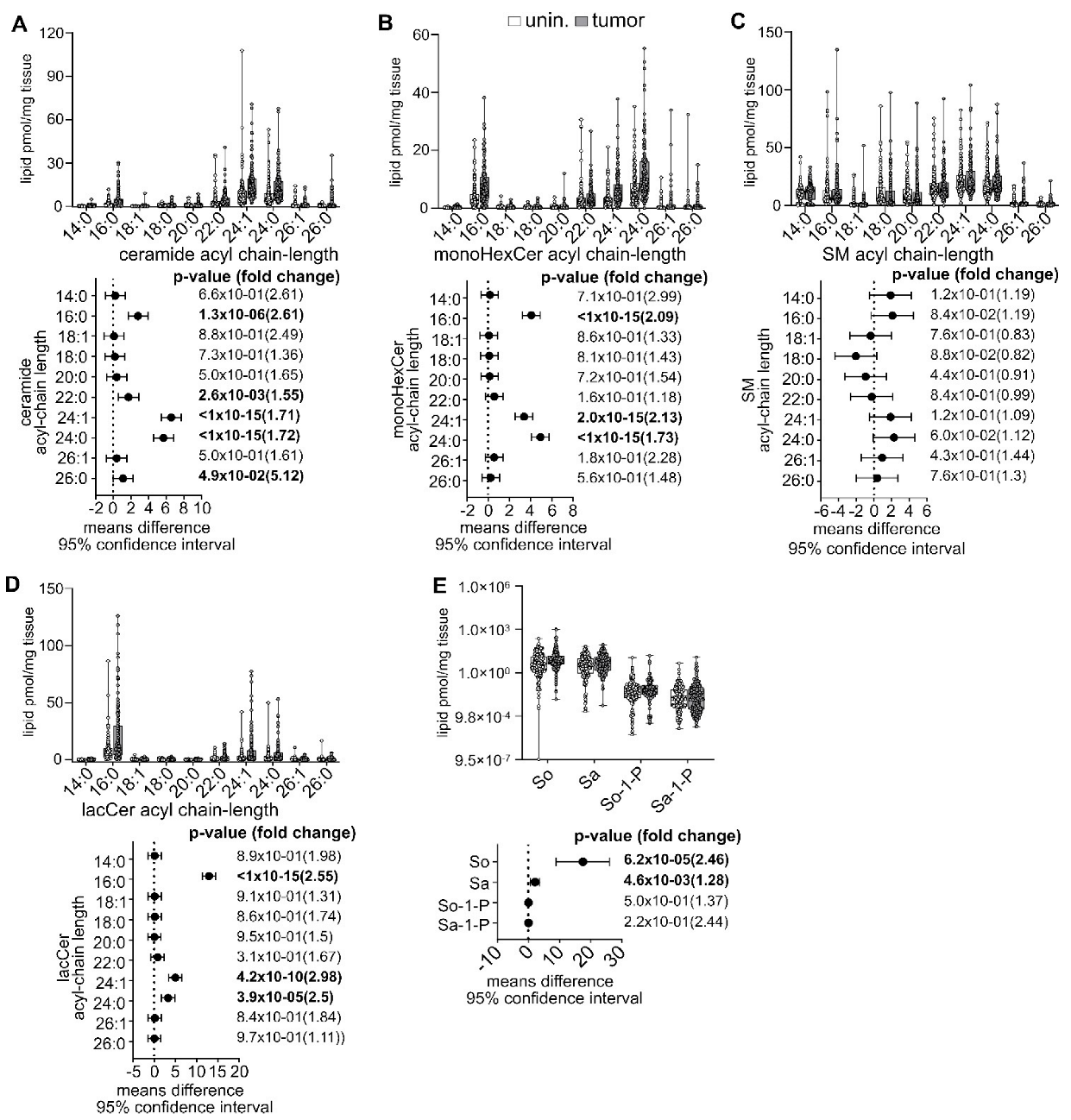
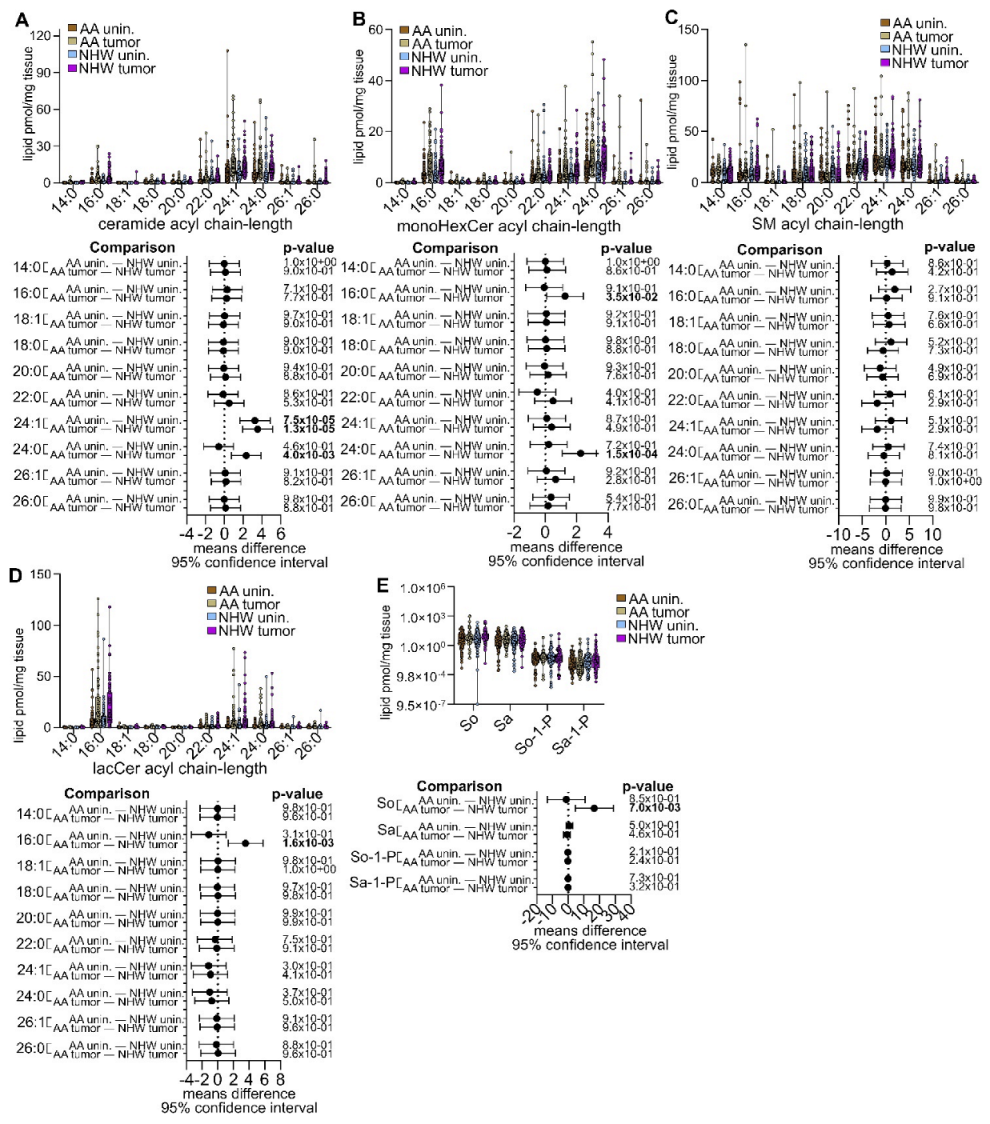
3.5. Pan-Cancer Analysis of Sphingolipid Metabolism Reprogramming
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kraft, M.L. Sphingolipid Organization in the Plasma Membrane and the Mechanisms That Influence It. Front. Cell Dev. Biol. 2016, 4, 154. [Google Scholar] [CrossRef] [PubMed]
- Breslow, D.K.; Weissman, J.S. Membranes in Balance: Mechanisms of Sphingolipid Homeostasis. Mol. Cell 2010, 40, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Lima, S.; Milstien, S.; Spiegel, S. Sphingosine and Sphingosine Kinase 1 Involvement in Endocytic Membrane Trafficking. J. Biol. Chem. 2017, 292, 3074–3088. [Google Scholar] [CrossRef] [PubMed]
- Lima, S.; Takabe, K.; Newton, J.; Saurabh, K.; Young, M.M.; Leopoldino, A.M.; Hait, N.C.; Roberts, J.L.; Wang, H.-G.; Dent, P.; et al. TP53 is required for BECN1- and ATG5-dependent cell death induced by sphingosine kinase 1 inhibition. Autophagy 2018, 14, 942–957. [Google Scholar] [CrossRef] [PubMed]
- Young, M.M.; Takahashi, Y.; Fox, T.E.; Yun, J.K.; Kester, M.; Wang, H.-G. Sphingosine Kinase 1 Cooperates with Autophagy to Maintain Endocytic Membrane Trafficking. Cell Rep. 2016, 17, 1532–1545. [Google Scholar] [CrossRef]
- Young, M.M.; Wang, H.-G. Sphingolipids as Regulators of Autophagy and Endocytic Trafficking. Adv. Cancer Res. 2018, 140, 27–60. [Google Scholar] [CrossRef]
- Shen, H.; Giordano, F.; Wu, Y.; Chan, J.; Zhu, C.; Milosevic, I.; Wu, X.; Yao, K.; Chen, B.; Baumgart, T.; et al. Coupling between endocytosis and sphingosine kinase 1 recruitment. Nature 2014, 16, 652–662. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175–191. [Google Scholar] [CrossRef]
- Russo, D.; Parashuraman, S.; D’Angelo, G. Glycosphingolipid–Protein Interaction in Signal Transduction. Int. J. Mol. Sci. 2016, 17, 1732. [Google Scholar] [CrossRef]
- Schiffmann, S.; Sandner, J.; Birod, K.; Wobst, I.; Angioni, C.; Ruckhäberle, E.; Kaufmann, M.; Ackermann, H.; Lötsch, J.; Schmidt, H.; et al. Ceramide Synthases and Ceramide Levels Are Increased in Breast Cancer Tissue. Carcinogenesis 2009, 30, 745–752. [Google Scholar] [CrossRef]
- Moro, K.; Kawaguchi, T.; Tsuchida, J.; Gabriel, E.; Qi, Q.; Yan, L.; Wakai, T.; Takabe, K.; Nagahashi, M. Ceramide species are elevated in human breast cancer and are associated with less aggressiveness. Oncotarget 2018, 9, 19874–19890. [Google Scholar] [CrossRef] [PubMed]
- Nagahashi, M.; Tsuchida, J.; Moro, K.; Hasegawa, M.; Tatsuda, K.; Woelfel, I.A.; Takabe, K.; Wakai, T. High levels of sphingolipids in human breast cancer. J. Surg. Res. 2016, 204, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Bhadwal, P.; Dahiya, D.; Shinde, D.; Vaiphei, K.; Math, R.G.H.; Randhawa, V.; Agnihotri, N. LC-HRMS based approach to identify novel sphingolipid biomarkers in breast cancer patients. Sci. Rep. 2020, 10, 4668. [Google Scholar] [CrossRef] [PubMed]
- Pakiet, A.; Sikora, K.; Kobiela, J.; Rostkowska, O.; Mika, A.; Sledzinski, T. Alterations in complex lipids in tumor tissue of patients with colorectal cancer. Lipids Health Dis. 2021, 20, 85. [Google Scholar] [CrossRef]
- Rohrbach, T.D.; Boyd, A.E.; Grizzard, P.J.; Spiegel, S.; Allegood, J.; Lima, S. A simple method for sphingolipid analysis of tissues embedded in optimal cutting temperature compound. J. Lipid Res. 2020, 61, 953–967. [Google Scholar] [CrossRef]
- Boyd, A.E.; Allegood, J.; Lima, S. Preparation of Human Tissues Embedded in Optimal Cutting Temperature Compound for Mass Spectrometry Analysis. J. Vis. Exp. 2021, 170, e62552. [Google Scholar]
- Karahatay, S.; Thomas, K.; Koybasi, S.; Senkal, C.E.; ElOjeimy, S.; Liu, X.; Bielawski, J.; Day, T.A.; Gillespie, M.B.; Sinha, D.; et al. Clinical relevance of ceramide metabolism in the pathogenesis of human head and neck squamous cell carcinoma (HNSCC): Attenuation of C18-ceramide in HNSCC tumors correlates with lymphovascular invasion and nodal metastasis. Cancer Lett. 2007, 256, 101–111. [Google Scholar] [CrossRef]
- Knapp, P.; Baranowski, M.; Knapp, M.; Zabielski, P.; Błachnio-Zabielska, A.U.; Górski, J. Altered sphingolipid metabolism in human endometrial cancer. Prostaglandins Other Lipid Mediat. 2010, 92, 62–66. [Google Scholar] [CrossRef]
- Li, Z.; Guan, M.; Lin, Y.; Cui, X.; Zhang, Y.; Zhao, Z.; Zhu, J. Aberrant Lipid Metabolism in Hepatocellular Carcinoma Revealed by Liver Lipidomics. Int. J. Mol. Sci. 2017, 18, 2550. [Google Scholar] [CrossRef]
- Chatterjee, S.B.; Hou, J.; Bandaru, V.V.R.; Pezhouh, M.K.; Mannan, A.A.S.R.; Sharma, R. Lactosylceramide Synthase Β-1,4-Galt-V: A Novel Target for the Diagnosis and Therapy of Human Colorectal Cancer. Biochem. Biophys. Res. Commun. 2019, 508, 380–386. [Google Scholar] [CrossRef]
- Janneh, A.H.; Ogretmen, B. Targeting Sphingolipid Metabolism as a Therapeutic Strategy in Cancer Treatment. Cancers 2022, 14, 2183. [Google Scholar] [CrossRef] [PubMed]
- Ogretmen, B. Sphingolipid metabolism in cancer signalling and therapy. Nat. Rev. Cancer 2017, 18, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Li, R.Z.; Wang, X.R.; Wang, J.; Xie, C.; Wang, X.X.; Pan, H.D.; Meng, W.Y.; Liang, T.L.; Li, J.X.; Yan, P.Y.; et al. The Key Role of Sphingolipid Metabolism in Cancer: New Therapeutic Targets, Diagnostic and Prognostic Values, and Anti-Tumor Immunotherapy Resistance. Front. Oncol. 2022, 12, 941643. [Google Scholar] [CrossRef]
- Cronin, K.A.; Scott, S.; Firth, A.U.; Sung, H.; Henley, S.J.; Sherman, R.L.; Siegel, R.L.; Anderson, R.N.; Kohler, B.A.; Benard, V.B.; et al. Annual Report to the Nation on the Status of Cancer, Part 1: National Cancer Statistics. Cancer 2022, 128, 4251–4284. [Google Scholar] [CrossRef] [PubMed]
- Whetstone, S.; Burke, W.; Sheth, S.S.; Brooks, R.; Cavens, A.; Huber-Keener, K.; Scott, D.M.; Worly, B.; Chelmow, D. Health Disparities in Uterine Cancer: Report from the Uterine Cancer Evidence Review Conference. Obstet. Gynecol. 2022, 139, 645–659. [Google Scholar] [CrossRef] [PubMed]
- Doll, K.M.; Khor, S.; Odem-Davis, K.; He, H.; Wolff, E.M.; Flum, D.R.; Ramsey, S.D.; Goff, B.A. Role of bleeding recognition and evaluation in Black-White disparities in endometrial cancer. Am. J. Obstet. Gynecol. 2018, 219, 593.e1–593.e14. [Google Scholar] [CrossRef]
- Williams, P.A.; Zaidi, S.K.; Sengupta, R. AACR Cancer Disparities Progress Report 2022. Cancer Epidemiol. Biomark. Prev. 2022, 31, 1249–1250. [Google Scholar] [CrossRef]
- Cancer Facts & Figures for African American/Black People 2022–2024; American Cancer Society: Atlanta, GA, USA, 2022.
- Zavala, V.A.; Bracci, P.M.; Carethers, J.M.; Carvajal-Carmona, L.; Coggins, N.B.; Cruz-Correa, M.R.; Davis, M.; de Smith, A.J.; Dutil, J.; Figueiredo, J.C.; et al. Cancer health disparities in racial/ethnic minorities in the United States. Br. J. Cancer 2021, 124, 315–332. [Google Scholar] [CrossRef]
- Fatumo, S.; Chikowore, T.; Choudhury, A.; Ayub, M.; Martin, A.R.; Kuchenbaecker, K. A roadmap to increase diversity in genomic studies. Nat. Med. 2022, 28, 243–250. [Google Scholar] [CrossRef]
- Nazha, B.; Mishra, M.; Pentz, R.; Owonikoko, T.K. Enrollment of Racial Minorities in Clinical Trials: Old Problem Assumes New Urgency in the Age of Immunotherapy. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 3–10. [Google Scholar] [CrossRef]
- Borrell, L.N.; Elhawary, J.R.; Fuentes-Afflick, E.; Witonsky, J.; Bhakta, N.; Wu, A.H.B.; Bibbins-Domingo, K.; Rodríguez-Santana, J.R.; Lenoir, M.A.; Gavin, J.R., 3rd; et al. Race and Genetic Ancestry in Medicine—A Time for Reckoning with Racism. N. Engl. J. Med. 2021, 384, 474–480. [Google Scholar] [CrossRef]
- Shaner, R.L.; Allegood, J.C.; Park, H.; Wang, E.; Kelly, S.; Haynes, C.A.; Sullards, M.C.; Merrill, A.H., Jr. Quantitative analysis of sphingolipids for lipidomics using triple quadrupole and quadrupole linear ion trap mass spectrometers. J. Lipid Res. 2009, 50, 1692–1707. [Google Scholar] [CrossRef]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, B.; Sun, Z. Spectrum of EGFR aberrations and potential clinical implications: Insights from integrative pan-cancer analysis. Cancer Commun. 2020, 40, 43–59. [Google Scholar] [CrossRef]
- Chen, F.; Zhang, Y.; Gibbons, D.L.; Deneen, B.; Kwiatkowski, D.J.; Ittmann, M.; Creighton, C.J. Pan-Cancer Molecular Classes Transcending Tumor Lineage across 32 Cancer Types, Multiple Data Platforms, and over 10,000 Cases. Clin. Cancer Res. 2018, 24, 2182–2193. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Mitchell, K.A.; Zingone, A.; Bowman, E.; Sinha, N.; Schäffer, A.A.; Lee, J.S.; Ruppin, E.; Ryan, B.M. Higher prevalence of homologous recombination deficiency in tumors from African Americans versus European Americans. Nat. Cancer 2020, 1, 112–121. [Google Scholar] [CrossRef]
- Ding, Y.C.; Adamson, A.W.; Steele, L.; Bailis, A.M.; John, E.M.; Tomlinson, G.; Neuhausen, S.L. Discovery of mutations in homologous recombination genes in African-American women with breast cancer. Fam. Cancer 2018, 17, 187–195. [Google Scholar] [CrossRef]
- Deveaux, A.E.; Allen, T.A.; Al Abo, M.; Qin, X.; Zhang, D.; Patierno, B.M.; Gu, L.; Gray, J.E.; Pecot, C.V.; Dressman, H.K.; et al. RNA splicing and aggregate gene expression differences in lung squamous cell carcinoma between patients of West African and European ancestry. Lung Cancer 2021, 153, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Yadav, S.; Anbalagan, M.; Baddoo, M.; Chellamuthu, V.K.; Mukhopadhyay, S.; Woods, C.; Jiang, W.; Moroz, K.; Flemington, E.K.; Makridakis, N. Somatic mutations in the DNA repairome in prostate cancers in African Americans and Caucasians. Oncogene 2020, 39, 4299–4311. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, K.A.; Nichols, N.; Tang, W.; Walling, J.; Stevenson, H.; Pineda, M.; Stefanescu, R.; Edelman, D.C.; Girvin, A.T.; Zingone, A.; et al. Recurrent PTPRT/JAK2 mutations in lung adenocarcinoma among African Americans. Nat. Commun. 2019, 10, 5735. [Google Scholar] [CrossRef]
- Bauml, J.; Mick, R.; Zhang, Y.; Watt, C.D.; Vachani, A.; Aggarwal, C.; Evans, T.; Langer, C. Frequency of EGFR and KRAS mutations in patients with non small cell lung cancer by racial background: Do disparities exist? Lung Cancer 2013, 81, 347–353. [Google Scholar] [CrossRef]
- Steuer, C.E.; Behera, M.; Berry, L.; Kim, S.; Rossi, M.; Sica, G.; Owonikoko, T.K.; Johnson, B.E.; Kris, M.G.; Bunn, P.A.; et al. Role of race in oncogenic driver prevalence and outcomes in lung adenocarcinoma: Results from the Lung Cancer Mutation Consortium. Cancer 2016, 122, 766–772. [Google Scholar] [CrossRef] [PubMed]
- Krishnaswamy, S.; Kanteti, R.; Duke-Cohan, J.S.; Loganathan, S.; Liu, W.; Ma, P.C.; Sattler, M.; Singleton, P.A.; Ramnath, N.; Innocenti, F.; et al. Ethnic Differences and Functional Analysis of MET Mutations in Lung Cancer. Clin. Cancer Res. 2009, 15, 5714–5723. [Google Scholar] [CrossRef]
- Kang, M.; Shen, X.J.; Kim, S.; Araujo-Perez, F.; Galanko, J.A.; Martin, C.F.; Sandler, R.S.; Keku, T.O. Somatic gene mutations in African Americans may predict worse outcomes in colorectal cancer. Cancer Biomark. 2013, 13, 359–366. [Google Scholar] [CrossRef]
- Bergers, G.; Fendt, S.-M. The metabolism of cancer cells during metastasis. Nat. Rev. Cancer 2021, 21, 162–180. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Reyes, I.; Chandel, N.S. Cancer metabolism: Looking forward. Nat. Rev. Cancer 2021, 21, 669–680. [Google Scholar] [CrossRef]
- Patel, Y.M.; Park, S.L.; Carmella, S.G.; Paiano, V.; Olvera, N.; Stram, D.O.; Haiman, C.A.; Le Marchand, L.; Hecht, S.S. Metabolites of the Polycyclic Aromatic Hydrocarbon Phenanthrene in the Urine of Cigarette Smokers from Five Ethnic Groups with Differing Risks for Lung Cancer. PLoS ONE 2016, 11, e0156203. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.E.; Park, S.-S.L.; Thompson, E.F.; Wilkens, L.R.; Patel, Y.; Stram, D.O.; Le Marchand, L. Nicotine N-glucuronidation relative to N-oxidation and C-oxidation and UGT2B10 genotype in five ethnic/racial groups. Carcinogenesis 2014, 35, 2526–2533. [Google Scholar] [CrossRef] [PubMed]
- Dator, R.; Villalta, P.W.; Thomson, N.; Jensen, J.; Hatsukami, D.K.; Stepanov, I.; Warth, B.; Balbo, S. Metabolomics Profiles of Smokers from Two Ethnic Groups with Differing Lung Cancer Risk. Chem. Res. Toxicol. 2020, 33, 2087–2098. [Google Scholar] [CrossRef]
- Ross, K.C.; Gubner, N.R.; Tyndale, R.F.; Hawk, L.W.; Lerman, C.; George, T.P.; Cinciripini, P.; Schnoll, R.A.; Benowitz, N.L. Racial differences in the relationship between rate of nicotine metabolism and nicotine intake from cigarette smoking. Pharmacol. Biochem. Behav. 2016, 148, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Chenoweth, M.J.; Ware, J.J.; Zhu, A.Z.X.; Cole, C.B.; Cox, L.S.; Nollen, N.; Ahluwalia, J.S.; Benowitz, N.L.; Schnoll, R.A.; Hawk, L.W.; et al. Genome-wide association study of a nicotine metabolism biomarker in African American smokers: Impact of chromosome 19 genetic influences. Addiction 2017, 113, 509–523. [Google Scholar] [CrossRef] [PubMed]
- Park, S.L.; Tiirikainen, M.I.; Patel, Y.M.; Wilkens, L.R.; Stram, D.O.; Le Marchand, L.; Murphy, S.E. Genetic determinants of CYP2A6 activity across racial/ethnic groups with different risks of lung cancer and effect on their smoking intensity. Carcinogenesis 2016, 37, 269–279. [Google Scholar] [CrossRef]
- Sun, Y.V.; Smith, A.K.; Conneely, K.N.; Chang, Q.; Li, W.; Lazarus, A.; Smith, J.; Almli, L.; Binder, E.B.; Klengel, T.; et al. Epigenomic association analysis identifies smoking-related DNA methylation sites in African Americans. Hum. Genet. 2013, 132, 1027–1037. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.E.; Park, S.L.; Balbo, S.; Haiman, C.A.; Hatsukami, D.K.; Patel, Y.; Peterson, L.A.; Stepanov, I.; Stram, D.O.; Tretyakova, N.; et al. Tobacco biomarkers and genetic/epigenetic analysis to investigate ethnic/racial differences in lung cancer risk among smokers. npj Precis. Oncol. 2018, 2, 17. [Google Scholar] [CrossRef]
- Kanaan, Y.M.; Sampey, B.P.; Beyene, D.; Esnakula, A.K.; Naab, T.J.; Ricks-Santi, L.J.; Dasi, S.; Day, A.; Blackman, K.W.; Frederick, W.; et al. Metabolic profile of triple-negative breast cancer in African-American women reveals potential biomarkers of aggressive disease. Cancer Genom.-Proteom. 2014, 11, 29–294. [Google Scholar]
- Vantaku, V.; Donepudi, S.R.; Piyarathna, D.W.B.; Amara, C.S.; Ambati, C.S.R.; Tang, W.; Putluri, V.; Chandrashekar, D.S.; Varambally, S.; Terris, M.K.; et al. Large-scale profiling of serum metabolites in African American and European American patients with bladder cancer reveals metabolic pathways associated with patient survival. Cancer 2019, 125, 921–932. [Google Scholar] [CrossRef]
- Zhao, H.; Shen, J.; Moore, S.C.; Ye, Y.; Wu, X.; Esteva, F.J.; Tripathy, D.; Chow, W.H. Breast Cancer Risk in Relation to Plasma Metabolites among Hispanic and African American Women. Breast Cancer Res. Treat. 2019, 176, 687–696. [Google Scholar] [CrossRef]
- Shen, J.; Yan, L.; Liu, S.; Ambrosone, C.B.; Zhao, H. Plasma Metabolomic Profiles in Breast Cancer Patients and Healthy Controls: By Race and Tumor Receptor Subtypes. Transl. Oncol. 2013, 6, 757–765. [Google Scholar] [CrossRef]
- Gohlke, J.H.; Lloyd, S.M.; Basu, S.; Putluri, V.; Vareed, S.K.; Rasaily, U.; Piyarathna, D.W.B.; Fuentes, H.; Rajendiran, T.M.; Dorsey, T.H.; et al. Methionine-Homocysteine Pathway in African-American Prostate Cancer. JNCI Cancer Spectr. 2019, 3, pkz019. [Google Scholar] [CrossRef]
- Rose, D.P.; Haffner, S.M.; Baillargeon, J. Adiposity, the Metabolic Syndrome, and Breast Cancer in African-American and White American Women. Endocr. Rev. 2007, 28, 763–777. [Google Scholar] [CrossRef]
- Aminov, Z.; Haase, R.; Olson, J.R.; Pavuk, M.; Carpenter, D.O. Racial differences in levels of serum lipids and effects of exposure to persistent organic pollutants on lipid levels in residents of Anniston, Alabama. Environ. Int. 2014, 73, 216–223. [Google Scholar] [CrossRef]
- Zhou, X.; Mei, H.; Agee, J.; Brown, T.; Mao, J. Racial differences in distribution of fatty acids in prostate cancer and benign prostatic tissues. Lipids Health Dis. 2019, 18, 189. [Google Scholar] [CrossRef]
- Purwaha, P.; Gu, F.; Piyarathna, D.W.B.; Rajendiran, T.; Ravindran, A.; Omilian, A.R.; Jiralerspong, S.; Das, G.; Morrison, C.; Ambrosone, C.; et al. Unbiased Lipidomic Profiling of Triple-Negative Breast Cancer Tissues Reveals the Association of Sphingomyelin Levels with Patient Disease-Free Survival. Metabolites 2018, 8, 41. [Google Scholar] [CrossRef] [PubMed]
- Ho, Q.W.C.; Zheng, X.; Ali, Y. Ceramide Acyl Chain Length and Its Relevance to Intracellular Lipid Regulation. Int. J. Mol. Sci. 2022, 23, 9697. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-R.; Lee, E.-J.; Shin, K.-O.; Kim, M.H.; Pewzner-Jung, Y.; Lee, Y.-M.; Park, J.-W.; Futerman, A.H.; Park, W.-J. Hepatic triglyceride accumulation via endoplasmic reticulum stress-induced SREBP-1 activation is regulated by ceramide synthases. Exp. Mol. Med. 2019, 51, 1–16. [Google Scholar] [CrossRef]
- Hartmann, D.; Wegner, M.-S.; Wanger, R.A.; Ferreirós, N.; Schreiber, Y.; Lucks, J.; Schiffmann, S.; Geisslinger, G.; Grösch, S. The equilibrium between long and very long chain ceramides is important for the fate of the cell and can be influenced by co-expression of CerS. Int. J. Biochem. Cell Biol. 2013, 45, 1195–1203. [Google Scholar] [CrossRef] [PubMed]
- Mesicek, J.; Lee, H.; Feldman, T.; Jiang, X.; Skobeleva, A.; Berdyshev, E.V.; Haimovitz-Friedman, A.; Fuks, Z.; Kolesnick, R. Ceramide synthases 2, 5, and 6 confer distinct roles in radiation-induced apoptosis in HeLa cells. Cell. Signal. 2010, 22, 1300–1307. [Google Scholar] [CrossRef]
- Zheng, X.; Ho, Q.W.C.; Chua, M.; Stelmashenko, O.; Yeo, X.Y.; Muralidharan, S.; Torta, F.; Chew, E.G.Y.; Lian, M.M.; Foo, J.N.; et al. Destabilization of β Cell FIT2 by saturated fatty acids alter lipid droplet numbers and contribute to ER stress and diabetes. Proc. Natl. Acad. Sci. USA 2022, 119, e2113074119. [Google Scholar] [CrossRef] [PubMed]
- Manukyan, L.; Ubhayasekera, S.J.K.A.; Bergquist, J.; Sargsyan, E.; Bergsten, P. Palmitate-Induced Impairments of β-Cell Function Are Linked with Generation of Specific Ceramide Species via Acylation of Sphingosine. Endocrinology 2015, 156, 802–812. [Google Scholar] [CrossRef]
- Aflaki, E.; Doddapattar, P.; Radović, B.; Povoden, S.; Kolb, D.; Vujić, N.; Wegscheider, M.; Koefeler, H.; Hornemann, T.; Graier, W.F.; et al. C16 Ceramide Is Crucial for Triacylglycerol-Induced Apoptosis in Macrophages. Cell Death Dis. 2012, 3, e280. [Google Scholar] [CrossRef]
- Rudd, A.K.; Devaraj, N.K. Traceless synthesis of ceramides in living cells reveals saturation-dependent apoptotic effects. Proc. Natl. Acad. Sci. USA 2018, 115, 7485–7490. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, V.; Perera, M.N.; Colombini, D.; Datskovskiy, D.; Chadha, K.; Colombini, M. Ceramide and activated Bax act synergistically to permeabilize the mitochondrial outer membrane. Apoptosis 2010, 15, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Fekry, B.; Jeffries, K.A.; Esmaeilniakooshkghazi, A.; Szulc, Z.M.; Knagge, K.J.; Kirchner, D.R.; Horita, D.A.; Krupenko, S.A.; Krupenko, N.I. C16-ceramide is a natural regulatory ligand of p53 in cellular stress response. Nat. Commun. 2018, 9, 4149. [Google Scholar] [CrossRef] [PubMed]
- Stiban, J.; Perera, M. Very long chain ceramides interfere with C16-ceramide-induced channel formation: A plausible mechanism for regulating the initiation of intrinsic apoptosis. Biochim. Biophys. Acta 2015, 1848, 561–567. [Google Scholar] [CrossRef]
- Reza, S.; Ugorski, M.; Suchański, J. Glucosylceramide and galactosylceramide, small glycosphingolipids with significant impact on health and disease. Glycobiology 2021, 31, 1416–1434. [Google Scholar] [CrossRef]
- Morad, S.A.F.; Cabot, M.C. Chapter Nine—The Onus of Sphingolipid Enzymes in Cancer Drug Resistance. In Advances in Cancer Research; Chalfant, C.E., Fisher, P.B., Eds.; Academic Press: Cambridge, MA, USA, 2018; pp. 235–263. [Google Scholar]
- Liu, J.C.; Egleston, B.L.; Blackman, E.; Ragin, C. Racial Survival Disparities in Head and Neck Cancer Clinical Trials. J. Natl. Cancer Inst. 2023, 115, 288–294. [Google Scholar] [CrossRef]
- Xu, S.; Zhang, S.; Chen, C.; Yan, J.; Cai, M.; Zhu, X.; Gu, J. Over-expression of beta-1,4-galactosyltransferase V increases the growth of astrocytoma cell line. J. Exp. Clin. Cancer Res. 2002, 21, 409–414. [Google Scholar]
- Jeong, H.Y.; Park, S.; Kim, H.; Moon, S.; Lee, S.; Lee, S.H.; Kim, S. B3GNT5 is a novel marker correlated with stem-like phenotype and poor clinical outcome in human gliomas. CNS Neurosci. Ther. 2020, 26, 1147–1154. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, B.; Xu, G.; Han, C.; Xing, G. Lncrna Mir4435-2hg Promotes the Progression of Liver Cancer by Upregulating B3gnt5 Expression. Mol. Med. Rep. 2022, 25, 38. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EEC | LUAD | HCC | HNSCC | COAD | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| AA (n = 10) | NHW (n = 12) | AA (Unin., n = 12; Tumor, n = 14) | NHW (Unin., n = 22; Tumor, n = 26) | AA (n = 10) | NHW (n = 11) | AA (Unin., n = 12; Tumor, n = 11) | NHW (n = 12) | AA (n = 30) | NHW (n = 24) | |
| Sex | Female | Male | Male | Male | Male | |||||
| Mean age (SD) | 60.7 (9.1) | 61.3 (14.4) | 60.8 (6.7) | 69.6 (9.1) | 62.2 (5.5) | 64.5 (16.8) | 62 (9.5) | 58.6 (11.6) | 63.5 (13.6) | 65.7 (10.5) |
| Minimum–maximum | 49–79 | 30–82 | 52–75 | 47–83 | 53–73 | 19–78 | 39–75 | 44–78 | 46–94 | 41–86 |
| p-value | 0.9176 | 0.0018 | 0.6901 | 0.4345 | 0.5214 | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boyd, A.E.; Grizzard, P.J.; Hylton Rorie, K.; Lima, S. Lipidomic Profiling Reveals Biological Differences between Tumors of Self-Identified African Americans and Non-Hispanic Whites with Cancer. Cancers 2023, 15, 2238. https://doi.org/10.3390/cancers15082238
Boyd AE, Grizzard PJ, Hylton Rorie K, Lima S. Lipidomic Profiling Reveals Biological Differences between Tumors of Self-Identified African Americans and Non-Hispanic Whites with Cancer. Cancers. 2023; 15(8):2238. https://doi.org/10.3390/cancers15082238
Chicago/Turabian StyleBoyd, April E., Pamela J. Grizzard, Katherine Hylton Rorie, and Santiago Lima. 2023. "Lipidomic Profiling Reveals Biological Differences between Tumors of Self-Identified African Americans and Non-Hispanic Whites with Cancer" Cancers 15, no. 8: 2238. https://doi.org/10.3390/cancers15082238
APA StyleBoyd, A. E., Grizzard, P. J., Hylton Rorie, K., & Lima, S. (2023). Lipidomic Profiling Reveals Biological Differences between Tumors of Self-Identified African Americans and Non-Hispanic Whites with Cancer. Cancers, 15(8), 2238. https://doi.org/10.3390/cancers15082238