
Surgical Management of Pancreatic Neuroendocrine Tumors


Abstract
Simple Summary
Abstract
1. Introduction
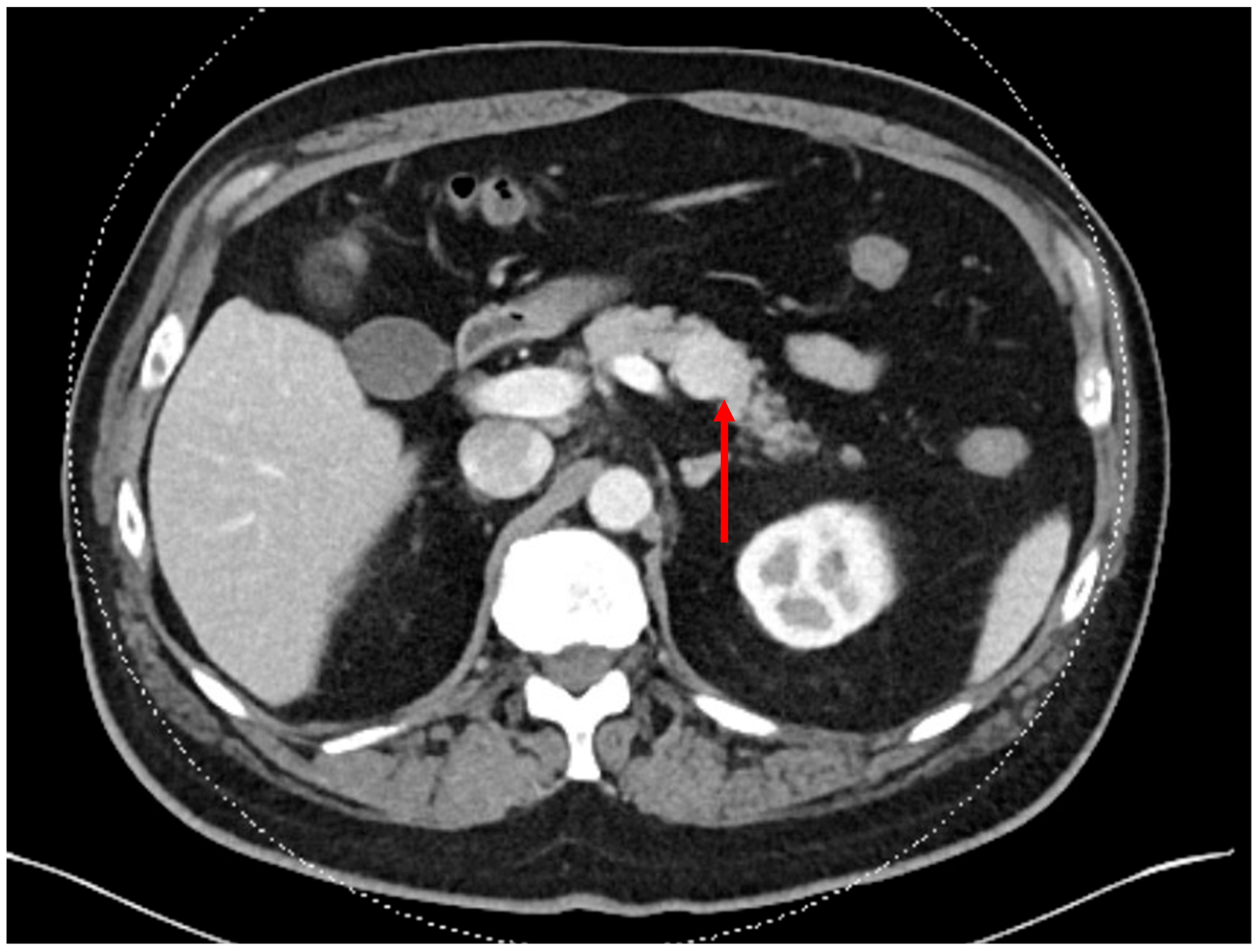
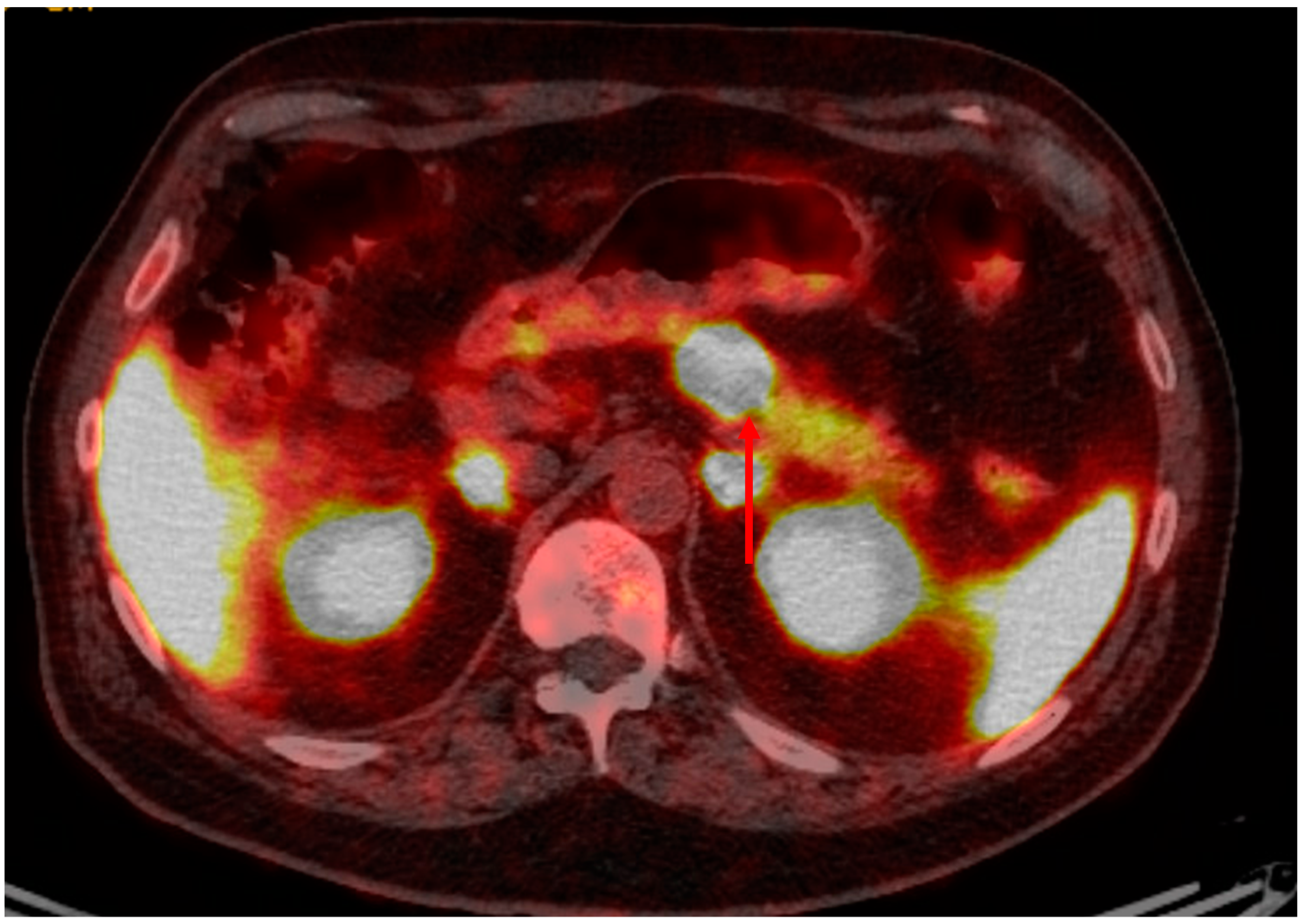
2. Presentation and Diagnosis of Pancreatic Neuroendocrine Tumors
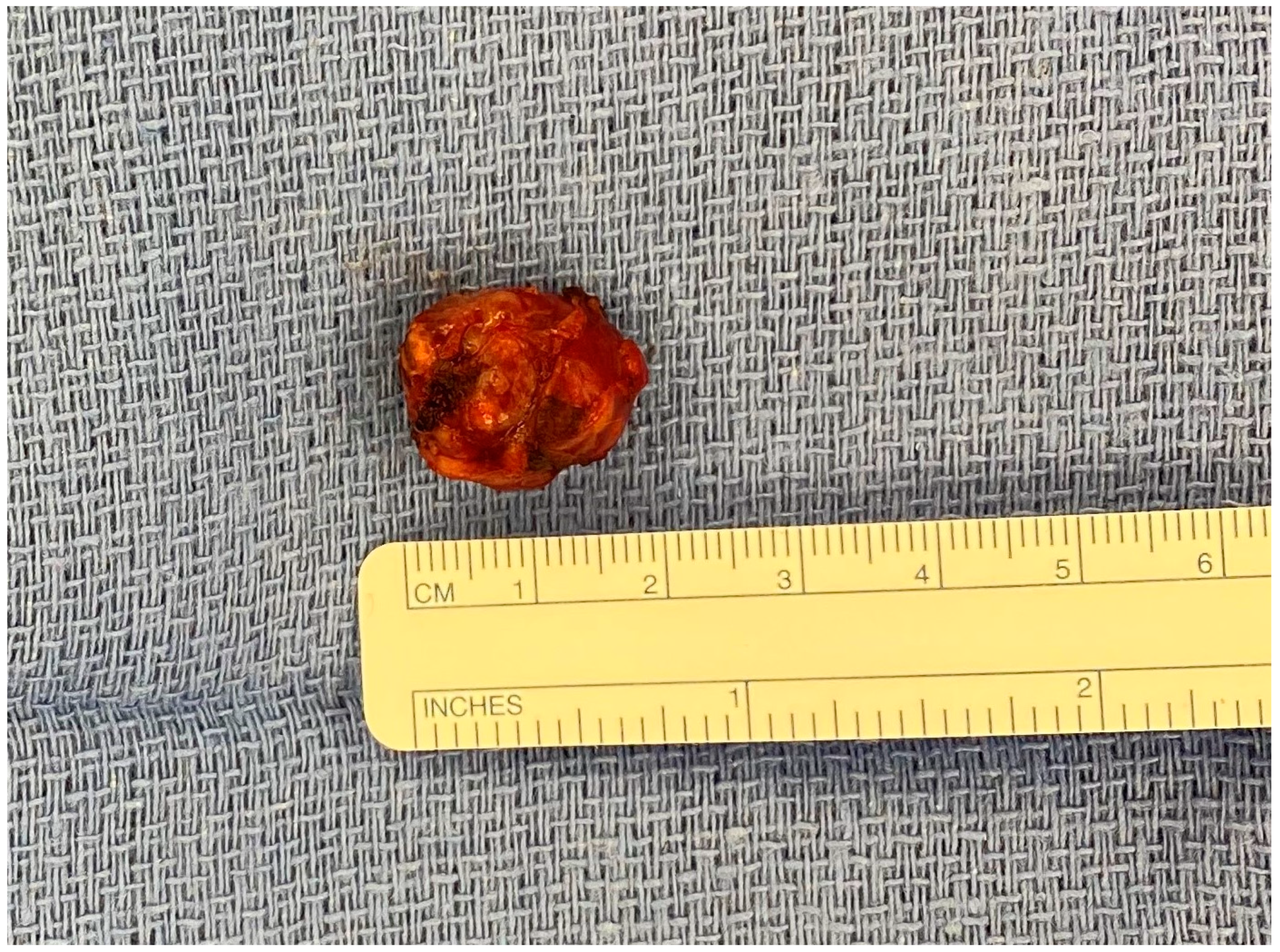
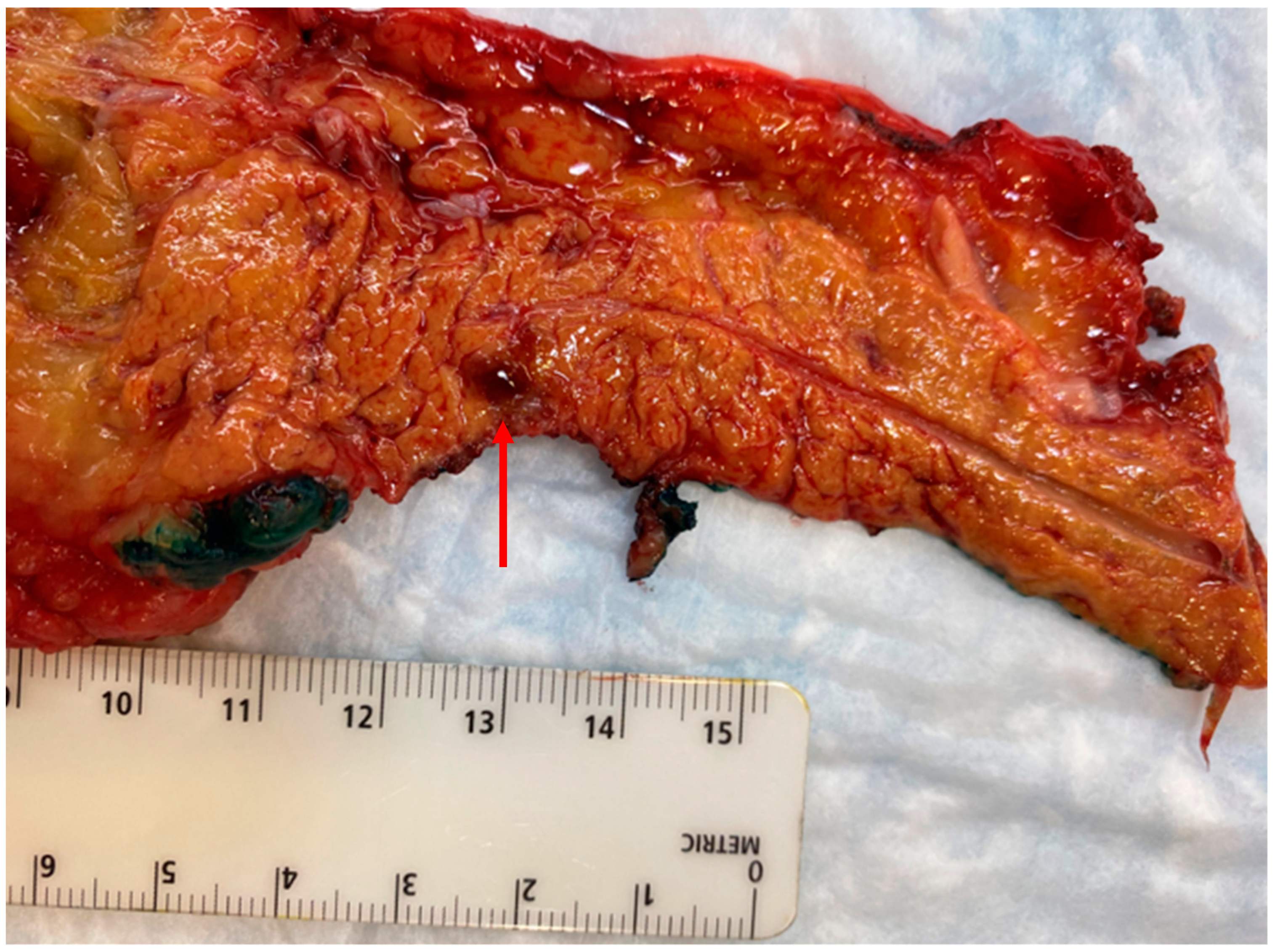
3. Surgical Management of Pancreatic Neuroendocrine Tumors
3.1. Functional PNETs
3.2. Nonfunctional PNETs
3.3. Genetic Disorders
3.4. Metastatic Disease
4. New Directions in the Treatment of Pancreatic Neuroendocrine Tumors
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dasari, A.; Shen, C.; Halperin, D.M.; Zhao, B.; Zhou, S.; Xu, Y.; Shih, T.; Yao, J.C. Trends in the incidence, prevalence, and survival outcomes in patients with neuroendocrine tumors in the united states. JAMA Oncol. 2017, 3, 1335–1342. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, L.; Dai, S.; Chen, M.; Li, F.; Sun, J.; Luo, F. Epidemiologic trends of and factors associated with overall survival for patients with gastroenteropancreatic neuroendocrine tumors in the United States. JAMA Netw. Open 2021, 4, e2124750. [Google Scholar] [CrossRef]
- Yao, J.C.; Hassan, M.M.; Phan, A.T.; Dagohoy, C.G.; Leary, C.C.; Mares, J.E.; Abdalla, E.K.; Fleming, J.B.; Vauthey, J.-N.; Rashid, A.; et al. One hundred years after “carcinoid”: Epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J. Clin. Oncol. 2008, 26, 3063–3072. [Google Scholar] [CrossRef]
- Perri, G.; Prakash, L.R.; Katz, M.H.G. Pancreatic neuroendocrine tumors. Curr. Opin. Gastroenterol. 2019, 35, 468–477. [Google Scholar] [CrossRef] [PubMed]
- Bilimoria, K.Y.; Tomlinson, J.S.; Merkow, R.P.; Stewart, A.K.; Ko, C.Y.; Talamonti, M.S.; Bentrem, D.J. Clinicopathologic features and treatment trends of pancreatic neuroendocrine tumors: Analysis of 9821 patients. J. Gastrointest. Surg. 2007, 11, 1460–1467; discussion 1467. [Google Scholar] [CrossRef] [PubMed]
- Sonbol, M.B.; Mazza, G.L.; Mi, L.; Oliver, T.; Starr, J.; Gudmundsdottir, H.; Cleary, S.P.; Hobday, T.; Halfdanarson, T.R. Survival and incidence patterns of pancreatic neuroendocrine tumors over the last 2 decades: A SEER database analysis. Oncologist 2022, 27, 573–578. [Google Scholar] [CrossRef]
- Kuo, E.J.; Salem, R.R. Population-level analysis of pancreatic neuroendocrine tumors 2 cm or less in size. Ann. Surg. Oncol. 2013, 20, 2815–2821. [Google Scholar] [CrossRef]
- Cloyd, J.M.; Poultsides, G.A. Non-functional neuroendocrine tumors of the pancreas: Advances in diagnosis and management. World J. Gastroenterol. 2015, 21, 9512–9525. [Google Scholar] [CrossRef]
- Singh, S.; Dey, C.; Kennecke, H.; Kocha, W.; Maroun, J.; Metrakos, P.; Mukhtar, T.; Pasieka, J.; Rayson, D.; Rowsell, C.; et al. Consensus Recommendations for the Diagnosis and Management of Pancreatic Neuroendocrine Tumors: Guidelines from a Canadian National Expert Group. Ann. Surg. Oncol. 2015, 22, 2685–2699. [Google Scholar] [CrossRef]
- Halfdanarson, T.R.; Strosberg, J.R.; Tang, L.; Bellizzi, A.M.; Bergsland, E.K.; O’Dorisio, T.M.; Halperin, D.M.; Fishbein, L.; Eads, J.; Hope, T.A.; et al. The north american neuroendocrine tumor society consensus guidelines for surveillance and medical management of pancreatic neuroendocrine tumors. Pancreas 2020, 49, 863–881. [Google Scholar] [CrossRef]
- Kulke, M.H.; Shah, M.H.; Benson, A.B., 3rd; Bergsland, E.; Berlin, J.D.; Blaszkowsky, L.S.; Emerson, L.; Engstrom, P.F.; Fanta, P.; Giordano, T.; et al. Neuroendocrine tumors, version 1.2015. J. Natl. Compr. Canc. Netw. 2015, 13, 78–108. [Google Scholar] [CrossRef]
- IARC (Ed.) WHO Classifocation of Tumours Editorial Board (2022); WHO: Basel, Switzerland, 2022. [Google Scholar]
- Rindi, G.; Mete, O.; Uccella, S.; Basturk, O.; La Rosa, S.; Brosens, L.A.A.; Ezzat, S.; de Herder, W.W.; Klimstra, D.S.; Papotti, M.; et al. Overview of the 2022 WHO classification of neuroendocrine neoplasms. Endocr. Pathol. 2022, 33, 115–154. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, M.Y.; Lopez-Aguiar, A.G.; Switchenko, J.M.; Lipscomb, J.; Andreasi, V.; Partelli, S.; Gamboa, A.C.; Lee, R.M.; Poultsides, G.A.; Dillhoff, M.; et al. A novel validated recurrence risk score to guide a pragmatic surveillance strategy after resection of pancreatic neuroendocrine tumors: An international study of 1006 patients. Ann. Surg. 2019, 270, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Heidsma, C.M.; Tsilimigras, D.I.; Rocha, F.; Abbott, D.E.; Fields, R.; Smith, P.M.; Poultsides, G.A.; Cho, C.; Van Eijck, C.; Van Dijkum, E.N.; et al. Clinical relevance of performing endoscopic ultrasound-guided fine-needle biopsy for pancreatic neuroendocrine tumors less than 2 cm. J. Surg. Oncol. 2020, 122, 1393–1400. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.; Ramos-Alvarez, I.; Jensen, R.T. Predictive Factors for Resistant Disease with Medical/Radiologic/Liver-Directed Anti-Tumor Treatments in Patients with Advanced Pancreatic Neuroendocrine Neoplasms: Recent Advances and Controversies. Cancers 2022, 14, 1250. [Google Scholar] [CrossRef] [PubMed]
- Gaujoux, S.; Menegaux, F. Going above and beyond the pancreatic neuroendocrine tumor classification. JCO Oncol. Pract. 2020, 16, 731–732. [Google Scholar] [CrossRef]
- Keutgen, X.M.; Nilubol, N.; Kebebew, E. Malignant-functioning neuroendocrine tumors of the pancreas: A survival analysis. Surgery 2016, 159, 1382–1389. [Google Scholar] [CrossRef]
- Galgano, S.J.; Morani, A.C.; Gopireddy, D.R.; Sharbidre, K.; Bates, D.D.B.; Goenka, A.H.; Arif-Tiwari, H.; Itani, M.; Iravani, A.; Javadi, S.; et al. Pancreatic neuroendocrine neoplasms: A 2022 update for radiologists. Abdom. Radiol. 2022, 47, 3962–3970. [Google Scholar] [CrossRef]
- Kim, J.H.; Eun, H.W.; Kim, Y.J.; Lee, J.M.; Han, J.K.; Choi, B.-I. Pancreatic neuroendocrine tumour (PNET): Staging accuracy of MDCT and its diagnostic performance for the differentiation of PNET with uncommon CT findings from pancreatic adenocarcinoma. Eur. Radiol. 2016, 26, 1338–1347. [Google Scholar] [CrossRef]
- Sundin, A.; Arnold, R.; Baudin, E.; Cwikla, J.B.; Eriksson, B.; Fanti, S.; Fazio, N.; Giammarile, F.; Hicks, R.J.; Kjaer, A.; et al. ENETS consensus guidelines for the standards of care in neuroendocrine tumors: Radiological, nuclear medicine & hybrid imaging. Neuroendocrinology 2017, 105, 212–244. [Google Scholar] [CrossRef]
- Bonds, M.; Rocha, F.G. Neuroendocrine tumors of the pancreatobiliary and gastrointestinal tracts. Surg. Clin. North Am. 2020, 100, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Canellas, R.; Lo, G.; Bhowmik, S.; Ferrone, C.; Sahani, D. Pancreatic neuroendocrine tumor: Correlations between MRI features, tumor biology, and clinical outcome after surgery. J. Magn. Reason. Imaging 2018, 47, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Treglia, G.; Castaldi, P.; Rindi, G.; Giordano, A.; Rufini, V. Diagnostic performance of Gallium-68 somatostatin receptor PET and PET/CT in patients with thoracic and gastroenteropancreatic neuroendocrine tumours: A meta-analysis. Endocrine 2012, 42, 80–87. [Google Scholar] [CrossRef]
- Zhou, J.; Zhao, R.; Pan, Y.; Ju, H.; Huang, X.; Jiang, Y.; Jin, J.; Zhang, Y. The Diagnostic and Grading Accuracy of 68Ga-DOTATATE and 18F-FDG PET/MR for Pancreatic Neuroendocrine Neoplasms. Front. Oncol. 2022, 12, 796391. [Google Scholar] [CrossRef]
- Deguelte, S.; de Mestier, L.; Hentic, O.; Cros, J.; Lebtahi, R.; Hammel, P.; Kianmanesh, R. Preoperative imaging and pathologic classification for pancreatic neuroendocrine tumors. J. Visc. Surg. 2018, 155, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Khashab, M.A.; Yong, E.; Lennon, A.M.; Shin, E.J.; Amateau, S.; Hruban, R.H.; Olino, K.; Giday, S.; Fishman, E.K.; Wolfgang, C.L.; et al. EUS is still superior to multidetector computerized tomography for detection of pancreatic neuroendocrine tumors. Gastrointest. Endosc. 2011, 73, 691–696. [Google Scholar] [CrossRef]
- James, P.D.; Tsolakis, A.V.; Zhang, M.; Belletrutti, P.J.; Mohamed, R.; Roberts, D.J.; Heitman, S.J. Incremental benefit of preoperative EUS for the detection of pancreatic neuroendocrine tumors: A meta-analysis. Gastrointest. Endosc. 2015, 81, 848–856.e1. [Google Scholar] [CrossRef]
- Huai, J.C.; Zhang, W.; Niu, H.O.; Su, Z.X.; McNamara, J.J.; Machi, J. Localization and surgical treatment of pancreatic insulinomas guided by intraoperative ultrasound. Am. J. Surg. 1998, 175, 18–21. [Google Scholar] [CrossRef]
- Menzel, J.; Domschke, W. Intraductal ultrasonography may localize islet cell tumours negative on endoscopic ultrasound. Scand. J. Gastroenterol. 1998, 33, 109–112. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, T.; Oohashi, K.; Yamao, K.; Naitoh, Y.; Hirooka, Y.; Taki, T.; Itoh, A.; Hayakawa, S.; Watanabe, Y.; Goto, H. Intraductal ultrasonography of the pancreas: Development and clinical potential. Endoscopy 1997, 29, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Nikfarjam, M.; Warshaw, A.L.; Axelrod, L.; Deshpande, V.; Thayer, S.P.; Ferrone, C.R.; Castillo, C.F.-D. Improved contemporary surgical management of insulinomas: A 25-year experience at the Massachusetts General Hospital. Ann. Surg. 2008, 247, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Service, F.J.; McMahon, M.M.; O’Brien, P.C.; Ballard, D.J. Functioning insulinoma--incidence, recurrence, and long-term survival of patients: A 60-year study. Mayo Clin. Proc. 1991, 66, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Thoeni, R.F.; Mueller-Lisse, U.G.; Chan, R.; Do, N.K.; Shyn, P.B. Detection of small, functional islet cell tumors in the pancreas: Selection of MR imaging sequences for optimal sensitivity. Radiology 2000, 214, 483–490. [Google Scholar] [CrossRef]
- Kisker, O.; Bartsch, D.; Weinel, R.J.; Joseph, K.; Welcke, U.H.; Zaraca, F.; Rothmund, M. The value of somatostatin-receptor scintigraphy in newly diagnosed endocrine gastroenteropancreatic tumors. J. Am. Coll. Surg. 1997, 184, 487–492. [Google Scholar]
- Morera, J.; Guillaume, A.; Courtheoux, P.; Palazzo, L.; Rod, A.; Joubert, M.; Reznik, Y. Preoperative localization of an insulinoma: Selective arterial calcium stimulation test performance. J. Endocrinol. Investig. 2016, 39, 455–463. [Google Scholar] [CrossRef]
- Stabile, B.E.; Morrow, D.J.; Passaro, E. The gastrinoma triangle: Operative implications. Am. J. Surg. 1984, 147, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Zollinger, R.M.; Ellison, E.H. Primary peptic ulcerations of the jejunum associated with islet cell tumors of the pancreas. Ann. Surg. 1955, 142, 709–723; discussion 724. [Google Scholar] [CrossRef]
- Wermers, R.A.; Fatourechi, V.; Wynne, A.G.; Kvols, L.K.; Lloyd, R.V. The glucagonoma syndrome. Clinical and pathologic features in 21 patients. Medicine 1996, 75, 53–63. [Google Scholar] [CrossRef]
- Verner, J.V.; Morrison, A.B. Islet cell tumor and a syndrome of refractory watery diarrhea and hypokalemia. Am. J. Med. 1958, 25, 374–380. [Google Scholar] [CrossRef]
- Ito, T.; Igarashi, H.; Jensen, R.T. Pancreatic neuroendocrine tumors: Clinical features, diagnosis and medical treatment: Advances. Best Pract. Res. Clin. Gastroenterol. 2012, 26, 737–753. [Google Scholar] [CrossRef]
- Zandee, W.T.; van Adrichem, R.C.; Kamp, K.; Feelders, R.A.; van Velthuysen, M.-L.F.; de Herder, W.W. Incidence and prognostic value of serotonin secretion in pancreatic neuroendocrine tumours. Clin. Endocrinol. 2017, 87, 165–170. [Google Scholar] [CrossRef]
- Kamp, K.; Feelders, R.A.; Van Adrichem, R.C.S.; De Rijke, Y.B.; Van Nederveen, F.H.; Kwekkeboom, D.J.; De Herder, W.W. Parathyroid hormone-related peptide (PTHrP) secretion by gastroenteropancreatic neuroendocrine tumors (GEP-NETs): Clinical features, diagnosis, management, and follow-up. J. Clin. Endocrinol. Metab. 2014, 99, 3060–3069. [Google Scholar] [CrossRef] [PubMed]
- Ruddy, M.C.; Atlas, S.A.; Salerno, F.G. Hypertension associated with a renin-secreting adenocarcinoma of the pancreas. N. Engl. J. Med. 1982, 307, 993–997. [Google Scholar] [CrossRef] [PubMed]
- Samyn, I.; Fontaine, C.; Van Tussenbroek, F.; Pipeleers-Marichal, M.; De Grève, J. Paraneoplastic syndromes in cancer: Case 1. Polycythemia as a result of ectopic erythropoietin production in metastatic pancreatic carcinoid tumor. J. Clin. Oncol. 2004, 22, 2240–2242. [Google Scholar] [CrossRef] [PubMed]
- Rehfeld, J.F.; Federspiel, B.; Bardram, L. A neuroendocrine tumor syndrome from cholecystokinin secretion. N. Engl. J. Med. 2013, 368, 1165–1166. [Google Scholar] [CrossRef]
- Chung, J.O.; Hong, S.I.; Cho, D.H.; Lee, J.H.; Chung, D.J.; Chung, M.Y. Hypoglycemia associated with the production of insulin-like growth factor II in a pancreatic islet cell tumor: A case report. Endocr. J. 2008, 55, 607–612. [Google Scholar] [CrossRef]
- Roberts, R.E.; Zhao, M.; Whitelaw, B.C.; Ramage, J.; Diaz-Cano, S.; Le Roux, C.W.; Quaglia, A.; Huang, G.C.; Aylwin, S.J.B.; Whitelaw, B. GLP-1 and glucagon secretion from a pancreatic neuroendocrine tumor causing diabetes and hyperinsulinemic hypoglycemia. J. Clin. Endocrinol. Metab. 2012, 97, 3039–3045. [Google Scholar] [CrossRef] [PubMed]
- Brignardello, E.; Manti, R.; Papotti, M.; Allìa, E.; Campra, D.; Isolato, G.; Cassinis, M.C.; Fronda, G.; Boccuzzi, G. Ectopic secretion of LH by an endocrine pancreatic tumor. J. Endocrinol. Investig. 2004, 27, 361–365. [Google Scholar] [CrossRef]
- Kondo, T.; Matsuyama, R.; Ashihara, H.; Matsuo, Y.; Sasaki, K.; Goto, R.; Ono, K.; Takaki, Y.; Honda, Y.; Iyama, K.-I.; et al. A case of ectopic adrenocorticotropic hormone-producing pancreatic neuroendocrine tumor with multiple liver metastases. Endocr. J. 2010, 57, 229–236. [Google Scholar] [CrossRef]
- Tadokoro, R.; Sato, S.; Otsuka, F.; Ueno, M.; Ohkawa, S.; Katakami, H.; Taniyama, M.; Nagasaka, S. Metastatic Pancreatic Neuroendocrine Tumor that Progressed to Ectopic Adrenocorticotropic Hormone (ACTH) Syndrome with Growth Hormone-releasing Hormone (GHRH) Production. Intern. Med. 2016, 55, 2979–2983. [Google Scholar] [CrossRef]
- Borson-Chazot, F.; Garby, L.; Raverot, G.; Claustrat, F.; Raverot, V.; Sassolas, G. Acromegaly induced by ectopic secretion of GHRH: A review 30 years after GHRH discovery. Ann. Endocrinol. 2012, 73, 497–502. [Google Scholar] [CrossRef]
- Finkelstein, P.; Sharma, R.; Picado, O.; Gadde, R.; Stuart, H.; Ripat, C.; Livingstone, A.S.; Sleeman, D.; Merchant, N.; Yakoub, D. Pancreatic neuroendocrine tumors (pannets): Analysis of overall survival of nonsurgical management versus surgical resection. J. Gastrointest. Surg. 2017, 21, 855–866. [Google Scholar] [CrossRef] [PubMed]
- Ilett, E.E.; Langer, S.W.; Olsen, I.H.; Federspiel, B.; Kjær, A.; Knigge, U. Neuroendocrine carcinomas of the gastroenteropancreatic system: A comprehensive review. Diagnostics 2015, 5, 119–176. [Google Scholar] [CrossRef] [PubMed]
- Xourafas, D.; Tavakkoli, A.; Clancy, T.E.; Ashley, S.W. Distal pancreatic resection for neuroendocrine tumors: Is laparoscopic really better than open? J. Gastrointest. Surg. 2015, 19, 831–840. [Google Scholar] [CrossRef] [PubMed]
- de Rooij, T.; van Hilst, J.; Boerma, D.; van Dam, R.; van Eijck, C.; Gerhards, M.; Klaase, J.; Kazemier, G.; Luyer, M.; Busch, O.; et al. Minimally invasive vs. open distal pancreatectomy (LEOPARD): Multicenter patient-blinded randomized controlled trial. HPB 2018, 20, S293–S294. [Google Scholar] [CrossRef]
- Howe, J.R.; Merchant, N.B.; Conrad, C.; Keutgen, X.M.; Hallet, J.; Drebin, J.A.; Minter, R.M.; Lairmore, T.C.; Tseng, J.F.; Zeh, H.J.; et al. The north american neuroendocrine tumor society consensus paper on the surgical management of pancreatic neuroendocrine tumors. Pancreas 2020, 49, 1–33. [Google Scholar] [CrossRef]
- Mehrabi, A.; Fischer, L.; Hafezi, M.; Dirlewanger, A.; Grenacher, L.; Diener, M.K.; Fonouni, H.; Golriz, M.; Garoussi, C.; Fard, N.; et al. A systematic review of localization, surgical treatment options, and outcome of insulinoma. Pancreas 2014, 43, 675–686. [Google Scholar] [CrossRef]
- Falconi, M.; Eriksson, B.; Kaltsas, G.; Bartsch, D.K.; Capdevila, J.; Caplin, M.; Kos-Kudla, B.; Kwekkeboom, D.; Rindi, G.; Klöppel, G.; et al. ENETS Consensus Guidelines Update for the Management of Patients with Functional Pancreatic Neuroendocrine Tumors and Non-Functional Pancreatic Neuroendocrine Tumors. Neuroendocrinology 2016, 103, 153–171. [Google Scholar] [CrossRef]
- Fendrich, V.; Waldmann, J.; Bartsch, D.K.; Langer, P. Surgical management of pancreatic endocrine tumors. Nat. Rev. Clin. Oncol. 2009, 6, 419–428. [Google Scholar] [CrossRef]
- Cauley, C.E.; Pitt, H.A.; Ziegler, K.M.; Nakeeb, A.; Schmidt, C.M.; Zyromski, N.J.; House, M.G.; Lillemoe, K.D. Pancreatic enucleation: Improved outcomes compared to resection. J. Gastrointest. Surg. 2012, 16, 1347–1353. [Google Scholar] [CrossRef]
- Iihara, M.; Kanbe, M.; Okamoto, T.; Ito, Y.; Obara, T. Laparoscopic ultrasonography for resection of insulinomas. Surgery 2001, 130, 1086–1091. [Google Scholar] [CrossRef] [PubMed]
- Hirshberg, B.; Libutti, S.K.; Alexander, R.H.; Bartlett, D.L.; Cochran, C.; Livi, A.; Chang, R.; Shawker, T.; Skarulis, M.C.; Gorden, P. Blind distal pancreatectomy for occult insulinoma, an inadvisable procedure. J. Am. Coll. Surg. 2002, 194, 761–764. [Google Scholar] [CrossRef] [PubMed]
- Fahmawi, Y.; Mehta, A.; Abdalhadi, H.; Merritt, L.; Mizrahi, M. Efficacy and safety of endoscopic ultrasound-guided radiofrequency ablation for management of pancreatic lesions: A systematic review and meta-analysis. Transl. Gastroenterol. Hepatol. 2022, 7, 30. [Google Scholar] [CrossRef]
- Garg, R.; Mohammed, A.; Singh, A.; Harnegie, M.; Rustagi, T.; Stevens, T.; Chahal, P. EUS-guided radiofrequency and ethanol ablation for pancreatic neuroendocrine tumors: A systematic review and meta-analysis. Endosc. Ultrasound 2022, 11, 170–185. [Google Scholar] [CrossRef] [PubMed]
- Weber, H.; Venzon, D.J.; Lin, J.-T.; Fishbein, V.A.; Orbuch, M.; Strader, D.B.; Gibril, F.; Metz, D.C.; Fraker, D.L.; Norton, J.A.; et al. Determinants of metastatic rate and survival in patients with Zollinger-Ellison syndrome: A prospective long-term study. Gastroenterology 1995, 108, 1637–1649. [Google Scholar] [CrossRef]
- Stabile, B.E.; Passaro, E. Benign and malignant gastrinoma. Am. J. Surg. 1985, 149, 144–150. [Google Scholar] [CrossRef]
- Clancy, T.E. Surgical management of pancreatic neuroendocrine tumors. Hematol. Oncol. Clin. N. Am. 2016, 30, 103–118. [Google Scholar] [CrossRef]
- Doherty, G.M. Rare endocrine tumours of the GI tract. Best Pract. Res. Clin. Gastroenterol. 2005, 19, 807–817. [Google Scholar] [CrossRef]
- Halfdanarson, T.R.; Rabe, K.G.; Rubin, J.; Petersen, G.M. Pancreatic neuroendocrine tumors (PNETs): Incidence, prognosis and recent trend toward improved survival. Ann. Oncol. 2008, 19, 1727–1733. [Google Scholar] [CrossRef]
- Mansour, J.C.; Chen, H. Pancreatic endocrine tumors. J. Surg. Res. 2004, 120, 139–161. [Google Scholar] [CrossRef]
- Mastoraki, A.; Schizas, D.; Papoutsi, E.; Ntella, V.; Kanavidis, P.; Sioulas, A.; Tsoli, M.; Charalampopoulos, G.; Vailas, M.; Felekouras, E. Clinicopathological data and treatment modalities for pancreatic somatostatinomas. In Vivo 2020, 34, 3573–3582. [Google Scholar] [CrossRef]
- Halfdanarson, T.R.; Rubin, J.; Farnell, M.B.; Grant, C.S.; Petersen, G.M. Pancreatic endocrine neoplasms: Epidemiology and prognosis of pancreatic endocrine tumors. Endocr. Relat. Cancer 2008, 15, 409–427. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Sun, C.; Li, E.; Wang, J.; He, X.; Yuan, R.; Yi, C.; Liao, W.; Wu, L. Non-functional pancreatic neuroendocrine tumours: Emerging trends in incidence and mortality. BMC Cancer 2019, 19, 334. [Google Scholar] [CrossRef]
- Metz, D.C.; Jensen, R.T. Gastrointestinal neuroendocrine tumors: Pancreatic endocrine tumors. Gastroenterology 2008, 135, 1469–1492. [Google Scholar] [CrossRef]
- Bettini, R.; Partelli, S.; Boninsegna, L.; Capelli, P.; Crippa, S.; Pederzoli, P.; Scarpa, A.; Falconi, M. Tumor size correlates with malignancy in nonfunctioning pancreatic endocrine tumor. Surgery 2011, 150, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Jeune, F.; Taibi, A.; Gaujoux, S. Update on the surgical treatment of pancreatic neuroendocrine tumors. Scand. J. Surg. 2020, 109, 42–52. [Google Scholar] [CrossRef]
- Genç, C.G.; Falconi, M.; Partelli, S.; Muffatti, F.; Van Eeden, S.; Doglioni, C.; Klümpen, H.J.; Van Eijck, C.H.J.; Nieveen Van Dijkum, E.J.M. Recurrence of pancreatic neuroendocrine tumors and survival predicted by ki67. Ann. Surg. Oncol. 2018, 25, 2467–2474. [Google Scholar] [CrossRef] [PubMed]
- Assi, H.A.; Mukherjee, S.; Kunz, P.L.; Machiorlatti, M.; Vesely, S.; Pareek, V.; Hatoum, H. Surgery Versus Surveillance for Well-Differentiated, Nonfunctional Pancreatic Neuroendocrine Tumors: An 11-Year Analysis of the National Cancer Database. Oncologist 2020, 25, e276–e283. [Google Scholar] [CrossRef]
- Fathi, A.H.; Romanyshyn, J.; Barati, M.; Choudhury, U.; Chen, A.; Sosa, J.A. Predicting Aggressive Behavior in Nonfunctional Pancreatic Neuroendocrine Tumors with Emphasis on Tumor Size Significance and Survival Trends: A Population-Based Analysis of 1787 Patients. Am. Surg. 2020, 86, 458–466. [Google Scholar] [CrossRef]
- Lee, L.C.; Grant, C.S.; Salomao, D.R.; Fletcher, J.G.; Takahashi, N.; Fidler, J.L.; Levy, M.J.; Huebner, M. Small, nonfunctioning, asymptomatic pancreatic neuroendocrine tumors (PNETs): Role for nonoperative management. Surgery 2012, 152, 965–974. [Google Scholar] [CrossRef]
- Sadot, E.; Reidy-Lagunes, D.L.; Tang, L.H.; Do, R.K.G.; Gonen, M.; D’Angelica, M.I.; DeMatteo, R.P.; Kingham, T.P.; Koerkamp, B.G.; Untch, B.R.; et al. Observation versus Resection for Small Asymptomatic Pancreatic Neuroendocrine Tumors: A Matched Case-Control Study. Ann. Surg. Oncol. 2016, 23, 1361–1370. [Google Scholar] [CrossRef]
- Gaujoux, S.; Partelli, S.; Maire, F.; D'Onofrio, M.; Larroque, B.; Tamburrino, D.; Sauvanet, A.; Falconi, M.; Ruszniewski, P. Observational study of natural history of small sporadic nonfunctioning pancreatic neuroendocrine tumors. J. Clin. Endocrinol. Metab. 2013, 98, 4784–4789. [Google Scholar] [CrossRef] [PubMed]
- Arra, D.A.S.M.; Ribeiro, H.S.C.; Henklain, G.; Barbosa, A.; Torres, S.M.; Diniz, A.L.; Godoy, A.L.; Farias, I.C.; Costa, W.L.; Coimbra, F.J.F. Surgery or active surveillance for pNETs < 2 cm: Preliminary results from a single center Brazilian cohort. J. Surg. Oncol. 2022, 126, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, A.M.; Friedmann, P.; Del Rivero, J.; Libutti, S.K.; Laird, A.M. Resection versus expectant management of small incidentally discovered nonfunctional pancreatic neuroendocrine tumors. Surgery 2016, 159, 302–309. [Google Scholar] [CrossRef]
- Barenboim, A.; Lahat, G.; Nachmany, I.; Nakache, R.; Goykhman, Y.; Geva, R.; Osher, E.; Scapa, E.; Wolf, I.; Orbach, L.; et al. Resection versus observation of small asymptomatic nonfunctioning pancreatic neuroendocrine tumors. J. Gastrointest. Surg. 2020, 24, 1366–1374. [Google Scholar] [CrossRef]
- Kurita, Y.; Hara, K.; Kuwahara, T.; Mizuno, N.; Okuno, N.; Haba, S.; Okuno, M.; Natsume, S.; Senda, Y.; Kubota, K.; et al. Comparison of prognosis between observation and surgical resection groups with small sporadic non-functional pancreatic neuroendocrine neoplasms without distant metastasis. J. Gastroenterol. 2020, 55, 543–552. [Google Scholar] [CrossRef]
- Haynes, A.; Deshpande, V.; Ingkakul, T.; Vagefi, P.A.; Szymonifka, J.; Thayer, S.P.; Ferrone, C.R.; Wargo, J.A.; Warshaw, A.L. Implications of incidentally discovered, nonfunctioning pancreatic endocrine tumors: Short-term and long-term patient outcomes. Arch. Surg. 2011, 146, 534–538. [Google Scholar] [CrossRef] [PubMed]
- Partelli, S.; Ramage, J.K.; Massironi, S.; Zerbi, A.; Kim, H.B.; Niccoli, P.; Panzuto, F.; Landoni, L.; Tomazic, A.; Ibrahim, T.; et al. Management of asymptomatic sporadic nonfunctioning pancreatic neuroendocrine neoplasms (ASPEN) ≤2 cm: Study protocol for a prospective observational study. Front. Med. 2020, 7, 598438. [Google Scholar] [CrossRef]
- Partelli, S.; Massironi, S.; Zerbi, A.; Niccoli, P.; Kwon, W.; Landoni, L.; Panzuto, F.; Tomazic, A.; Bongiovanni, A.; Kaltsas, G.; et al. Management of asymptomatic sporadic non-functioning pancreatic neuroendocrine neoplasms no larger than 2 cm: Interim analysis of prospective ASPEN trial. Br. J. Surg. 2022, 109, 1186–1190. [Google Scholar] [CrossRef]
- Krogh, S.; Grønbæk, H.; Knudsen, A.R.; Kissmeyer-Nielsen, P.; Hummelshøj, N.E.; Dam, G. Predicting progression, recurrence, and survival in pancreatic neuroendocrine tumors: A single center analysis of 174 patients. Front. Endocrinol. 2022, 13, 925632. [Google Scholar] [CrossRef]
- Lillemoe, K.D.; Kaushal, S.; Cameron, J.L.; Sohn, T.A.; Pitt, H.A.; Yeo, C.J. Distal pancreatectomy: Indications and outcomes in 235 patients. Ann Surg. 1999, 229, 693–698; discussion 698. [Google Scholar] [CrossRef] [PubMed]
- Fong, Z.V.; Alvino, D.M.; Castillo, C.F.-D.; Nipp, R.D.; Traeger, L.N.; Ruddy, M.; Lubitz, C.C.; Johnson, C.D.; Chang, D.C.; Warshaw, A.L.; et al. Health-related Quality of Life and Functional Outcomes in 5-year Survivors After Pancreaticoduodenectomy. Ann. Surg. 2017, 266, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Braga, M.; Capretti, G.; Pecorelli, N.; Balzano, G.; Doglioni, C.; Ariotti, R.; Di Carlo, V. A prognostic score to predict major complications after pancreaticoduodenectomy. Ann. Surg. 2011, 254, 702–707; discussion 707. [Google Scholar] [CrossRef]
- Crippa, S.; Bassi, C.; Warshaw, A.L.; Falconi, M.; Partelli, S.; Thayer, S.P.; Pederzoli, P.; Castillo, C.F.-D. Middle pancreatectomy: Indications, short- and long-term operative outcomes. Ann. Surg. 2007, 246, 69–76. [Google Scholar] [CrossRef]
- Sato, N.; Yamaguchi, K.; Chijiiwa, K.; Tanaka, M. Risk analysis of pancreatic fistula after pancreatic head resection. Arch. Surg. 1998, 133, 1094–1098. [Google Scholar] [CrossRef]
- Bromley-Dulfano, R.; August, A.T.; Li, A.Y.; Park, W.; Visser, B. Characterizing gastrointestinal dysfunction after pancreatic resection: A single-center retrospective study. BMC Gastroenterol. 2022, 22, 488. [Google Scholar] [CrossRef]
- Mesleh, M.G.; Stauffer, J.A.; Asbun, H.J. Minimally invasive surgical techniques for pancreatic cancer: Ready for prime time? J. Hepatobiliary Pancreat. Sci. 2013, 20, 578–582. [Google Scholar] [CrossRef]
- Joyce, D.; Morris-Stiff, G.; Falk, G.A.; El-Hayek, K.; Chalikonda, S.; Walsh, R.M. Robotic surgery of the pancreas. World J. Gastroenterol. 2014, 20, 14726–14732. [Google Scholar] [CrossRef]
- Zheng, J.; Pulvirenti, A.; A Javed, A.; Michelakos, T.; Paniccia, A.M.; Lee, K.K.M.; Ferrone, C.R.M.; Wei, A.C.M.; He, J.M.; Zureikat, A.H.M.; et al. Minimally Invasive vs Open Pancreatectomy for Pancreatic Neuroendocrine Tumors: Multi-Institutional 10-Year Experience of 1023 Patients. J. Am. Coll. Surg. 2022, 235, 315–330. [Google Scholar] [CrossRef]
- Nießen, A.; Bechtiger, F.A.; Hinz, U.; Lewosinska, M.; Billmann, F.; Hackert, T.; Büchler, M.W.; Schimmack, S. Enucleation Is a Feasible Procedure for Well-Differentiated pNEN-A Matched Pair Analysis. Cancers 2022, 14, 2570. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, F.; Li, Z.; Ye, Z.; Zhuo, Q.; Xu, W.; Liu, W.; Liu, M.; Fan, G.; Qin, Y.; et al. Value of lymphadenectomy in patients with surgically resected pancreatic neuroendocrine tumors. BMC Surg. 2022, 22, 160. [Google Scholar] [CrossRef]
- Hashim, Y.M.; Trinkaus, K.M.; Linehan, D.C.; Strasberg, S.S.; Fields, R.C.; Cao, D.; Hawkins, W.G. Regional lymphadenectomy is indicated in the surgical treatment of pancreatic neuroendocrine tumors (PNETs). Ann. Surg. 2014, 259, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Ellison, T.A.; Wolfgang, C.L.; Shi, C.; Cameron, J.L.; Murakami, P.; Mun, L.J.; Singhi, A.D.; Cornish, T.; Olino, K.; Meriden, Z.; et al. A single institution’s 26-year experience with nonfunctional pancreatic neuroendocrine tumors: A validation of current staging systems and a new prognostic nomogram. Ann. Surg. 2014, 259, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Gratian, L.; Pura, J.; Dinan, M.; Roman, S.; Reed, S.; Sosa, J.A. Impact of extent of surgery on survival in patients with small nonfunctional pancreatic neuroendocrine tumors in the United States. Ann. Surg. Oncol. 2014, 21, 3515–3521. [Google Scholar] [CrossRef] [PubMed]
- Mao, R.; Zhao, H.; Li, K.; Luo, S.; Turner, M.; Cai, J.-Q.; Blazer, D. Outcomes of Lymph Node Dissection for Non-metastatic Pancreatic Neuroendocrine Tumors: A Propensity Score-Weighted Analysis of the National Cancer Database. Ann. Surg. Oncol. 2019, 26, 2722–2729. [Google Scholar] [CrossRef]
- Powell, A.C.; Libutti, S.K. Multiple endocrine neoplasia type 1: Clinical manifestations and management. Cancer Treat. Res. 2010, 153, 287–302. [Google Scholar] [CrossRef]
- Blansfield, J.A.; Choyke, L.; Morita, S.Y.; Choyke, P.L.; Pingpank, J.F.; Alexander, H.R.; Seidel, G.; Shutack, Y.; Yuldasheva, N.; Eugeni, M.; et al. Clinical, genetic and radiographic analysis of 108 patients with von Hippel-Lindau disease (VHL) manifested by pancreatic neuroendocrine neoplasms (PNETs). Surgery 2007, 142, 814–818; discussion 818.e1. [Google Scholar] [CrossRef]
- Laks, S.; Leeuwaarde, R.; Patel, D.; Keutgen, X.M.; Hammel, P.; Nilubol, N.; Links, T.P.; Halfdanarson, T.R.; Daniels, A.B.; Tirosh, A.; et al. Management recommendations for pancreatic manifestations of von Hippel-Lindau disease. Cancer 2022, 128, 435–446. [Google Scholar] [CrossRef]
- Ito, T.; Igarashi, H.; Uehara, H.; Berna, M.J.; Jensen, R.T. Causes of death and prognostic factors in multiple endocrine neoplasia type 1: A prospective study: Comparison of 106 MEN1/Zollinger-Ellison syndrome patients with 1613 literature MEN1 patients with or without pancreatic endocrine tumors. Medicine 2013, 92, 135–181. [Google Scholar] [CrossRef]
- Jensen, R.T.; Norton, J.A. Treatment of pancreatic neuroendocrine tumors in multiple endocrine neoplasia type 1: Some clarity but continued controversy. Pancreas 2017, 46, 589–594. [Google Scholar] [CrossRef]
- Triponez, F.; Goudet, P.; Dosseh, D.; Cougard, P.; Bauters, C.; Murat, A.; Cadiot, G.; Niccoli-Sire, P.; Calender, A.; Proye, C.A.G.; et al. Is surgery beneficial for MEN1 patients with small (< or = 2 cm), nonfunctioning pancreaticoduodenal endocrine tumor? An analysis of 65 patients from the GTE. World J. Surg. 2006, 30, 654–662; discussion 663. [Google Scholar] [CrossRef] [PubMed]
- Sadowski, S.M.; Pieterman, C.R.C.; Perrier, N.D.; Triponez, F.; Valk, G.D. Prognostic factors for the outcome of nonfunctioning pancreatic neuroendocrine tumors in MEN1: A systematic review of literature. Endocr. Relat. Cancer. 2020, 27, R145–R161. [Google Scholar] [CrossRef] [PubMed]
- Bergquist, J.R.; Shariq, O.A.; Li, A.Y.; Worth, P.J.; Chatzizacharias, N.; Soonawalla, Z.; Athanasopoulos, P.; Toumpanakis, C.; Hansen, P.; Parks, R.W.; et al. Clinical features and postoperative survival in patients with sporadic versus multiple endocrine neoplasia type 1-related pancreatic neuroendocrine tumors: An international cohort study. Surgery 2022, 172, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Tsoli, M.; Spei, M.-E.; Wallin, G.; Kaltsas, G.; Daskalakis, K. Association of a Palliative Surgical Approach to Stage IV Pancreatic Neuroendocrine Neoplasms with Survival: A Systematic Review and Meta-Analysis. Cancers 2020, 12, 2246. [Google Scholar] [CrossRef]
- Saxena, A.; Chua, T.C.; Perera, M.; Chu, F.; Morris, D.L. Surgical resection of hepatic metastases from neuroendocrine neoplasms: A systematic review. Surg. Oncol. 2012, 21, e131–e141. [Google Scholar] [CrossRef]
- Mou, Y.; Wang, Z.-Y.; Tan, C.-L.; Chen, Y.-H.; Liu, X.-B.; Ke, N.-W. The role of primary tumor resection in patients with pancreatic neuroendocrine tumors with liver metastases. Front. Oncol. 2022, 12, 838103. [Google Scholar] [CrossRef]
- Chawla, A.; Williams, R.T.; Sich, N.; Clancy, T.; Wang, J.; Ashley, S.; Pezzi, C.; Swanson, R. Pancreaticoduodenectomy and metastasectomy for metastatic pancreatic neuroendocrine tumors. J. Surg. Oncol. 2018, 118, 983–990. [Google Scholar] [CrossRef]
- Fairweather, M.; Swanson, R.; Brais, L.K.; Dutton, T.; Kulke, M.H.; Wang, J.; Clancy, T.E. Management of Neuroendocrine Tumor Liver Metastases: Long-Term Outcomes and Prognostic Factors from a Large Prospective Database. Ann. Surg. Oncol. 2017, 24, 2319–2325. [Google Scholar] [CrossRef]
- Tsilimigras, D.I.; Hyer, J.M.; Paredes, A.Z.; Ejaz, A.; Cloyd, J.M.; Beane, J.D.; Dillhoff, M.; Tsung, A.; Pawlik, T.M. Resection of Primary Gastrointestinal Neuroendocrine Tumor Among Patients with Non-Resected Metastases Is Associated with Improved Survival: A SEER-Medicare Analysis. J. Gastrointest. Surg. 2021, 25, 2368–2376. [Google Scholar] [CrossRef]
- Kjaer, J.; Clancy, T.E.; Thornell, A.M.; Andersson, N.; Hellman, P.M.; Crona, J.M.; Welin, S.M.; Sulciner, M.; Powell, B.; Brais, L.M.; et al. Benefit of primary tumor resection in stage IV, grade 1 and 2, pancreatic neuroendocrine tumors. Ann. Surg. Open 2022, 3, e151. [Google Scholar] [CrossRef]
- Mayo, S.C.; de Jong, M.C.; Pulitano, C.; Clary, B.M.; Reddy, S.K.; Gamblin, T.C.; Celinksi, S.A.; Kooby, D.A.; Staley, C.A.; Stokes, J.B.; et al. Surgical management of hepatic neuroendocrine tumor metastasis: Results from an international multi-institutional analysis. Ann. Surg. Oncol. 2010, 17, 3129–3136. [Google Scholar] [CrossRef] [PubMed]
- Maire, F.; Lombard-Bohas, C.; O’Toole, D.; Vullierme, M.-P.; Rebours, V.; Couvelard, A.; Pelletier, A.L.; Zappa, M.; Pilleul, F.; Hentic, O.; et al. Hepatic arterial embolization versus chemoembolization in the treatment of liver metastases from well-differentiated midgut endocrine tumors: A prospective randomized study. Neuroendocrinology 2012, 96, 294–300. [Google Scholar] [CrossRef] [PubMed]
- Mohan, H.; Nicholson, P.; Winter, D.C.; O’Shea, D.; O’Toole, D.; Geoghegan, J.; Maguire, D.; Hoti, E.; Traynor, O.; Cantwell, C.P. Radiofrequency ablation for neuroendocrine liver metastases: A systematic review. J. Vasc. Interv. Radiol. 2015, 26, 935–942.e1. [Google Scholar] [CrossRef] [PubMed]
- Dermine, S.; Palmieri, L.-J.; Lavolé, J.; Barré, A.; Dohan, A.; Ali, E.A.; Cottereau, A.-S.; Gaujoux, S.; Brezault, C.; Chaussade, S.; et al. Non-Pharmacological Therapeutic Options for Liver Metastases in Advanced Neuroendocrine Tumors. J. Clin. Med. 2019, 8, 1970. [Google Scholar] [CrossRef] [PubMed]
- Kulke, M.H.; Anthony, L.B.; Bushnell, D.; de Herder, W.W.; Goldsmith, S.J.; Klimstra, D.S.; Marx, S.J.; Pasieka, J.L.; Pommier, R.F.; Yao, J.C.; et al. NANETS treatment guidelines: Well-differentiated neuroendocrine tumors of the stomach and pancreas. Pancreas 2010, 39, 735–752. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Yao, J.C.; Ahrar, K.; Wallace, M.J.; Morello, F.A.; Madoff, D.C.; Murthy, R.; Hicks, M.E.; Ajani, J.A. Hepatic artery embolization and chemoembolization for treatment of patients with metastatic carcinoid tumors: The M.D. Anderson experience. Cancer J. 2003, 9, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, A.; Bester, L.; Salem, R.; Sharma, R.A.; Parks, R.W.; Ruszniewski, P. Role of hepatic intra-arterial therapies in metastatic neuroendocrine tumours (NET): Guidelines from the NET-Liver-Metastases Consensus Conference. HPB 2015, 17, 29–37. [Google Scholar] [CrossRef]
- Mazzaglia, P.J.; Berber, E.; Milas, M.; Siperstein, A.E. Laparoscopic radiofrequency ablation of neuroendocrine liver metastases: A 10-year experience evaluating predictors of survival. Surgery 2007, 142, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Chen, O.T.; Dojki, F.K.; Weber, S.M.; Hinshaw, J.L. Percutaneous Microwave Ablation of an Insulinoma in a Patient with Refractory Symptomatic Hypoglycemia. J. Gastrointest. Surg. 2015, 19, 1378–1381. [Google Scholar] [CrossRef]
- Egorov, A.V.; Vasilyev, I.A.; Musayev, G.H.; Mironova, A.V. The role of microwave ablation in management of functioning pancreatic neuroendocrine tumors. Gland. Surg. 2019, 8, 766–772. [Google Scholar] [CrossRef]
- Koshy, A.A.; Gordon, I.O.; Van Ha, T.G.; Kaplan, E.L.; Philipson, L.H. Metastatic insulinoma following resection of nonsecreting pancreatic islet cell tumor: A case report and review of the literature. J. Investig. Med. High Impact Case Rep. 2013, 1, 2324709612473274. [Google Scholar] [CrossRef] [PubMed]
- Cozzi, P.J.; Englund, R.; Morris, D.L. Cryotherapy treatment of patients with hepatic metastases from neuroendocrine tumors. Cancer 1995, 76, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Virdee, P.S.; Moschandreas, J.; Gebski, V.; Love, S.B.; Francis, E.A.; Wasan, H.S.; Van Hazel, G.; Gibbs, P.; A Sharma, R. Protocol for Combined Analysis of FOXFIRE, SIRFLOX, and FOXFIRE-Global Randomized Phase III Trials of Chemotherapy +/− Selective Internal Radiation Therapy as First-Line Treatment for Patients with Metastatic Colorectal Cancer. JMIR Res. Protoc. 2017, 6, e43. [Google Scholar] [CrossRef] [PubMed]
- Zubiri, L.; Bilbao, J.I.; Rodríguez, J.; Sangro, B. Selective internal radiation therapy: An effective treatment for hormonal syndromes in pancreatic neuroendocrine tumors. Hepat. Oncol. 2018, 5, HEP09. [Google Scholar] [CrossRef]
- Grozinsky-Glasberg, S.; Kaltsas, G.; Kaltsatou, M.; Lev-Cohain, N.; Klimov, A.; Vergadis, V.; Uri, I.; Bloom, A.I.; Gross, D.J. Hepatic intra-arterial therapies in metastatic neuroendocrine tumors: Lessons from clinical practice. Endocrine 2018, 60, 499–509. [Google Scholar] [CrossRef]
- Le Treut, Y.P.; Grégoire, E.; Klempnauer, J.; Belghiti, J.; Jouve, E.; Lerut, J.; Castaing, D.; Soubrane, O.; Boillot, O.; Mantion, G.; et al. Liver transplantation for neuroendocrine tumors in Europe-results and trends in patient selection: A 213-case European liver transplant registry study. Ann. Surg. 2013, 257, 807–815. [Google Scholar] [CrossRef]
- Gedaly, R.; Daily, M.F.; Davenport, D.; McHugh, P.P.; Koch, A.; Angulo, P.; Hundley, J.C. Liver transplantation for the treatment of liver metastases from neuroendocrine tumors: An analysis of the UNOS database. Arch Surg. 2011, 146, 953–958. [Google Scholar] [CrossRef]
- Mazzaferro, V.; Pulvirenti, A.; Coppa, J. Neuroendocrine tumors metastatic to the liver: How to select patients for liver transplantation? J. Hepatol. 2007, 47, 460–466. [Google Scholar] [CrossRef]
- Titan, A.L.; Norton, J.A.; Fisher, A.T.; Foster, D.S.; Harris, E.J.; Worhunsky, D.J.; Worth, P.J.; Dua, M.M.; Visser, B.C.; Poultsides, G.A.; et al. Evaluation of outcomes following surgery for locally advanced pancreatic neuroendocrine tumors. JAMA Netw. Open 2020, 3, e2024318. [Google Scholar] [CrossRef]
- Squires, M.H.; Worth, P.; Konda, B.; Shah, M.H.; Dillhoff, M.E.; Abdel-Misih, S.; Norton, J.A.; Visser, B.C.; Dua, M.; Pawlik, T.M.; et al. Neoadjuvant capecitabine/temozolomide for locally advanced or metastatic pancreatic neuroendocrine tumors. Pancreas 2020, 49, 355–360. [Google Scholar] [CrossRef]
- Sato, A.; Masui, T.; Sankoda, N.; Nakano, K.; Uchida, Y.; Anazawa, T.; Takaori, K.; Kawaguchi, Y.; Uemoto, S. A case of successful conversion from everolimus to surgical resection of a giant pancreatic neuroendocrine tumor. Surg. Case Rep. 2017, 3, 82. [Google Scholar] [CrossRef] [PubMed]
- Devata, S.; Kim, E.J. Neoadjuvant Chemotherapy with Capecitabine and Temozolomide for Unresectable Pancreatic Neuroendocrine Tumor. Case Rep. Oncol. 2012, 5, 622–626. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.D.; Katz, M.H.G. Preoperative therapy for pancreatic adenocarcinoma-precision beyond anatomy. Cancer 2022, 128, 3041–3056. [Google Scholar] [CrossRef]
- Michelakos, T.; Pergolini, I.; Castillo, C.F.-D.; Honselmann, K.C.; Cai, L.; Deshpande, V.; Wo, J.Y.; Ryan, D.P.; Allen, J.N.; Blaszkowsky, L.S.; et al. Predictors of resectability and survival in patients with borderline and locally advanced pancreatic cancer who underwent neoadjuvant treatment with FOLFIRINOX. Ann. Surg. 2019, 269, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Cloyd, J.M.; Omichi, K.; Mizuno, T.; Kawaguchi, Y.; Tzeng, C.-W.D.; Conrad, C.; Chun, Y.S.; Aloia, T.A.; Katz, M.H.G.; Lee, J.E.; et al. Preoperative fluorouracil, doxorubicin, and streptozocin for the treatment of pancreatic neuroendocrine liver metastases. Ann. Surg. Oncol. 2018, 25, 1709–1715. [Google Scholar] [CrossRef]
- Raymond, E.; Dahan, L.; Raoul, J.-L.; Bang, Y.-J.; Borbath, I.; Lombard-Bohas, C.; Valle, J.; Metrakos, P.; Smith, D.; Vinik, A.; et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N. Engl. J. Med. 2011, 364, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.; Lepage, C.; Vicaut, E.; Dominguez, S.; Coriat, R.; Dubreuil, O.; Lecomte, T.; Baudin, E.; Bouvet, L.V.; Samalin, E.; et al. Observational Study in a Real-World Setting of Targeted Therapy in the Systemic Treatment of Progressive Unresectable or Metastatic Well-Differentiated Pancreatic Neuroendocrine Tumors (pNETs) in France: OPALINE Study. Adv. Ther. 2022, 39, 2731–2748. [Google Scholar] [CrossRef]
- Ribas, A.; Hamid, O.; Daud, A.; Hodi, F.S.; Wolchok, J.D.; Kefford, R.; Joshua, A.M.; Patnaik, A.; Hwu, W.-J.; Weber, J.S.; et al. Association of pembrolizumab with tumor response and survival among patients with advanced melanoma. JAMA 2016, 315, 1600–1609. [Google Scholar] [CrossRef]
- Emens, L.A.; Cruz, C.; Eder, J.P.; Braiteh, F.; Chung, C.; Tolaney, S.M.; Kuter, I.; Nanda, R.; Cassier, P.A.; Delord, J.-P.; et al. Long-term Clinical Outcomes and Biomarker Analyses of Atezolizumab Therapy for Patients with Metastatic Triple-Negative Breast Cancer: A Phase 1 Study. JAMA Oncol. 2019, 5, 74–82. [Google Scholar] [CrossRef]
- Cercek, A.; Lumish, M.; Sinopoli, J.; Weiss, J.; Shia, J.; Lamendola-Essel, M.; El Dika, I.H.; Segal, N.; Shcherba, M.; Sugarman, R.; et al. PD-1 Blockade in Mismatch Repair-Deficient, Locally Advanced Rectal Cancer. N. Engl. J. Med. 2022, 386, 2363–2376. [Google Scholar] [CrossRef]
- Strosberg, J.R.; Mizuno, N.; Doi, T.; Grande, E.; Delord, J.-P.; Shapira-Frommer, R.; Bergsland, E.K.; Shah, M.H.; Fakih, M.; Takahashi, S.; et al. Efficacy and Safety of Pembrolizumab in Previously Treated Advanced Neuroendocrine Tumors: Results from the Phase II KEYNOTE-158 Study. Clin. Cancer Res. 2020, 26, 2124–2130. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Hu-Lieskovan, S. What does PD-L1 positive or negative mean? J. Exp. Med. 2016, 213, 2835–2840. [Google Scholar] [CrossRef] [PubMed]
- Strosberg, J.R.; E Caplin, M.; Kunz, P.L.; Ruszniewski, P.B.; Bodei, L.; Hendifar, A.; Mittra, E.; Wolin, E.M.; Yao, J.C.; Pavel, M.E.; et al. 177Lu-Dotatate plus long-acting octreotide versus high-dose long-acting octreotide in patients with midgut neuroendocrine tumours (NETTER-1): Final overall survival and long-term safety results from an open-label, randomised, controlled, phase 3 trial. Lancet Oncol. 2021, 22, 1752–1763. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Well-Differentiated | |||
| Ki67 Proliferation Index | Mitotic Index per High Power Field | ||
| Grade 1 | <3% | <2 mitoses/2 mm2 | |
| Grade 2 | 3–20% | 2–20 mitoses/2 mm2 | |
| Grade 3 | >20% | >20 mitoses/2 mm2 | |
| Poorly Differentiated | |||
| Ki Proliferation Index | Mitotic Index per High Power Field | Cell Cytomorphology | |
| Small cell | >20% | >20 mitoses/2 mm2 | Small |
| Large cell | >20% | >20 mitoses/2 mm2 | Large |
| Study Authors | Year | Type of Study | Number of Patients (n) | Size Cutoff | Frequency of Serial Imaging | Outcome |
|---|---|---|---|---|---|---|
| Lee et al. | 2012 | Retrospective cohort, single institution | 133 (56 surgery, 77 active surveillance) | <4 cm | Variable, CT or MRI | No difference in disease specific survival |
| Sadot et al. | 2016 | Retrospective cohort, single institution | 181 (77 surgery, 104 active surveillance) | <3 cm | Variable | No difference in disease specific survival |
| Rosenberg et al. | 2016 | Retrospective cohort, single institution | 35 (20 surgery, 15 active surveillance) | <2 cm | Every 6 months, CT or MRI | No difference in disease specific survival |
| Regenet et al. | 2016 | Retrospective cohort, multi-institution | 80 (66 surgery, 14 active surveillance) | <1.7 cm | Variable | No difference in disease free survival |
| Kurita et al. | 2020 | Retrospective cohort, single institution | 75 (52 surgery, 23 active surveillance) | ≤2 cm | Every 6 months, CT and EUS (for first 5 years) | No difference in overall survival |
| Barenboim et al. | 2020 | Retrospective cohort, single institution | 99 (55 surgery, 44 active surveillance) | <2 cm | Every 6 months, CT; Every 12 months, Gallium [67]. DOTATOC-PET | No difference in disease specific survival |
| Arra et al. | 2022 | Retrospective cohort, single institution | 64 (41 surgery, 23 active surveillance) | <2 cm | Every 6 months, CT or MRI (for first 2 years) | No difference in disease progression rate |
| North American Neuroendocrine Tumor Society (NANTS) | National Comprehensive Cancer Network (NCCN) | Canadian National Expert Group | European Neuroendocrine Tumor Society (ENETS) | |
|---|---|---|---|---|
| Active Surveillance | Size 1 cm or less Asymptomatic | Size less than 1 cm preferred, but can selectively be observed if less than 2 cm Asymptomatic Low-grade Incidentally discovered | Size less than 2 cm Solitary lesion with no evidence of invasive disease Low Ki67 Stability on serial surveillance | Size less than or equal to 2 cm Asymptomatic Low-grade No evidence of malignant potential |
| Consideration of Resection | Tumors 1–2 cm * | Invasive Node-positive tumors | Progression on surveillance | Symptomatic Higher grade (G2) Patient preference |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sulciner, M.L.; Clancy, T.E. Surgical Management of Pancreatic Neuroendocrine Tumors. Cancers 2023, 15, 2006. https://doi.org/10.3390/cancers15072006
Sulciner ML, Clancy TE. Surgical Management of Pancreatic Neuroendocrine Tumors. Cancers. 2023; 15(7):2006. https://doi.org/10.3390/cancers15072006
Chicago/Turabian StyleSulciner, Megan L., and Thomas E. Clancy. 2023. "Surgical Management of Pancreatic Neuroendocrine Tumors" Cancers 15, no. 7: 2006. https://doi.org/10.3390/cancers15072006
APA StyleSulciner, M. L., & Clancy, T. E. (2023). Surgical Management of Pancreatic Neuroendocrine Tumors. Cancers, 15(7), 2006. https://doi.org/10.3390/cancers15072006