

Euchromatic Histone Lysine Methyltransferase 2 Inhibition Enhances Carfilzomib Sensitivity and Overcomes Drug Resistance in Multiple Myeloma Cell Lines
 , , , ,
, , , , 
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture Conditions and Reagents
2.2. Cell-Growth Luminescence Assay
2.3. Drug Screening
2.4. Analysis of Apoptosis
2.5. Proteasome-Inhibitor-Resistant (PIR) Cell Reconditioning Protocol
2.6. 3D Co-Culture
2.7. Purification of Total RNA and Reverse Transcription–Quantitative Polymerase Chain Reaction (RT-qPCR)
2.8. Western Blotting
2.9. Gene Expression Data
2.10. Statistical Analysis
3. Results
3.1. Drug-Library Screening in Multiple Myeloma Cell Lines Identifies EHMT2 Inhibition as Synthetic Lethal to the Proteasome Inhibitor Carfilzomib
3.2. EHMT2 Expression Is Increased in Bortezomib-Resistant Multiple Myeloma Patients and Correlates to Worse Survival
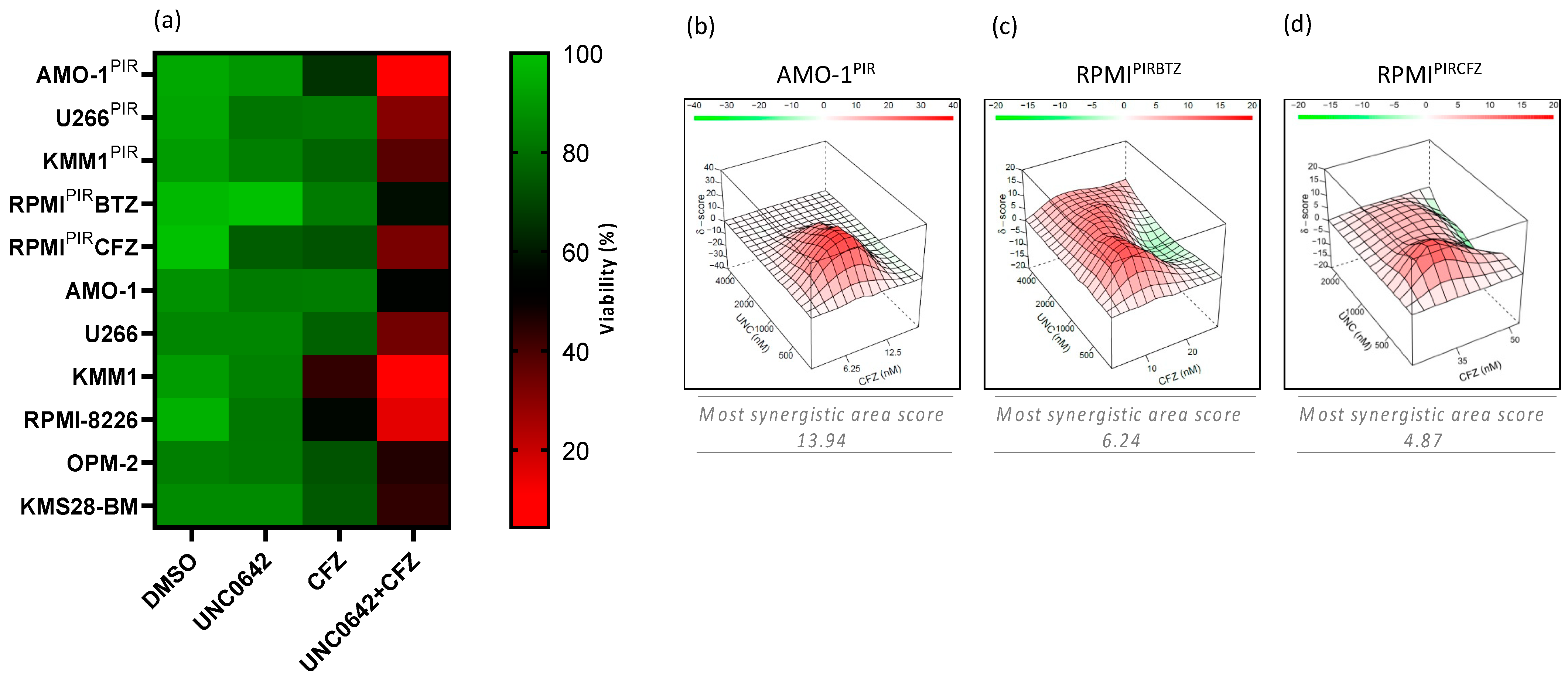
3.3. EHMT2 Inhibition Synergically Increases Proteasome Inhibitor-Mediated Cell Death in PI-Resistant and PI-Sensitive Multiple Myeloma Cell Lines
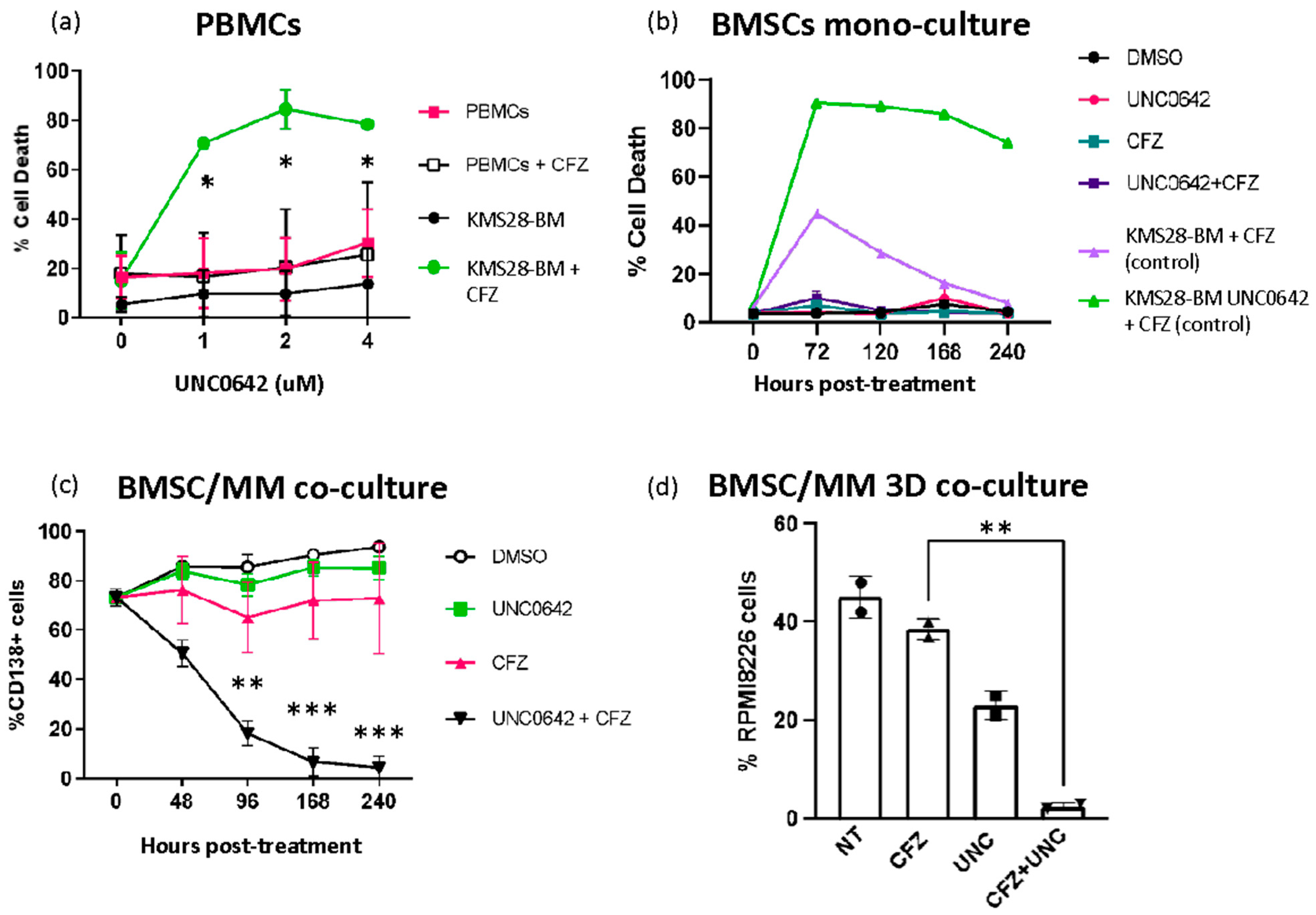
3.4. Combining EHMT2 and Proteasome Inhibition Is Not Toxic to Normal Cells and It Is Effective in the Presence of Bone Marrow Milieu
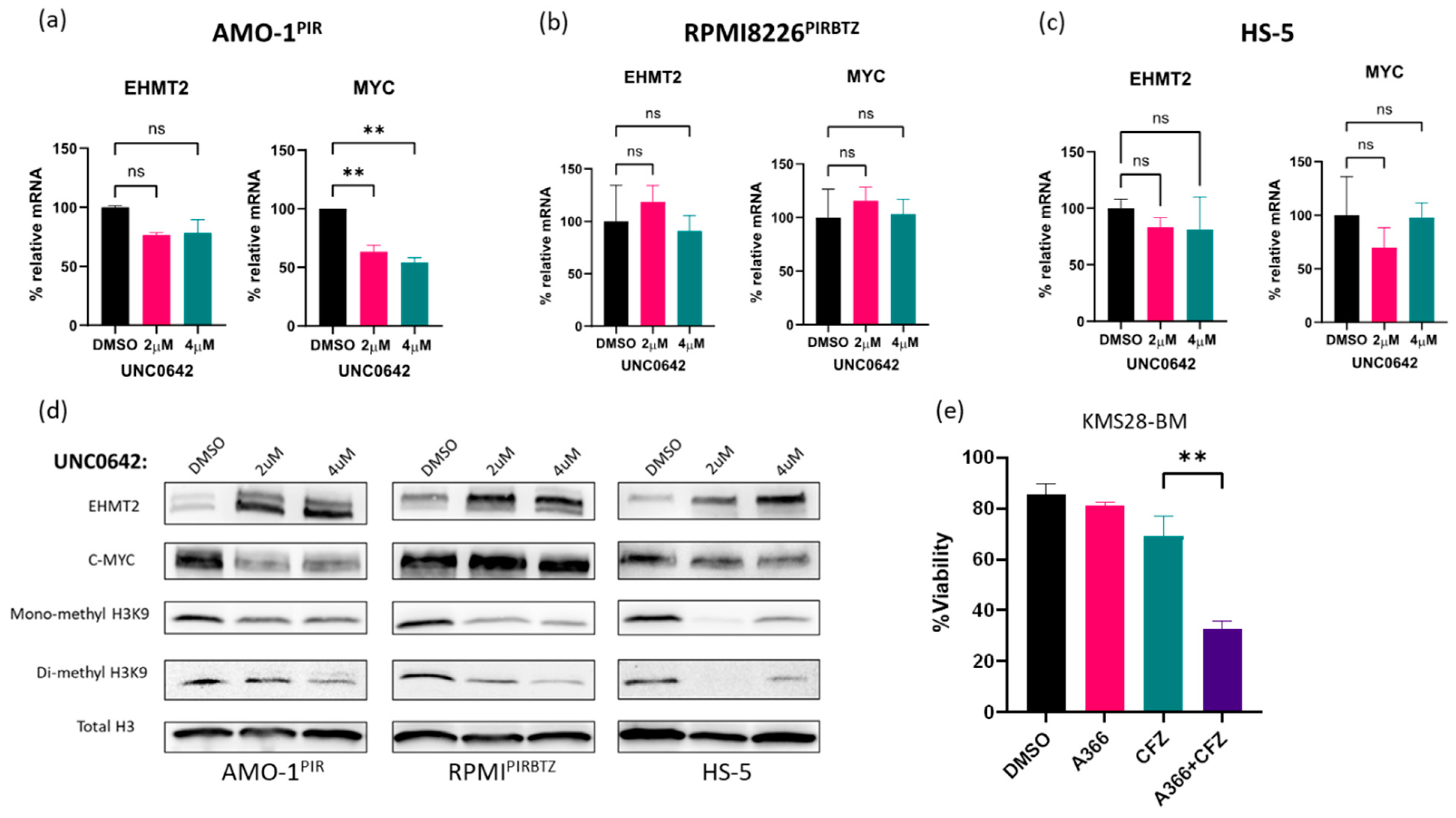
3.5. Pathway Specificity: Investigation of Molecular Markers Associated with EHMT2 Inhibition
3.6. UNC0642/CFZ Synergistic Interaction Is Not Dependent on Cell-Cycle Perturbation
3.7. Autophagy and DNA Damage Pathways Are Activated by UNC0642/CFZ Combinatorial Treatment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Van de Donk, N.W.C.J.; Pawlyn, C.; Yong, K.L. Multiple myeloma. Lancet 2021, 397, 410–427. [Google Scholar] [CrossRef]
- Kumar, S.K.; Rajkumar, V.; Kyle, R.A.; Van Duin, M.; Sonneveld, P.; Mateos, M.V.; Gay, F.; Anderson, K.C. Multiple myeloma. Nat. Rev. Dis. Prim. 2017, 3, 17046. [Google Scholar] [CrossRef]
- Ito, S. Proteasome inhibitors for the treatment of multiple myeloma. Cancers 2020, 12, 265. [Google Scholar] [CrossRef] [PubMed]
- Alsina, M.; Trudel, S.; Furman, R.R.; Rosen, P.J.; O’Connor, O.A.; Comenzo, R.L.; Wong, A.; Kunkel, L.A.; Molineaux, C.J.; Goy, A. A phase I single-agent study of twice-weekly consecutive-day dosing of the proteasome inhibitor carfilzomib in patients with relapsed or refractory multiple myeloma or lymphoma. Clin. Cancer Res. 2012, 18, 4830–4840. [Google Scholar] [CrossRef] [PubMed]
- Manasanch, E.E.; Orlowski, R.Z. Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2017, 14, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Paradzik, T.; Bandini, C.; Mereu, E.; Labrador, M.; Taiana, E.; Amodio, N.; Neri, A.; Piva, R. The landscape of signaling pathways and proteasome inhibitors combinations in multiple myeloma. Cancers 2021, 13, 1235. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V.; Kumar, S. Multiple myeloma current treatment algorithms. Blood Cancer J. 2020, 10, 94. [Google Scholar] [CrossRef]
- Liu, F.; Barsyte-Lovejoy, D.; Li, F.; Xiong, Y.; Korboukh, V.; Huang, X.P.; Allali-Hassani, A.; Janzen, W.P.; Roth, B.L.; Frye, S.V.; et al. Discovery of an in vivo chemical probe of the lysine methyltransferases G9a and GLP. J. Med. Chem. 2013, 56, 8931–8942. [Google Scholar] [CrossRef]
- Casciello, F.; Windloch, K.; Gannon, F.; Lee, J.S. Functional role of G9a histone methyltransferase in cancer. Front. Immunol. 2015, 6, 3–9. [Google Scholar] [CrossRef]
- Jan, S.; Dar, M.I.; Wani, R.; Sandey, J.; Mushtaq, I.; Lateef, S.; Syed, S.H. Targeting EHMT2/G9a for cancer therapy: Progress and perspective. Eur. J. Pharmacol. 2021, 893, 173827. [Google Scholar] [CrossRef]
- Nachiyappan, A.; Gupta, N.; Taneja, R. EHMT1/EHMT2 in EMT, cancer stemness and drug resistance: Emerging evidence and mechanisms. FEBS J. 2022, 289, 1329–1351. [Google Scholar] [CrossRef]
- Poulard, C.; Bittencourt, D.; Wu, D.Y.; Hu, Y.; Gerke, D.S.; Stallcup, M.R. A post-translational modification switch controls coactivator function of histone methyltransferases G9a and GLP. EMBO Rep. 2017, 18, 1442–1459. [Google Scholar] [CrossRef]
- Chae, Y.C.; Kim, J.Y.; Park, J.W.; Kim, K.B.; Oh, H.; Lee, K.H.; Seo, S.B. FOXO1 degradation via G9a-mediated methylation promotes cell proliferation in colon cancer. Nucleic Acids Res. 2019, 47, 1692–1705. [Google Scholar] [CrossRef]
- Kim, C.; Park, K.; Lee, S. G9a/GLP methyltransferases inhibit autophagy by methylation- mediated ATG12 protein degradation. Biorxiv Prepr. Serv. Biol. 2021. [Google Scholar] [CrossRef]
- Rada, M.; Vasileva, E.; Lezina, L.; Marouco, D.; Antonov, A.V.; MacIp, S.; Melino, G.; Barlev, N.A. Human EHMT2/G9a activates p53 through methylation-independent mechanism. Oncogene 2017, 36, 922–932. [Google Scholar] [CrossRef] [PubMed]
- Sampath, S.C.; Marazzi, I.; Yap, K.L.; Sampath, S.C.; Krutchinsky, A.N.; Mecklenbräuker, I.; Viale, A.; Rudensky, E.; Zhou, M.M.; Chait, B.T.; et al. Methylation of a Histone Mimic within the Histone Methyltransferase G9a Regulates Protein Complex Assembly. Mol. Cell 2007, 27, 596–608. [Google Scholar] [CrossRef]
- Soriano, G.P.; Besse, L.; Li, N.; Kraus, M.; Besse, A.; Meeuwenoord, N.; Bader, J.; Everts, B.; Den Dulk, H.; Overkleeft, H.S.; et al. Proteasome inhibitor-adapted myeloma cells are largely independent from proteasome activity and show complex proteomic changes, in particular in redox and energy metabolism. Leukemia 2016, 30, 2198–2207. [Google Scholar] [CrossRef]
- Niepel, M.; Hafner, M.; Duan, Q.; Wang, Z.; Paull, E.O.; Chung, M.; Lu, X.; Stuart, J.M.; Golub, T.R.; Subramanian, A.; et al. Common and cell-type specific responses to anti-cancer drugs revealed by high throughput transcript profiling. Nat. Commun. 2017, 8, 1186. [Google Scholar] [CrossRef] [PubMed]
- Belloni, D.; Ferrarini, M.; Ferrero, E.; Guzzeloni, V.; Barbaglio, F.; Ghia, P.; Scielzo, C. Protocol for generation of 3D bone marrow surrogate microenvironments in a rotary cell culture system. STAR Protoc. 2022, 3, 101601. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Bergaggio, E.; Riganti, C.; Garaffo, G.; Vitale, N.; Mereu, E.; Bandini, C.; Pellegrino, E.; Pullano, V.; Omedè, P.; Todoerti, K.; et al. IDH2 inhibition enhances proteasome inhibitor responsiveness in hematological malignancies. Blood 2019, 133, 156–167. [Google Scholar] [CrossRef]
- Yu, C.; Friday, B.B.; Lai, J.P.; Yang, L.; Sarkaria, J.; Kay, N.E.; Carter, C.A.; Roberts, L.R.; Kaufmann, S.H.; Adjei, A.A. Cytotoxic synergy between the multikinase inhibitor sorafenib and the proteasome inhibitor bortezomib in vitro: Induction of apoptosis through Akt and c-Jun NH2-terminal kinase pathways. Mol. Cancer Ther. 2006, 5, 2378–2387. [Google Scholar] [CrossRef]
- Hu, Z.; Pan, X.F.; Wu, F.Q.; Ma, L.Y.; Liu, D.P.; Feng, T.T.; Meng, F.Y.; Liu, X.L.; Jiang, Q.L.; Chen, X.Q.; et al. Synergy between proteasome inhibitors and imatinib mesylate in chronic myeloid leukemia. PLoS ONE 2009, 4, e6257. [Google Scholar] [CrossRef]
- Yu, L.; Mohamed, A.J.; Simonson, O.E.; Vargas, L.; Blomberg, K.E.M.; Björkstrand, B.; Arteaga, H.J.; Nore, B.F.; Smith, C.I.E. Proteasome-dependent autoregulation of bruton tyrosine kinase (Btk) promoter via NF-κB. Blood 2008, 111, 4617–4626. [Google Scholar] [CrossRef] [PubMed]
- Taiana, E.; Ronchetti, D.; Favasuli, V.; Todoerti, K.; Manzoni, M.; Amodio, N.; Tassone, P.; Agnelli, L.; Neri, A. Long non-coding RNA NEAT1 shows high expression unrelated to molecular features and clinical outcome in multiple myeloma. Haematologica 2019, 104, e72. [Google Scholar] [CrossRef]
- Ronchetti, D.; Favasuli, V.K.; Silvestris, I.; Todoerti, K.; Torricelli, F.; Bolli, N.; Ciarrocchi, A.; Taiana, E.; Neri, A. Expression levels of NONO, a nuclear protein primarily involved in paraspeckles function, are associated with several deregulated molecular pathways and poor clinical outcome in multiple myeloma. Discov. Oncol. 2022, 13, 124. [Google Scholar] [CrossRef]
- Belloni, D.; Heltai, S.; Ponzoni, M.; Villa, A.; Vergani, B.; Pecciarini, L.; Marcatti, M.; Girlanda, S.; Tonon, G.; Ciceri, F.; et al. Modeling multiple myeloma-bone marrow interactions and response to drugs in a 3d surrogate microenvironment. Haematologica 2018, 103, 707–716. [Google Scholar] [CrossRef]
- Zhang, N.; Shang, M.; Li, H.; Wu, L.; Dong, M.; Huang, B.; Lu, J.; Zhang, Y. Dual Inhibition of H3K9me2 and H3K27me3 Promotes Tumor Cell Senescence without Triggering the Secretion of SASP. Int. J. Mol. Sci. 2022, 23, 3911. [Google Scholar] [CrossRef] [PubMed]
- de Smedt, E.; Devin, J.; Muylaert, C.; Robert, N.; Requirand, G.; Vlummens, P.; Vincent, L.; Cartron, G.; Maes, K.; Moreaux, J.; et al. G9a/GLP targeting in MM promotes autophagy-associated apoptosis and boosts proteasome inhibitor-mediated cell death. Blood Adv. 2021, 5, 2325–2338. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu, H.; Ge, S.; Xue, C.; Li, H.; Jing, X.; Liang, K.; Zhang, X.; Zhang, C. EHMT2 (G9a) activation in mantle cell lymphoma and its associated DNA methylation and gene expression. Cancer Biol. Med. 2021, 18, 836. [Google Scholar] [CrossRef]
- Milite, C.; Feoli, A.; Horton, J.R.; Rescigno, D.; Cipriano, A.; Pisapia, V.; Viviano, M.; Pepe, G.; Amendola, G.; Novellino, E.; et al. Discovery of a Novel Chemotype of Histone Lysine Methyltransferase EHMT1/2 (GLP/G9a) Inhibitors: Rational Design, Synthesis, Biological Evaluation, and Co-crystal Structure. J. Med. Chem. 2019, 62, 2666–2689. [Google Scholar] [CrossRef] [PubMed]
- Feoli, A.; Viviano, M.; Cipriano, A.; Milite, C.; Castellano, S.; Sbardella, G. Lysine methyltransferase inhibitors: Where we are now. RSC Chem. Biol. 2021, 3, 359–406. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.; Ke, X.; Zhang, R.; Hou, J.; Dong, Z.; Wang, F.; Zhang, K.; Zhong, X.; Yang, L.; Cui, H. G9a promotes cell proliferation and suppresses autophagy in gastric cancer by directly activating mTOR. FASEB J. 2019, 33, 14036–14050. [Google Scholar] [CrossRef]
- Lee, S.H.; Hyeon, D.Y.; Yoon, S.H.; Jeong, J.H.; Han, S.M.; Jang, J.W.; Nguyen, M.P.; Chi, X.Z.; An, S.; Hyun, K.G.; et al. RUNX3 methylation drives hypoxia-induced cell proliferation and antiapoptosis in early tumorigenesis. Cell Death Differ. 2021, 28, 1251–1269. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, K.; Kitajima, H.; Niinuma, T.; Maruyama, R.; Nishiyama, N.; Ohtani, H.; Sudo, G.; Toyota, M.; Sasaki, H.; Yamamoto, E.; et al. Dual EZH2 and G9a inhibition suppresses multiple myeloma cell proliferation by regulating the interferon signal and IRF4-MYC axis. Cell Death Discov. 2021, 7, 7. [Google Scholar] [CrossRef]
- Li, Z.; Jiao, X.; Di Sante, G.; Ertel, A.; Casimiro, M.C.; Wang, M.; Katiyar, S.; Ju, X.; Klopfenstein, D.V.; Tozeren, A.; et al. Cyclin D1 integrates G9a-mediated histone methylation. Oncogene 2019, 38, 4232–4249. [Google Scholar] [CrossRef]
- Park, S.E.; Yi, H.J.; Suh, N.; Park, Y.Y.; Koh, J.Y.; Jeong, S.Y.; Cho, D.H.; Kim, C.S.; Hwang, J.J. Correction: Inhibition of EHMT2/G9a epigenetically increases the transcription of Beclin-1 via an increase in ROS and activation of NF-κB. Oncotarget 2019, 10, 4348–4349. [Google Scholar] [CrossRef] [PubMed]
- Artal-Martinez de Narvajas, A.; Gomez, T.S.; Zhang, J.-S.; Mann, A.O.; Taoda, Y.; Gorman, J.A.; Herreros-Villanueva, M.; Gress, T.M.; Ellenrieder, V.; Bujanda, L.; et al. Epigenetic Regulation of Autophagy by the Methyltransferase G9a. Mol. Cell. Biol. 2013, 33, 3983–3993. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Guan, K. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 2, 25–32. [Google Scholar] [CrossRef]
- Yang, Q.; Zhu, Q.; Lu, X.; Du, Y.; Cao, L.; Shen, C.; Hou, T.; Li, M.; Li, Z.; Liu, C.; et al. G9a coordinates with the RPA complex to promote DNA damage repair and cell survival. Proc. Natl. Acad. Sci. USA 2017, 114, E6054–E6063. [Google Scholar] [CrossRef]
- Watanabe, S.; Iimori, M.; Chan, D.V.; Hara, E.; Kitao, H.; Maehara, Y. MDC1 methylation mediated by lysine methyltransferases EHMT1 and EHMT2 regulates active ATM accumulation flanking DNA damage sites. Sci. Rep. 2018, 8, 10888. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wang, H.Y.; Zhao, X.; Duan, H.; Cheng, B.; Liu, Y.; Zhao, M.; Shu, W.; Mei, Y.; Wen, Z.; et al. A methylation-phosphorylation switch determines Plk1 kinase activity and function in DNA damage repair. Sci. Adv. 2019, 5, eaau7566. [Google Scholar] [CrossRef] [PubMed]
- Von Stechow, L. Cancer Systems Biology: Methods and Protocols; Springer: New York, NY, USA, 2011; Chapter 17; Volume 1711, ISBN 9781493974931. [Google Scholar]
- Lin, A.; Sheltzer, J.M. Discovering and validating cancer genetic dependencies: Approaches and pitfalls. Nat. Rev. Genet. 2020, 21, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Pattanayak, V.; Lin, S.; Guilinger, J.P.; Ma, E.; Doudna, J.A.; Liu, D.R. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat. Biotechnol. 2013, 31, 839–843. [Google Scholar] [CrossRef]
- Rahman, Z.; Bazaz, M.R.; Devabattula, G.; Khan, M.A.; Godugu, C. Targeting H3K9 methyltransferase G9a and its related molecule GLP as a potential therapeutic strategy for cancer. J. Biochem. Mol. Toxicol. 2021, 35, e22674. [Google Scholar] [CrossRef]
- Shen, S.; Vagner, S.; Robert, C. Persistent Cancer Cells: The Deadly Survivors. Cell 2020, 183, 860–874. [Google Scholar] [CrossRef]
- Rehman, S.K.; Haynes, J.; Collignon, E.; Brown, K.R.; Wang, Y.; Nixon, A.M.L.; Bruce, J.P.; Wintersinger, J.A.; Singh Mer, A.; Lo, E.B.L.; et al. Colorectal Cancer Cells Enter a Diapause-like DTP State to Survive Chemotherapy. Cell 2021, 184, 226–242.e21. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Sequence |
|---|---|
| MYC | Fw: GGACCCGCTTCTCTGAAAGG Rw: TAACGTTGAGGGGCATCGTC |
| EHMT2 | Fw: CTGTCAGAGGAGTTAGGTTCTGC Rw: CTTGCTGTCGGAGTCCACG |
| EHMT1 | Fw: CCTCGACTCGGAAAAACCCA Rw: AGTTGGGGTCAATTCCGTCC |
| GAPDH | Fw: TCTTTTGCGTCGCCAGCCGAG Rw: TGACCAGGCGCCCAATACGAC |
| Antibody | Species | Source |
|---|---|---|
| Vinculin | mouse | SAB4200080, Sigma |
| α-tubulin | mouse | clone B-5-1-2, T5168, Sigma-Aldrich |
| β-Actin | mouse | #sc69879, SCB |
| P27/Kip1 | mouse | #610241, BD Biosciences |
| P53 | mouse | #2524S, Cell Signaling Technology |
| G9a/EHMT2 | rabbit | #3306S, Cell Signaling Technology |
| Histone H3 | rabbit | #4499T Cell Signaling Technology |
| Mono-methyl-histone H3 (Lys9) | rabbit | #14186T Cell Signaling Technology |
| Di-methyl-histone H3 (Lys9) | rabbit | #4658T Cell Signaling Technology |
| Cleaved PARP | rabbit | #9532 Cell Signaling Technology |
| ATG12 | rabbit | #4180T Cell Signaling Technology |
| LC3A/B | rabbit | #12741T Cell Signaling Technology |
| 53BP1 | rabbit | #4937S Cell Signaling Technology |
| p-P70-S6 Kinase (Thr389) | rabbit | #9234T Cell Signaling Technology |
| P70-S6 Kinase | rabbit | #9202L Cell Signaling Technology |
| p-mTOR (Ser2448) | rabbit | #5536T Cell Signaling Technology |
| mTOR | rabbit | #2983T Cell Signaling Technology |
| p-4E-BP1 (Thr37/46) | rabbit | #2855T Cell Signaling Technology |
| 4E-BP1 | rabbit | #9644L Cell Signaling Technology |
| p-H2A.X (Ser139) | rabbit | #9718T Cell Signaling Technology |
| H2A.X | rabbit | #2595 Cell Signaling Technology |
| p-ATM (Ser1981) | rabbit | #5883T Cell Signaling Technology |
| ATM | rabbit | #2873 Cell Signaling Technology |
| c-Myc | rabbit | #5605S Cell Signaling Technology |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mereu, E.; Abbo, D.; Paradzik, T.; Cumerlato, M.; Bandini, C.; Labrador, M.; Maccagno, M.; Ronchetti, D.; Manicardi, V.; Neri, A.; et al. Euchromatic Histone Lysine Methyltransferase 2 Inhibition Enhances Carfilzomib Sensitivity and Overcomes Drug Resistance in Multiple Myeloma Cell Lines. Cancers 2023, 15, 2199. https://doi.org/10.3390/cancers15082199
Mereu E, Abbo D, Paradzik T, Cumerlato M, Bandini C, Labrador M, Maccagno M, Ronchetti D, Manicardi V, Neri A, et al. Euchromatic Histone Lysine Methyltransferase 2 Inhibition Enhances Carfilzomib Sensitivity and Overcomes Drug Resistance in Multiple Myeloma Cell Lines. Cancers. 2023; 15(8):2199. https://doi.org/10.3390/cancers15082199
Chicago/Turabian StyleMereu, Elisabetta, Damiano Abbo, Tina Paradzik, Michela Cumerlato, Cecilia Bandini, Maria Labrador, Monica Maccagno, Domenica Ronchetti, Veronica Manicardi, Antonino Neri, and et al. 2023. "Euchromatic Histone Lysine Methyltransferase 2 Inhibition Enhances Carfilzomib Sensitivity and Overcomes Drug Resistance in Multiple Myeloma Cell Lines" Cancers 15, no. 8: 2199. https://doi.org/10.3390/cancers15082199
APA StyleMereu, E., Abbo, D., Paradzik, T., Cumerlato, M., Bandini, C., Labrador, M., Maccagno, M., Ronchetti, D., Manicardi, V., Neri, A., & Piva, R. (2023). Euchromatic Histone Lysine Methyltransferase 2 Inhibition Enhances Carfilzomib Sensitivity and Overcomes Drug Resistance in Multiple Myeloma Cell Lines. Cancers, 15(8), 2199. https://doi.org/10.3390/cancers15082199