
CT Image-Based Biopsy to Aid Prediction of HOPX Expression Status and Prognosis for Non-Small Cell Lung Cancer Patients

Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
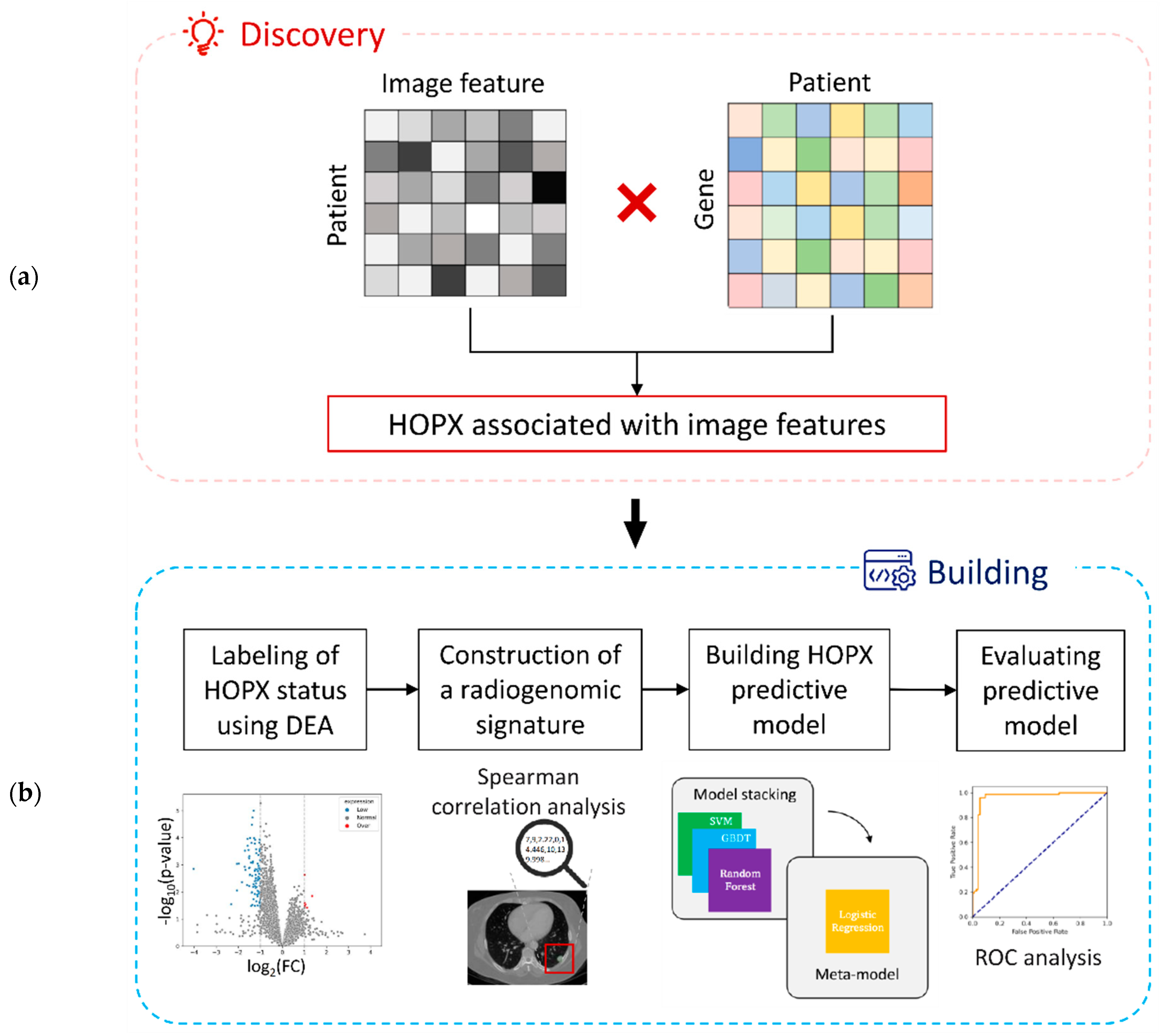
2.1. Study Worflow
2.2. Clinical Cases
2.3. Calculation of Image Features
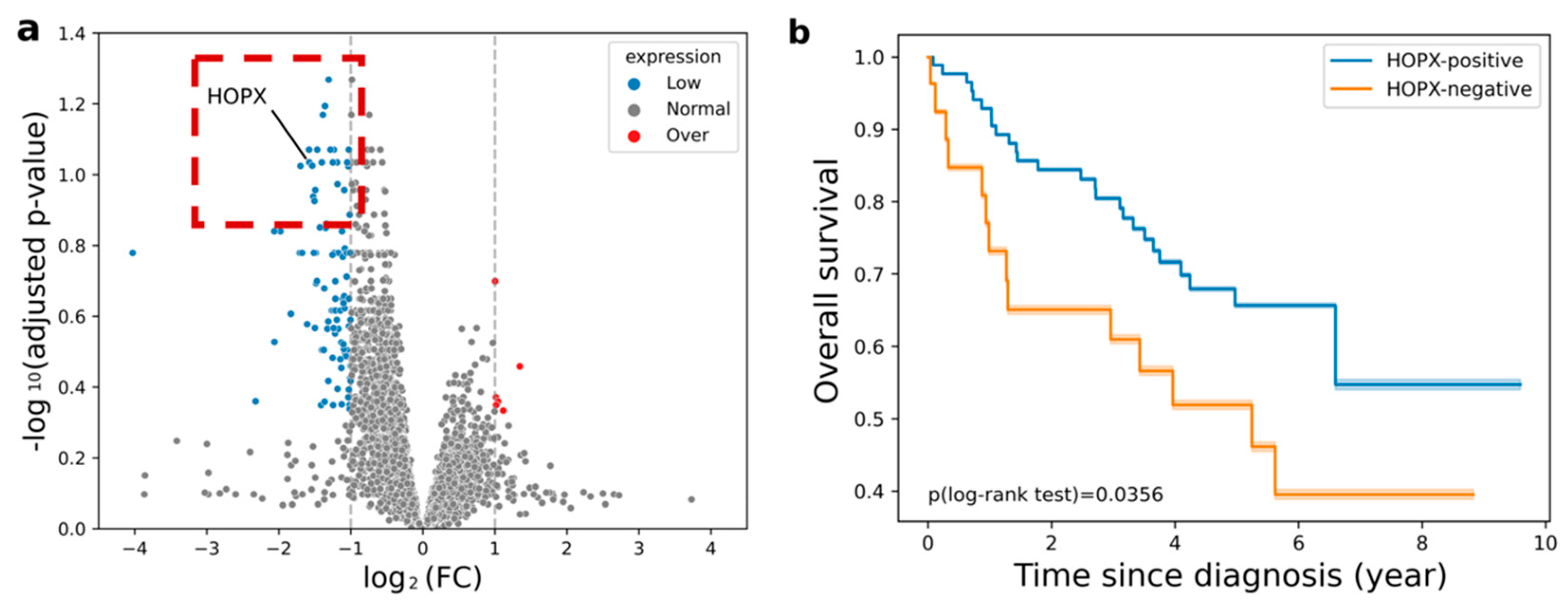
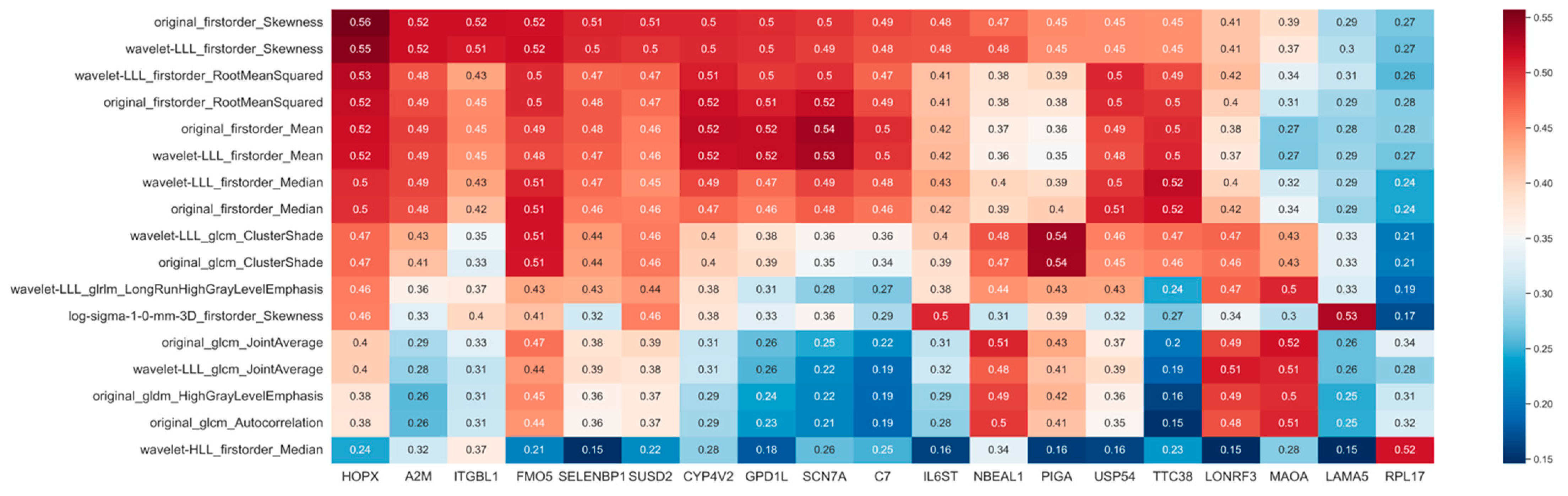
2.4. Exploration of Significant Genes Associated with Image Features
2.5. Labeling of HOPX Status Using Differential Expression Analysis
2.6. Construction of the Radiogenomic Signature
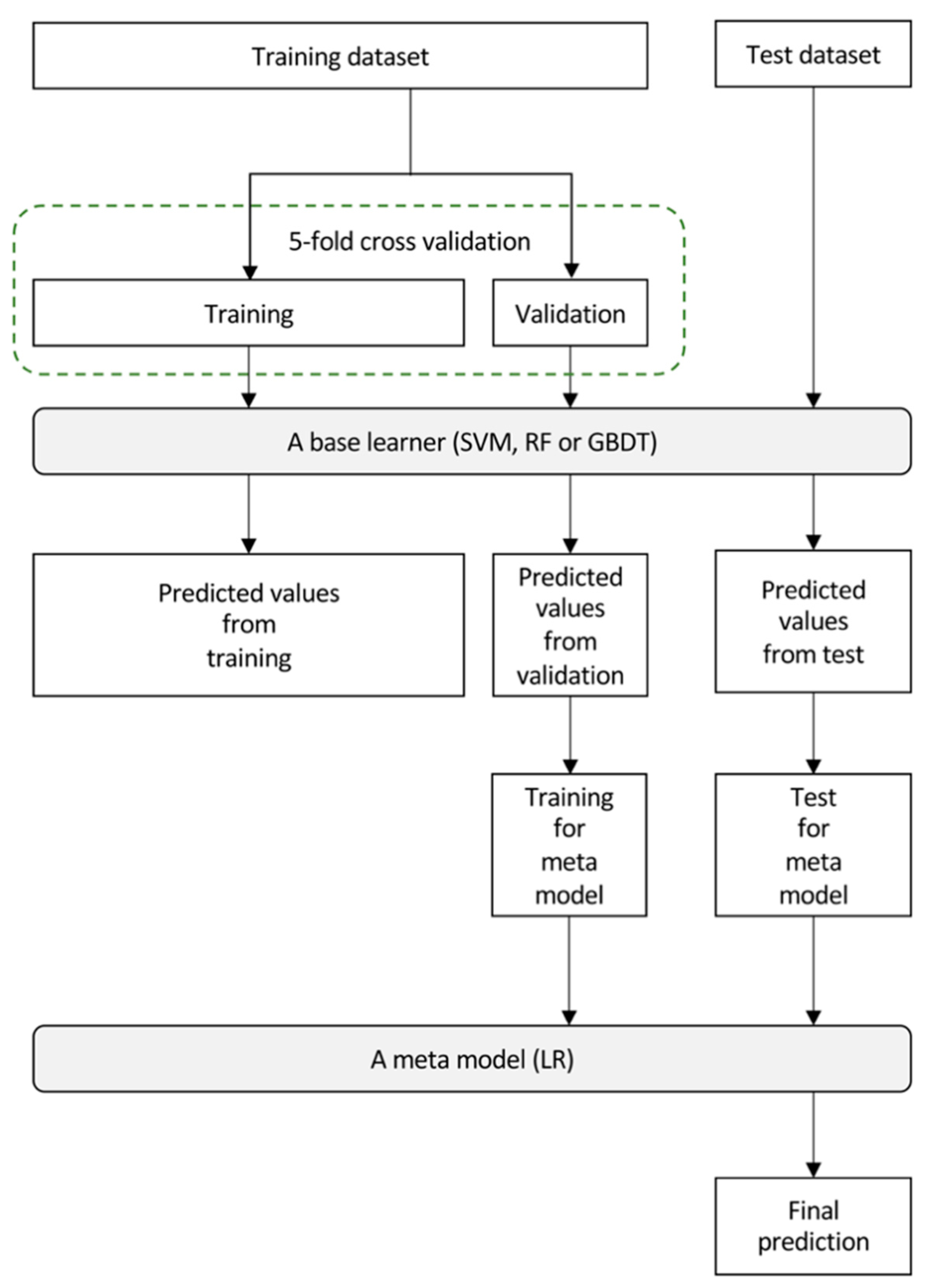
2.7. Building of the Imaging Biopsy Model
2.8. Evaluation of the Imaging Biopsy Model
3. Results
3.1. Radiogenomic Features
3.2. Prediction Power of HOPX Expression Status and Prognosis
3.3. Final Image Biopsy with Radiogenomic Signature
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive Molecular Profiling of Lung Adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Byron, S.A.; Van Keuren-Jensen, K.R.; Engelthaler, D.M.; Carpten, J.D.; Craig, D.W. Translating RNA Sequencing into Clinical Diagnostics: Opportunities and Challenges. Nat. Rev. Genet. 2016, 17, 257–271. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Fillmore, C.M.; Hammerman, P.S.; Kim, C.F.; Wong, K.-K. Non-Small-Cell Lung Cancers: A Heterogeneous Set of Diseases. Nat. Rev. Cancer 2014, 14, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Lambin, P.; Rios-Velazquez, E.; Leijenaar, R.; Carvalho, S.; van Stiphout, R.G.P.M.; Granton, P.; Zegers, C.M.L.; Gillies, R.; Boellard, R.; Dekker, A.; et al. Radiomics: Extracting More Information from Medical Images Using Advanced Feature Analysis. Eur. J. Cancer 2012, 48, 441–446. [Google Scholar] [CrossRef]
- Aerts, H.J.W.L.; Velazquez, E.R.; Leijenaar, R.T.H.; Parmar, C.; Grossmann, P.; Carvalho, S.; Cavalho, S.; Bussink, J.; Monshouwer, R.; Haibe-Kains, B.; et al. Decoding Tumour Phenotype by Noninvasive Imaging Using a Quantitative Radiomics Approach. Nat. Commun. 2014, 5, 4006. [Google Scholar] [CrossRef]
- Yip, S.S.F.; Liu, Y.; Parmar, C.; Li, Q.; Liu, S.; Qu, F.; Ye, Z.; Gillies, R.J.; Aerts, H.J.W.L. Associations between Radiologist-Defined Semantic and Automatically Computed Radiomic Features in Non-Small Cell Lung Cancer. Sci. Rep. 2017, 7, 3519. [Google Scholar] [CrossRef]
- Valentin, J. The 2007 Recommendations of the International Commission on Radiological Protection; ICRP Publication 103; Elsevier: Oxford, UK, 2007; Volume 37, pp. 1–332. [Google Scholar] [CrossRef]
- Wang, X.; Kong, C.; Xu, W.; Yang, S.; Shi, D.; Zhang, J.; Du, M.; Wang, S.; Bai, Y.; Zhang, T.; et al. Decoding Tumor Mutation Burden and Driver Mutations in Early Stage Lung Adenocarcinoma Using CT-Based Radiomics Signature. Thorac. Cancer 2019, 10, 1904–1912. [Google Scholar] [CrossRef]
- Zou, J.; Lv, T.; Zhu, S.; Lu, Z.; Shen, Q.; Xia, L.; Wu, J.; Song, Y.; Liu, H. Computed Tomography and Clinical Features Associated with Epidermal Growth Factor Receptor Mutation Status in Stage I/II Lung Adenocarcinoma. Thorac. Cancer 2017, 8, 260–270. [Google Scholar] [CrossRef]
- Zhou, M.; Leung, A.; Echegaray, S.; Gentles, A.; Shrager, J.B.; Jensen, K.C.; Berry, G.J.; Plevritis, S.K.; Rubin, D.L.; Napel, S.; et al. Non-Small Cell Lung Cancer Radiogenomics Map Identifies Relationships between Molecular and Imaging Phenotypes with Prognostic Implications. Radiology 2018, 286, 307–315. [Google Scholar] [CrossRef]
- Bodalal, Z.; Trebeschi, S.; Nguyen-Kim, T.D.L.; Schats, W.; Beets-Tan, R. Radiogenomics: Bridging Imaging and Genomics. Abdom. Radiol. 2019, 44, 1960–1984. [Google Scholar] [CrossRef] [PubMed]
- Nair, J.K.R.; Saeed, U.A.; McDougall, C.C.; Sabri, A.; Kovacina, B.; Raidu, B.V.S.; Khokhar, R.A.; Probst, S.; Hirsh, V.; Chankowsky, J.; et al. Radiogenomic Models Using Machine Learning Techniques to Predict EGFR Mutations in Non-Small Cell Lung Cancer. Can. Assoc. Radiol. J. 2021, 72, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Kirienko, M.; Sollini, M.; Corbetta, M.; Voulaz, E.; Gozzi, N.; Interlenghi, M.; Gallivanone, F.; Castiglioni, I.; Asselta, R.; Duga, S.; et al. Radiomics and Gene Expression Profile to Characterise the Disease and Predict Outcome in Patients with Lung Cancer. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 3643–3655. [Google Scholar] [CrossRef] [PubMed]
- Ninomiya, K.; Arimura, H.; Chan, W.Y.; Tanaka, K.; Mizuno, S.; Muhammad Gowdh, N.F.; Yaakup, N.A.; Liam, C.-K.; Chai, C.-S.; Ng, K.H. Robust Radiogenomics Approach to the Identification of EGFR Mutations among Patients with NSCLC from Three Different Countries Using Topologically Invariant Betti Numbers. PLoS ONE 2021, 16, e0244354. [Google Scholar] [CrossRef] [PubMed]
- Gierada, D.S.; Black, W.C.; Chiles, C.; Pinsky, P.F.; Yankelevitz, D.F. Low-Dose CT Screening for Lung Cancer: Evidence from 2 Decades of Study. Radiol. Imaging Cancer 2020, 2, e190058. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Li, M.; Zhang, H.; Hua, S.; Meng, F.; Yang, H.; Li, X.; Cao, D. A Novel Radiomic Nomogram for Predicting Epidermal Growth Factor Receptor Mutation in Peripheral Lung Adenocarcinoma. Phys. Med. Biol. 2020, 65, 055012. [Google Scholar] [CrossRef]
- Qi, Y.; Zhao, T.; Han, M. The Application of Radiomics in Predicting Gene Mutations in Cancer. Eur. Radiol. 2022, 32, 4014–4024. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, Q.; Zhu, L.; Xie, S.; Tu, L.; Yang, Y.; Wu, K.; Zhao, Y.; Wang, Y.; Xu, Y.; et al. SERPINE2/PN-1 Regulates the DNA Damage Response and Radioresistance by Activating ATM in Lung Cancer. Cancer Lett. 2022, 524, 268–283. [Google Scholar] [CrossRef]
- Scott, J.G.; Berglund, A.; Schell, M.J.; Mihaylov, I.; Fulp, W.J.; Yue, B.; Welsh, E.; Caudell, J.J.; Ahmed, K.; Strom, T.S.; et al. A Genome-Based Model for Adjusting Radiotherapy Dose (GARD): A Retrospective, Cohort-Based Study. Lancet Oncol. 2017, 18, 202–211. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, L.; Cui, T.; Pacyna-Gengelbach, M.; Petersen, I. HOPX Is Methylated and Exerts Tumour-Suppressive Function through Ras-Induced Senescence in Human Lung Cancer. J. Pathol. 2015, 235, 397–407. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, W. The Role of HOPX in Normal Tissues and Tumor Progression. Biosci. Rep. 2020, 40, BSR20191953. [Google Scholar] [CrossRef]
- Mariotto, A.; Pavlova, O.; Park, H.-S.; Huber, M.; Hohl, D. HOPX: The Unusual Homeodomain-Containing Protein. J. Investig. Dermatol. 2016, 136, 905–911. [Google Scholar] [CrossRef]
- Cheung, W.K.C.; Zhao, M.; Liu, Z.; Stevens, L.E.; Cao, P.D.; Fang, J.E.; Westbrook, T.F.; Nguyen, D.X. Control of Alveolar Differentiation by the Lineage Transcription Factors GATA6 and HOPX Inhibits Lung Adenocarcinoma Metastasis. Cancer Cell 2013, 23, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Bakr, S.; Gevaert, O.; Echegaray, S.; Ayers, K.; Zhou, M.; Shafiq, M.; Zheng, H.; Benson, J.A.; Zhang, W.; Leung, A.N.C.; et al. A Radiogenomic Dataset of Non-Small Cell Lung Cancer. Sci. Data 2018, 5, 180202. [Google Scholar] [CrossRef]
- van Griethuysen, J.J.M.; Fedorov, A.; Parmar, C.; Hosny, A.; Aucoin, N.; Narayan, V.; Beets-Tan, R.G.H.; Fillion-Robin, J.-C.; Pieper, S.; Aerts, H.J.W.L. Computational Radiomics System to Decode the Radiographic Phenotype. Cancer Res. 2017, 77, e104–e107. [Google Scholar] [CrossRef]
- Hemyari, P. Robustness of the Quartiles of Survival Time and Survival Probability. J. Biopharm. Stat. 2000, 10, 299–318. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Auer, P.L.; Doerge, R.W. Statistical Design and Analysis of RNA Sequencing Data. Genetics 2010, 185, 405–416. [Google Scholar] [CrossRef]
- Wasserstein, R.L.; Lazar, N.A. The ASA Statement on P-Values: Context, Process, and Purpose. Am. Stat. 2016, 70, 129–133. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, J.; Yang, G.; Wu, D.; Jiang, L.; Wen, Z.; Li, M. Investigating the Concordance of Gene Ontology Terms Reveals the Intra- and Inter-Platform Reproducibility of Enrichment Analysis. BMC Bioinform. 2013, 14, 143. [Google Scholar] [CrossRef]
- Liang, M.; Chang, T.; An, B.; Duan, X.; Du, L.; Wang, X.; Miao, J.; Xu, L.; Gao, X.; Zhang, L.; et al. A Stacking Ensemble Learning Framework for Genomic Prediction. Front. Genet. 2021, 12, 600040. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Bai, Y.; Garcia, E.A.; Li, S. ADASYN: Adaptive Synthetic Sampling Approach for Imbalanced Learning. In Proceedings of the 2008 IEEE International Joint Conference on Neural Networks (IEEE World Congress on Computational Intelligence), Hong Kong, China, 1–8 June 2008; pp. 1322–1328. [Google Scholar] [CrossRef]
- Chawla, N.V.; Bowyer, K.W.; Hall, L.O.; Kegelmeyer, W.P. SMOTE: Synthetic Minority over-sampling Technique. J. Artif. Intell. Res. 2002, 16, 321–357. Available online: https://www.jair.org/index.php/jair/article/view/10302 (accessed on 1 June 2002). [CrossRef]
- Snoek, J.; Larochelle, H.; Adams, R.P. Practical Bayesian Optimization of Machine Learning Algorithms. In Advances in Neural Information Processing Systems; Curran Associates, Inc: Red Hook, NY, USA, 2012; Volume 25. [Google Scholar]
- Yin, Z.; Gonzales, L.; Kolla, V.; Rath, N.; Zhang, Y.; Lu, M.M.; Kimura, S.; Ballard, P.L.; Beers, M.F.; Epstein, J.A.; et al. Hop Functions Downstream of Nkx2.1 and GATA6 to Mediate HDAC-Dependent Negative Regulation of Pulmonary Gene Expression. Am. J. Physiol. Lung Cell Mol. Physiol. 2006, 291, L191–L199. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Meyerson, M. Hopping between Differentiation States in Lung Adenocarcinoma. Cancer Cell 2013, 23, 707–709. [Google Scholar] [CrossRef]
- Desai, T.J.; Brownfield, D.G.; Krasnow, M.A. Alveolar Progenitor and Stem Cells in Lung Development, Renewal and Cancer. Nature 2014, 507, 190–194. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Pu, Q.; Zhang, Q.; Liu, Y.; Ma, Y.; Yuan, Y.; Liu, L.; Zhu, W. Selenium-Binding Protein 1 Inhibits Malignant Progression and Induces Apoptosis via Distinct Mechanisms in Non-Small-Cell Lung Cancer. Cancer Med. 2022. [Google Scholar] [CrossRef]
- Guo, W.; Shao, F.; Sun, S.; Song, P.; Guo, L.; Xue, X.; Zhang, G.; Zhang, H.; Gao, Y.; Qiu, B.; et al. Loss of SUSD2 Expression Correlates with Poor Prognosis in Patients with Surgically Resected Lung Adenocarcinoma. J. Cancer 2020, 11, 1648–1656. [Google Scholar] [CrossRef]
- Fortin, J.-P.; Cullen, N.; Sheline, Y.I.; Taylor, W.D.; Aselcioglu, I.; Cook, P.A.; Adams, P.; Cooper, C.; Fava, M.; McGrath, P.J.; et al. Harmonization of Cortical Thickness Measurements across Scanners and Sites. Neuroimage 2018, 167, 104–120. [Google Scholar] [CrossRef]
- Chen, T.; Guestrin, C. XGBoost: A Scalable Tree Boosting System. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining; KDD ’16. Association for Computing Machinery: New York, NY, USA, 2016; pp. 785–794. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| All | Training Dataset | Test Dataset | p-Value (Method) | ||
|---|---|---|---|---|---|
| Number of Cases | (n = 116) | (n = 92) | (n = 24) | ||
| Age | <60 | 17 | 12 (70.59%) | 5 (29.41%) | 0.976 |
| ≥60 | 99 | 80 (80.81) | 19 (19.19%) | (Mann-Whitney) | |
| Gender | Male | 87 | 67 (77.01%) | 20 (22.99%) | 0.427 |
| Female | 29 | 25 (86.21%) | 4 (13.79%) | (Chi-squared) | |
| Histology | LUAD 1 | 88 | 70 (79.55%) | 18 (24.45%) | 0.621 |
| LUSC 2 | 25 | 19 (76.00%) | 6 (24.00%) | (Chi-squared) | |
| NOS 3 | 3 | 3 (100.00%) | 0 (0.00%) | ||
| Stage | 0 | 5 | 4 (80.00%) | 1 (20.00%) | 0.386 |
| Ia | 44 | 38 (86.36%) | 6 (13.64%) | (Chi-squared) | |
| Ib | 27 | 21 (77.78%) | 6 (22.22%) | ||
| IIa | 11 | 7 (63.64%) | 4 (36.36%) | ||
| IIb | 9 | 5 (55.56%) | 4 (44.44%) | ||
| IIIa | 15 | 12 (80.00%) | 3 (20.00%) | ||
| IIIb | 1 | 1 (100.00%) | 0 (0.00%) | ||
| IV | 4 | 4 (100.00%) | 0 (0.00%) | ||
| HOPX status | HOPX-negative | 27 | 20 (74.07%) | 7 (25.93%) | 0.620 |
| HOPX-positive | 89 | 72 (80.90%) | 17 (19.10%) | (Chi-squared) |
| Training Dataset | Test Dataset | |||||||
|---|---|---|---|---|---|---|---|---|
| AUC | Accuracy | Specificity | Sensitivity | AUC | Accuracy | Specificity | Sensitivity | |
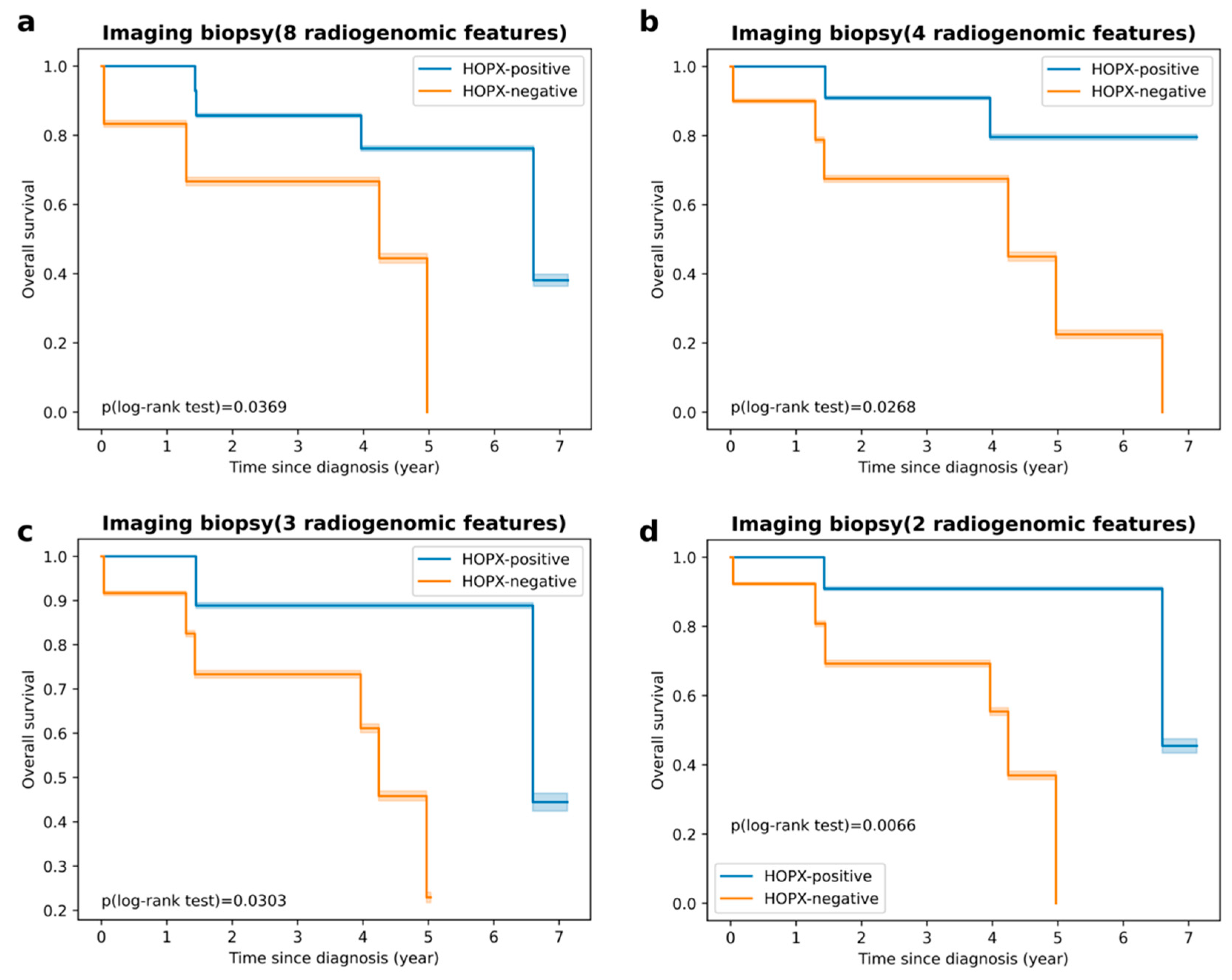
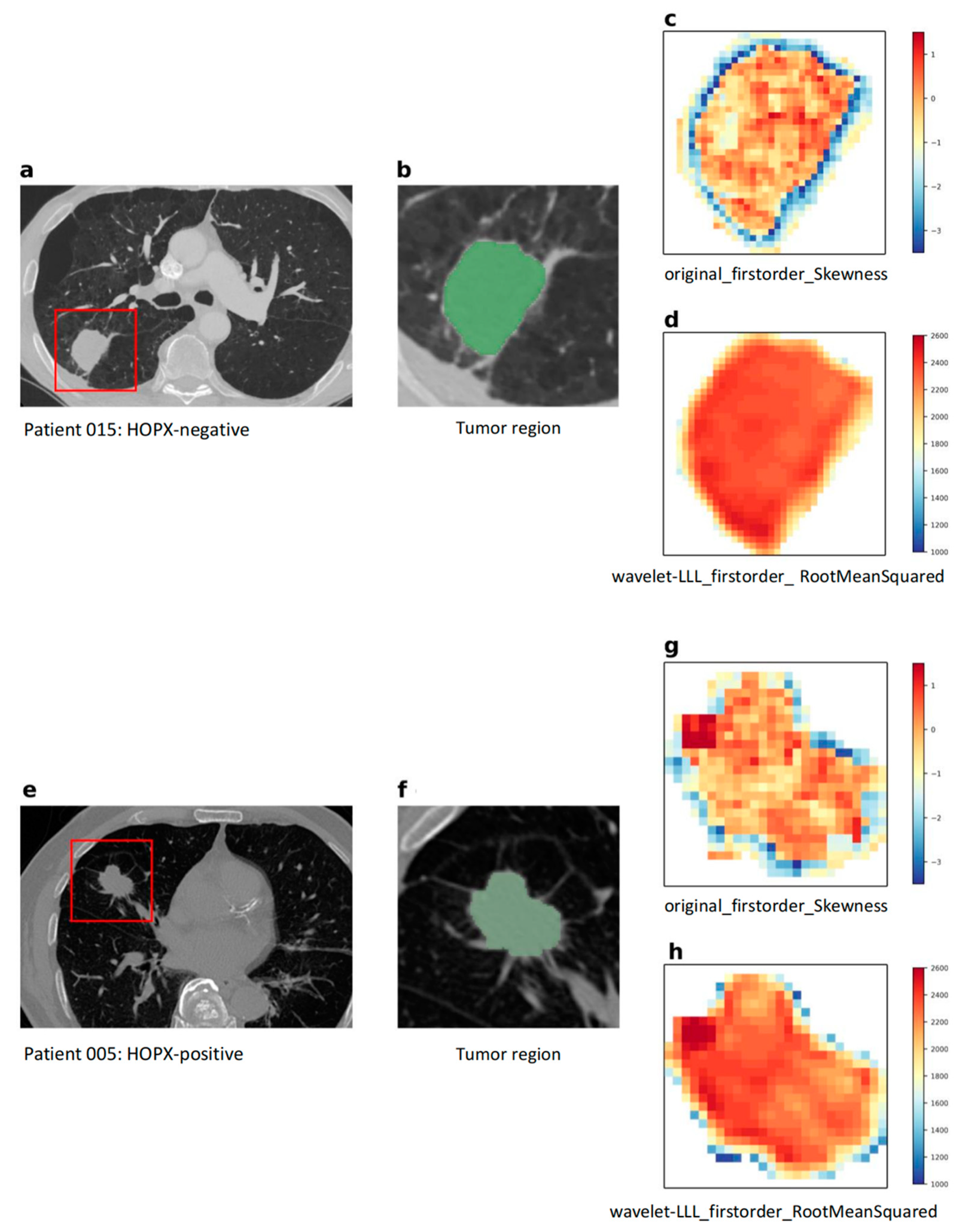
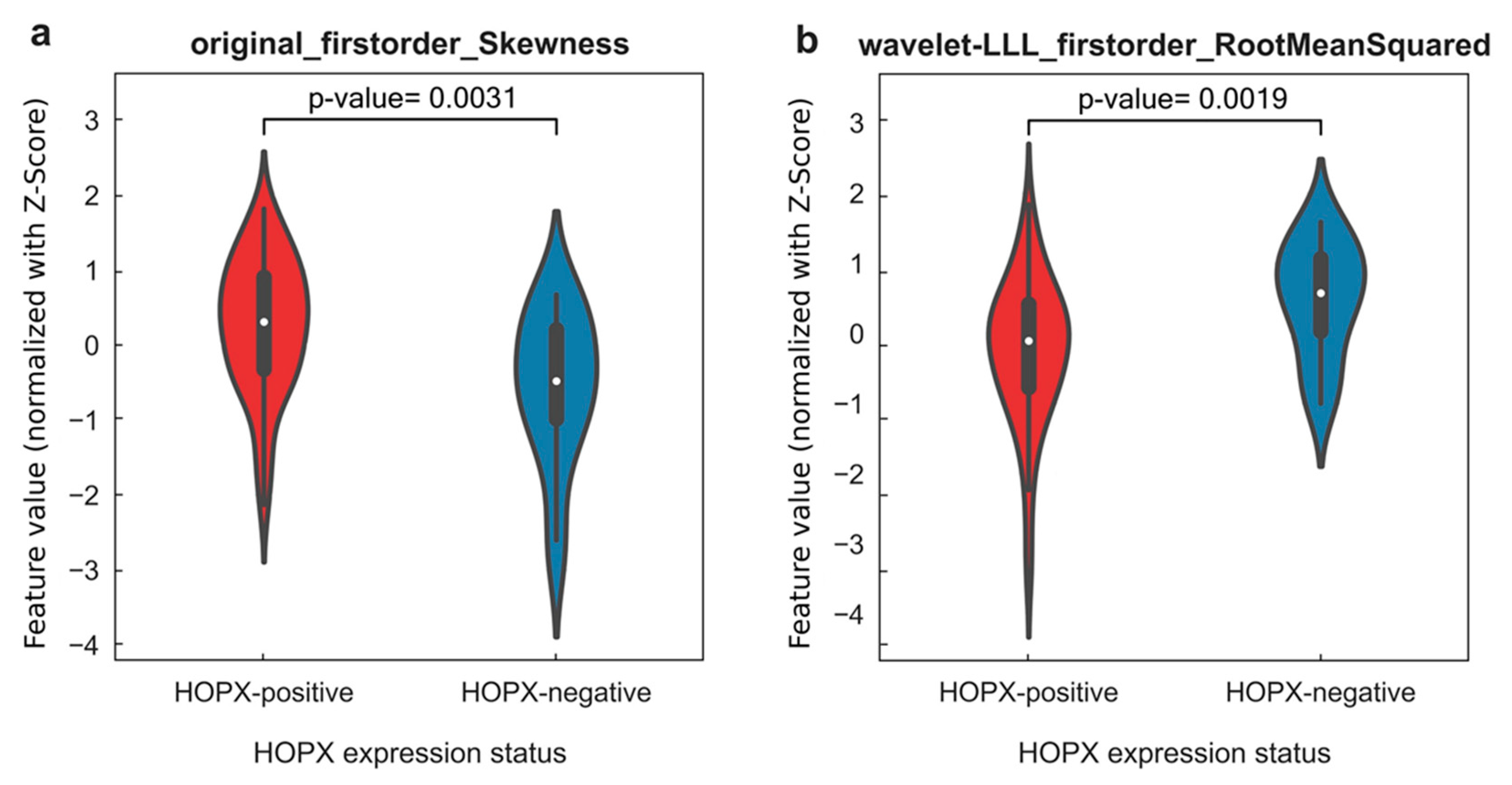
| Imaging biopsy A, consisting of 8 radiogenomic features: original_firstorder_Skewness original_firstorder_Median original_firstorder_Mean original_firstorder_RootMeanSquared wavelet-LLL_firstorder_Skewness wavelet-LLL_firstorder_Median wavelet-LLL_firstorder_Mean wavelet-LLL_firstorder_RootMeanSquared | 0.995 | 0.939 | 0.985 | 0.920 | 0.664 | 0.625 | 0.286 | 0.764 |
| Imaging biopsy B, consisting of 4 radiogenomic features: original_firstorder_Skewness original_firstorder_Mean wavelet-LLL_firstorder_Median wavelet-LLL_firstorder_RootMeanSquared | 0.998 | 0.986 | 0.705 | 0.714 | 0.672 | 0.708 | 0.706 | 0.714 |
| Imaging biopsy C, consisting of 3 radiogenomic features: original_firstorder_Skewness wavelet-LLL_firstorder_Median wavelet-LLL_firstorder_RootMeanSquared | 0.953 | 0.890 | 0.904 | 0.706 | 0.706 | 0.625 | 0.588 | 0.714 |
| Imaging biopsy D, consisting of 2 radiogenomic features: original_firstorder_Skewness wavelet-LLL_firstorder_RootMeanSquared | 0.965 | 0.876 | 0.877 | 0.877 | 0.873 | 0.750 | 0.647 | 1.000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jin, Y.; Arimura, H.; Cui, Y.; Kodama, T.; Mizuno, S.; Ansai, S. CT Image-Based Biopsy to Aid Prediction of HOPX Expression Status and Prognosis for Non-Small Cell Lung Cancer Patients. Cancers 2023, 15, 2220. https://doi.org/10.3390/cancers15082220
Jin Y, Arimura H, Cui Y, Kodama T, Mizuno S, Ansai S. CT Image-Based Biopsy to Aid Prediction of HOPX Expression Status and Prognosis for Non-Small Cell Lung Cancer Patients. Cancers. 2023; 15(8):2220. https://doi.org/10.3390/cancers15082220
Chicago/Turabian StyleJin, Yu, Hidetaka Arimura, YunHao Cui, Takumi Kodama, Shinichi Mizuno, and Satoshi Ansai. 2023. "CT Image-Based Biopsy to Aid Prediction of HOPX Expression Status and Prognosis for Non-Small Cell Lung Cancer Patients" Cancers 15, no. 8: 2220. https://doi.org/10.3390/cancers15082220
APA StyleJin, Y., Arimura, H., Cui, Y., Kodama, T., Mizuno, S., & Ansai, S. (2023). CT Image-Based Biopsy to Aid Prediction of HOPX Expression Status and Prognosis for Non-Small Cell Lung Cancer Patients. Cancers, 15(8), 2220. https://doi.org/10.3390/cancers15082220