

Cancer-Associated Fibroblasts: Master Tumor Microenvironment Modifiers


Abstract
Simple Summary
Abstract
1. Introduction
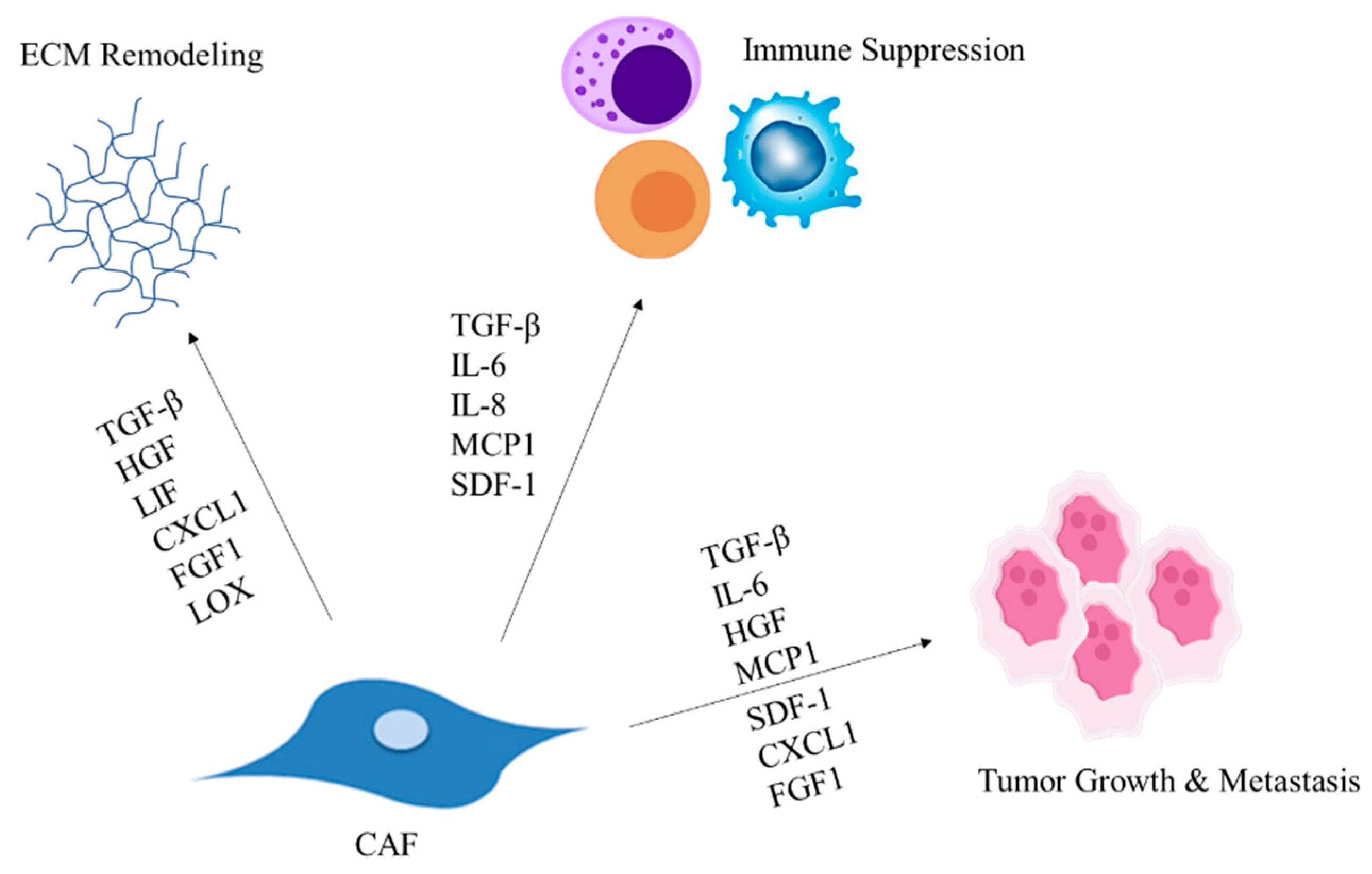
2. CAFs Modulate the ECM Composition
3. CAF-Secreted Factors Facilitate Tumor Progression
4. CAFs Modulate Immune Cell Recruitment Leading to Suppressed Immune Response

{kind=link}
| CAF Subtype | Factor | Effect | Cancer Type | Reference |
|---|---|---|---|---|
| eCAF | Macrophages | M2 polarization | GC | [103] |
| Not determined | Monocytes | Differentiation into macrophages and recruitment to tumors | CRC and breast | [72,73] |
| Not determined | Mast cells | Mast cell degranulation | GC | [104] |
| Not determined | Natural killer cells | Reduction in NK cytotoxicity | PDAC | [105] |
| Not determined | Neutrophils | Neutrophil chemotaxis | HCC | [106] |
| apCAF | Tregs | T-cell differentiation into Tregs | PDAC | [107] |
| High-CAF | Helper T cells | Reduced helper T population | HNSCC | [108] |
| apCAFs | Cytotoxic T cells | Suppression of cytotoxic T cells | PDAC | [12] |
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Knowles, L.M.; Stabile, L.P.; Egloff, A.M.; Rothstein, M.E.; Thomas, S.M.; Gubish, C.T.; Lerner, E.C.; Seethala, R.R.; Suzuki, S.; Quesnelle, K.M.; et al. Hgf and c-met participate in paracrine tumorigenic pathways in head and neck squamous cell cancer. Clin. Cancer Res. 2009, 15, 3740–3750. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Shinde, A.V.; Humeres, C.; Frangogiannis, N.G. The role of alpha-smooth muscle actin in fibroblast-mediated matrix contraction and remodeling. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 298–309. [Google Scholar] [CrossRef]
- Kumar, D.; New, J.; Vishwakarma, V.; Joshi, R.; Enders, J.; Lin, F.; Dasari, S.; Gutierrez, W.R.; Leef, G.; Ponnurangam, S.; et al. Cancer-associated fibroblasts drive glycolysis in a targetable signaling loop implicated in head and neck squamous cell carcinoma progression. Cancer Res. 2018, 78, 3769–3782. [Google Scholar] [CrossRef]
- Kuzet, S.E.; Gaggioli, C. Fibroblast activation in cancer: When seed fertilizes soil. Cell Tissue Res. 2016, 365, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F. Tumors: Wounds that do not heal-redux. Cancer Immunol. Res. 2015, 3, 1–11. [Google Scholar] [CrossRef]
- Gascard, P.; Tlsty, T.D. Carcinoma-associated fibroblasts: Orchestrating the composition of malignancy. Genes Dev. 2016, 30, 1002–1019. [Google Scholar] [CrossRef]
- Miyai, Y.; Esaki, N.; Takahashi, M.; Enomoto, A. Cancer-associated fibroblasts that restrain cancer progression: Hypotheses and perspectives. Cancer Sci. 2020, 111, 1047–1057. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef]
- Costa, A.; Kieffer, Y.; Scholer-Dahirel, A.; Pelon, F.; Bourachot, B.; Cardon, M.; Sirven, P.; Magagna, I.; Fuhrmann, L.; Bernard, C.; et al. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell 2018, 33, 463–479.e410. [Google Scholar] [CrossRef] [PubMed]
- Ohlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef] [PubMed]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef] [PubMed]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell 2017, 171, 1611–1624.e1624. [Google Scholar] [CrossRef]
- Grout, J.A.; Sirven, P.; Leader, A.M.; Maskey, S.; Hector, E.; Puisieux, I.; Steffan, F.; Cheng, E.; Tung, N.; Maurin, M.; et al. Spatial positioning and matrix programs of cancer-associated fibroblasts promote t-cell exclusion in human lung tumors. Cancer Discov. 2022, 12, 2606–2625. [Google Scholar] [CrossRef]
- Li, H.; Courtois, E.T.; Sengupta, D.; Tan, Y.; Chen, K.H.; Goh, J.J.L.; Kong, S.L.; Chua, C.; Hon, L.K.; Tan, W.S.; et al. Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat. Genet. 2017, 49, 708–718. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Rick, J.W.; Joshi, R.S.; Beniwal, A.; Spatz, J.; Gill, S.; Chang, A.C.; Choudhary, N.; Nguyen, A.T.; Sudhir, S.; et al. Single-cell rna sequencing and spatial transcriptomics reveal cancer-associated fibroblasts in glioblastoma with protumoral effects. J. Clin. Investig. 2023, 133, e147087. [Google Scholar] [CrossRef]
- Muncie, J.M.; Weaver, V.M. The physical and biochemical properties of the extracellular matrix regulate cell fate. Curr. Top Dev. Biol. 2018, 130, 1–37. [Google Scholar]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 2020, 11, 5120. [Google Scholar] [CrossRef]
- Nissen, N.I.; Karsdal, M.; Willumsen, N. Collagens and Cancer Associated Fibroblasts in the Reactive Stroma and Its Relation to Cancer Biology. J. Exp. Clin. Cancer Res. 2019, 38, 115. [Google Scholar] [CrossRef]
- Mittal, R.; Patel, A.P.; Debs, L.H.; Nguyen, D.; Patel, K.; Grati, M.; Mittal, J.; Yan, D.; Chapagain, P.; Liu, X.Z. Intricate functions of matrix metalloproteinases in physiological and pathological conditions. J. Cell. Physiol. 2016, 231, 2599–2621. [Google Scholar] [CrossRef]
- Jackson, H.W.; Defamie, V.; Waterhouse, P.; Khokha, R. Timps: Versatile extracellular regulators in cancer. Nat. Rev. Cancer 2017, 17, 38–53. [Google Scholar] [CrossRef]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef] [PubMed]
- Acerbi, I.; Cassereau, L.; Dean, I.; Shi, Q.; Au, A.; Park, C.; Chen, Y.Y.; Liphardt, J.; Hwang, E.S.; Weaver, V.M. Human breast cancer invasion and aggression correlates with ecm stiffening and immune cell infiltration. Integr. Biol. (Camb) 2015, 7, 1120–1134. [Google Scholar] [CrossRef] [PubMed]
- Najafi, M.; Farhood, B.; Mortezaee, K. Extracellular matrix (ecm) stiffness and degradation as cancer drivers. J. Cell. Biochem. 2019, 120, 2782–2790. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Yu, F.X. Gpcr-hippo signaling in cancer. Cells 2019, 8, 426. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, S.; Saito, A.; Nagase, T. Yap/taz signaling as a molecular link between fibrosis and cancer. Int. J. Mol. Sci. 2018, 19, 3674. [Google Scholar] [CrossRef]
- Meng, Z.; Qiu, Y.; Lin, K.C.; Kumar, A.; Placone, J.K.; Fang, C.; Wang, K.C.; Lu, S.; Pan, M.; Hong, A.W.; et al. Rap2 mediates mechanoresponses of the hippo pathway. Nature 2018, 560, 655–660. [Google Scholar] [CrossRef]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and yap-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef]
- Ferrari, N.; Ranftl, R.; Chicherova, I.; Slaven, N.D.; Moeendarbary, E.; Farrugia, A.J.; Lam, M.; Semiannikova, M.; Westergaard, M.C.W.; Tchou, J.; et al. Dickkopf-3 links hsf1 and yap/taz signalling to control aggressive behaviours in cancer-associated fibroblasts. Nat. Commun. 2019, 10, 130. [Google Scholar] [CrossRef]
- Sorushanova, A.; Delgado, L.M.; Wu, Z.; Shologu, N.; Kshirsagar, A.; Raghunath, R.; Mullen, A.M.; Bayon, Y.; Pandit, A.; Raghunath, M.; et al. The collagen suprafamily: From biosynthesis to advanced biomaterial development. Adv. Mater. 2019, 31, e1801651. [Google Scholar] [CrossRef]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef]
- Liu, J.; Shen, J.X.; Wu, H.T.; Li, X.L.; Wen, X.F.; Du, C.W.; Zhang, G.J. Collagen 1a1 (col1a1) promotes metastasis of breast cancer and is a potential therapeutic target. Discov. Med. 2018, 25, 211–223. [Google Scholar] [PubMed]
- Pickup, M.W.; Laklai, H.; Acerbi, I.; Owens, P.; Gorska, A.E.; Chytil, A.; Aakre, M.; Weaver, V.M.; Moses, H.L. Stromally derived lysyl oxidase promotes metastasis of transforming growth factor-beta-deficient mouse mammary carcinomas. Cancer Res. 2013, 73, 5336–5346. [Google Scholar] [CrossRef]
- Cordes, N.; Ney, M.; Beleites, T.; Aust, D.; Baretton, G.; Thames, H.; Baumann, M.; Krause, M.; Lock, S.; Appold, S. Retrospective investigation of the prognostic value of the beta1 integrin expression in patients with head and neck squamous cell carcinoma receiving primary radio(chemo)therapy. PLoS ONE 2018, 13, e0209479. [Google Scholar] [CrossRef]
- Hanley, C.J.; Noble, F.; Ward, M.; Bullock, M.; Drifka, C.; Mellone, M.; Manousopoulou, A.; Johnston, H.E.; Hayden, A.; Thirdborough, S.; et al. A subset of myofibroblastic cancer-associated fibroblasts regulate collagen fiber elongation, which is prognostic in multiple cancers. Oncotarget 2016, 7, 6159–6174. [Google Scholar] [CrossRef]
- Li, Q.; Zhu, C.C.; Ni, B.; Zhang, Z.Z.; Jiang, S.H.; Hu, L.P.; Wang, X.; Zhang, X.X.; Huang, P.Q.; Yang, Q.; et al. Lysyl oxidase promotes liver metastasis of gastric cancer via facilitating the reciprocal interactions between tumor cells and cancer associated fibroblasts. EBioMedicine 2019, 49, 157–171. [Google Scholar] [CrossRef]
- Brisson, B.K.; Mauldin, E.A.; Lei, W.; Vogel, L.K.; Power, A.M.; Lo, A.; Dopkin, D.; Khanna, C.; Wells, R.G.; Pure, E.; et al. Type iii collagen directs stromal organization and limits metastasis in a murine model of breast cancer. Am. J. Pathol. 2015, 185, 1471–1486. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; He, X.; Zhang, Y.; Hosaka, K.; Andersson, P.; Wu, J.; Wu, J.; Jing, X.; Du, Q.; Hui, X.; et al. Inflammatory cell-derived cxcl3 promotes pancreatic cancer metastasis through a novel myofibroblast-hijacked cancer escape mechanism. Gut 2022, 71, 129–147. [Google Scholar] [CrossRef] [PubMed]
- Graf, F.; Horn, P.; Ho, A.D.; Boutros, M.; Maercker, C. The extracellular matrix proteins type i collagen, type iii collagen, fibronectin, and laminin 421 stimulate migration of cancer cells. FASEB J. 2021, 35, e21692. [Google Scholar] [CrossRef]
- Huang, G.; Ge, G.; Izzi, V.; Greenspan, D.S. A3 chains of type v collagen regulate breast tumour growth via glypican-1. Nat. Commun. 2017, 8, 14351. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Palmero, I.; Torres, S.; Bartolome, R.A.; Pelaez-Garcia, A.; Larriba, M.J.; Lopez-Lucendo, M.; Pena, C.; Escudero-Paniagua, B.; Munoz, A.; Casal, J.I. Twist1-induced activation of human fibroblasts promotes matrix stiffness by upregulating palladin and collagen alpha1(vi). Oncogene 2016, 35, 5224–5236. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Cescon, M.; Bonaldo, P. Collagen vi in cancer and its biological mechanisms. Trends Mol. Med. 2013, 19, 410–417. [Google Scholar] [CrossRef]
- Raglow, Z.; Thomas, S.M. Tumor matrix protein collagen xiα1 in cancer. Cancer Lett. 2015, 357, 448–453. [Google Scholar] [CrossRef] [PubMed]
- Sok, J.C.; Lee, J.A.; Dasari, S.; Joyce, S.; Contrucci, S.C.; Egloff, A.M.; Trevelline, B.K.; Joshi, R.; Kumari, N.; Grandis, J.R.; et al. Collagen type xi α1 facilitates head and neck squamous cell cancer growth and invasion. Br. J. Cancer 2013, 109, 3049–3056. [Google Scholar] [CrossRef]
- Misawa, K.; Kanazawa, T.; Imai, A.; Endo, S.; Mochizuki, D.; Fukushima, H.; Misawa, Y.; Mineta, H. Prognostic value of type xxii and xxiv collagen mrna expression in head and neck cancer patients. Mol. Clin. Oncol. 2014, 2, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Ries, C. Cytokine functions of timp-1. Cell Mol. Life Sci. 2014, 71, 659–672. [Google Scholar] [CrossRef]
- Dittmer, A.; Dittmer, J. A caf-fueled timp-1/cd63/itgb1/stat3 feedback loop promotes migration and growth of breast cancer cells. Cancers 2022, 14, 4983. [Google Scholar] [CrossRef] [PubMed]
- Miyake, M.; Hori, S.; Morizawa, Y.; Tatsumi, Y.; Nakai, Y.; Anai, S.; Torimoto, K.; Aoki, K.; Tanaka, N.; Shimada, K.; et al. Cxcl1-mediated interaction of cancer cells with tumor-associated macrophages and cancer-associated fibroblasts promotes tumor progression in human bladder cancer. Neoplasia 2016, 18, 636–646. [Google Scholar] [CrossRef]
- Li, Y.Y.; Zhou, C.X.; Gao, Y. Interaction between oral squamous cell carcinoma cells and fibroblasts through tgf-β1 mediated by podoplanin. Exp. Cell Res. 2018, 369, 43–53. [Google Scholar] [CrossRef]
- Zhang, J.; Li, S.; Zhao, Y.; Ma, P.; Cao, Y.; Liu, C.; Zhang, X.; Wang, W.; Chen, L.; Li, Y. Cancer-associated fibroblasts promote the migration and invasion of gastric cancer cells via activating il-17a/jak2/stat3 signaling. Ann. Transl. Med. 2020, 8, 877. [Google Scholar] [CrossRef]
- Attieh, Y.; Clark, A.G.; Grass, C.; Richon, S.; Pocard, M.; Mariani, P.; Elkhatib, N.; Betz, T.; Gurchenkov, B.; Vignjevic, D.M. Cancer-associated fibroblasts lead tumor invasion through integrin-β3-dependent fibronectin assembly. J. Cell Biol. 2017, 216, 3509–3520. [Google Scholar] [CrossRef]
- Glentis, A.; Oertle, P.; Mariani, P.; Chikina, A.; El Marjou, F.; Attieh, Y.; Zaccarini, F.; Lae, M.; Loew, D.; Dingli, F.; et al. Cancer-associated fibroblasts induce metalloprotease-independent cancer cell invasion of the basement membrane. Nat. Commun. 2017, 8, 924. [Google Scholar] [CrossRef] [PubMed]
- Gaggioli, C.; Hooper, S.; Hidalgo-Carcedo, C.; Grosse, R.; Marshall, J.F.; Harrington, K.; Sahai, E. Fibroblast-led collective invasion of carcinoma cells with differing roles for rhogtpases in leading and following cells. Nat. Cell Biol. 2007, 9, 1392–1400. [Google Scholar] [CrossRef] [PubMed]
- Waldeland, J.O.; Polacheck, W.J.; Evje, S. Collective tumor cell migration in the presence of fibroblasts. J. Biomech. 2020, 100, 109568. [Google Scholar] [CrossRef] [PubMed]
- Neri, S.; Ishii, G.; Hashimoto, H.; Kuwata, T.; Nagai, K.; Date, H.; Ochiai, A. Podoplanin-expressing cancer-associated fibroblasts lead and enhance the local invasion of cancer cells in lung adenocarcinoma. Int. J. Cancer 2015, 137, 784–796. [Google Scholar] [CrossRef]
- Sanz-Moreno, V.; Gaggioli, C.; Yeo, M.; Albrengues, J.; Wallberg, F.; Viros, A.; Hooper, S.; Mitter, R.; Féral, C.C.; Cook, M.; et al. Rock and jak1 signaling cooperate to control actomyosin contractility in tumor cells and stroma. Cancer Cell 2011, 20, 229–245. [Google Scholar] [CrossRef]
- Corsa, C.A.; Brenot, A.; Grither, W.R.; Van Hove, S.; Loza, A.J.; Zhang, K.; Ponik, S.M.; Liu, Y.; DeNardo, D.G.; Eliceiri, K.W.; et al. The action of discoidin domain receptor 2 in basal tumor cells and stromal cancer-associated fibroblasts is critical for breast cancer metastasis. Cell Rep. 2016, 15, 2510–2523. [Google Scholar] [CrossRef]
- McAndrews, K.M.; Vazquez-Arreguin, K.; Kwak, C.; Sugimoto, H.; Zheng, X.; Li, B.; Kirtley, M.L.; LeBleu, V.S.; Kalluri, R. Alphasma(+) fibroblasts suppress lgr5(+) cancer stem cells and restrain colorectal cancer progression. Oncogene 2021, 40, 4440–4452. [Google Scholar] [CrossRef]
- Sperb, N.; Tsesmelis, M.; Wirth, T. Crosstalk between tumor and stromal cells in pancreatic ductal adenocarcinoma. Int. J. Mol. Sci 2020, 21, 5486. [Google Scholar] [CrossRef]
- Mizutani, Y.; Kobayashi, H.; Iida, T.; Asai, N.; Masamune, A.; Hara, A.; Esaki, N.; Ushida, K.; Mii, S.; Shiraki, Y.; et al. Meflin-positive cancer-associated fibroblasts inhibit pancreatic carcinogenesis. Cancer Res. 2019, 79, 5367–5381. [Google Scholar] [CrossRef]
- Fabregat, I.; Caballero-Diaz, D. Transforming growth factor-beta-induced cell plasticity in liver fibrosis and hepatocarcinogenesis. Front. Oncol. 2018, 8, 357. [Google Scholar] [CrossRef] [PubMed]
- Kojima, Y.; Acar, A.; Eaton, E.N.; Mellody, K.T.; Scheel, C.; Ben-Porath, I.; Onder, T.T.; Wang, Z.C.; Richardson, A.L.; Weinberg, R.A.; et al. Autocrine tgf-beta and stromal cell-derived factor-1 (sdf-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc. Natl. Acad. Sci. USA 2010, 107, 20009–20014. [Google Scholar] [CrossRef]
- Varadaraj, A.; Jenkins, L.M.; Singh, P.; Chanda, A.; Snider, J.; Lee, N.Y.; Amsalem-Zafran, A.R.; Ehrlich, M.; Henis, Y.I.; Mythreye, K. Tgf-beta triggers rapid fibrillogenesis via a novel tbetarii-dependent fibronectin-trafficking mechanism. Mol. Biol. Cell 2017, 28, 1195–1207. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.C.; Tse, A.P.; Huang, Y.P.; Zhu, Y.T.; Chiu, D.K.; Lai, R.K.; Au, S.L.; Kai, A.K.; Lee, J.M.; Wei, L.L.; et al. Lysyl oxidase-like 2 is critical to tumor microenvironment and metastatic niche formation in hepatocellular carcinoma. Hepatology 2014, 60, 1645–1658. [Google Scholar] [CrossRef]
- Yu, Y.; Xiao, C.H.; Tan, L.D.; Wang, Q.S.; Li, X.Q.; Feng, Y.M. Cancer-associated fibroblasts induce epithelial-mesenchymal transition of breast cancer cells through paracrine tgf-beta signalling. Br. J. Cancer 2014, 110, 724–732. [Google Scholar] [CrossRef]
- Suzuki, J.; Aokage, K.; Neri, S.; Sakai, T.; Hashimoto, H.; Su, Y.; Yamazaki, S.; Nakamura, H.; Tane, K.; Miyoshi, T.; et al. Relationship between podoplanin-expressing cancer-associated fibroblasts and the immune microenvironment of early lung squamous cell carcinoma. Lung Cancer 2021, 153, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.Y.; Chan, M.K.; Li, J.S.; Chan, A.S.; Tang, P.C.; Leung, K.T.; To, K.F.; Lan, H.Y.; Tang, P.M. Tgf-beta signaling: From tissue fibrosis to tumor microenvironment. Int. J. Mol. Sci. 2021, 22, 7575. [Google Scholar] [CrossRef]
- Kesh, K.; Garrido, V.T.; Dosch, A.; Durden, B.; Gupta, V.K.; Sharma, N.S.; Lyle, M.; Nagathihalli, N.; Merchant, N.; Saluja, A.; et al. Stroma secreted il6 selects for “stem-like” population and alters pancreatic tumor microenvironment by reprogramming metabolic pathways. Cell Death Dis. 2020, 11, 967. [Google Scholar] [CrossRef]
- Nagasaki, T.; Hara, M.; Nakanishi, H.; Takahashi, H.; Sato, M.; Takeyama, H. Interleukin-6 released by colon cancer-associated fibroblasts is critical for tumour angiogenesis: Anti-interleukin-6 receptor antibody suppressed angiogenesis and inhibited tumour-stroma interaction. Br. J. Cancer 2014, 110, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Qi, F.; Zhao, F.; Li, G.; Shao, S.; Zhang, X.; Yuan, L.; Feng, Y. Cancer-associated fibroblasts enhance tumor-associated macrophages enrichment and suppress nk cells function in colorectal cancer. Cell Death Dis. 2019, 10, 273. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Li, X.; Zhao, L.; Yang, Y.; Xue, C.; Gao, Y.; Li, J.; Han, Q.; Sun, Z.; Bai, C.; et al. The role of pkm2 nuclear translocation in the constant activation of the nf-kappab signaling pathway in cancer-associated fibroblasts. Cell Death Dis. 2021, 12, 291. [Google Scholar] [CrossRef]
- Gok Yavuz, B.; Gunaydin, G.; Gedik, M.E.; Kosemehmetoglu, K.; Karakoc, D.; Ozgur, F.; Guc, D. Cancer associated fibroblasts sculpt tumour microenvironment by recruiting monocytes and inducing immunosuppressive pd-1+ tams. Sci. Rep. 2019, 9, 3172. [Google Scholar] [CrossRef]
- Min, A.; Zhu, C.; Wang, J.; Peng, S.; Shuai, C.; Gao, S.; Tang, Z.; Su, T. Focal adhesion kinase knockdown in carcinoma-associated fibroblasts inhibits oral squamous cell carcinoma metastasis via downregulating mcp-1/ccl2 expression. J. Biochem. Mol. Toxicol. 2015, 29, 70–76. [Google Scholar] [CrossRef]
- Sun, C.; Li, X.; Guo, E.; Li, N.; Zhou, B.; Lu, H.; Huang, J.; Xia, M.; Shan, W.; Wang, B.; et al. Mcp-1/ccr-2 axis in adipocytes and cancer cell respectively facilitates ovarian cancer peritoneal metastasis. Oncogene 2020, 39, 1681–1695. [Google Scholar] [CrossRef]
- Deying, W.; Feng, G.; Shumei, L.; Hui, Z.; Ming, L.; Hongqing, W. Caf-derived hgf promotes cell proliferation and drug resistance by up-regulating the c-met/pi3k/akt and grp78 signalling in ovarian cancer cells. Biosci. Rep. 2017, 37, BSR20160470. [Google Scholar] [CrossRef] [PubMed]
- Konstorum, A.; Lowengrub, J.S. Activation of the hgf/c-met axis in the tumor microenvironment: A multispecies model. J. Theor. Biol. 2018, 439, 86–99. [Google Scholar] [CrossRef] [PubMed]
- Albrengues, J.; Bourget, I.; Pons, C.; Butet, V.; Hofman, P.; Tartare-Deckert, S.; Feral, C.C.; Meneguzzi, G.; Gaggioli, C. Lif mediates proinvasive activation of stromal fibroblasts in cancer. Cell Rep. 2014, 7, 1664–1678. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Gao, W.; Lytle, N.K.; Huang, P.; Yuan, X.; Dann, A.M.; Ridinger-Saison, M.; DelGiorno, K.E.; Antal, C.E.; Liang, G.; et al. Targeting lif-mediated paracrine interaction for pancreatic cancer therapy and monitoring. Nature 2019, 569, 131–135. [Google Scholar] [CrossRef]
- Wang, Y.; Lan, W.; Xu, M.; Song, J.; Mao, J.; Li, C.; Du, X.; Jiang, Y.; Li, E.; Zhang, R.; et al. Cancer-associated fibroblast-derived sdf-1 induces epithelial-mesenchymal transition of lung adenocarcinoma via cxcr4/beta-catenin/ppardelta signalling. Cell Death Dis. 2021, 12, 214. [Google Scholar] [CrossRef]
- Cheng, W.L.; Wang, C.S.; Huang, Y.H.; Tsai, M.M.; Liang, Y.; Lin, K.H. Overexpression of cxcl1 and its receptor cxcr2 promote tumor invasion in gastric cancer. Ann. Oncol. 2011, 22, 2267–2276. [Google Scholar] [CrossRef]
- Lefler, J.E.; MarElia-Bennett, C.B.; Thies, K.A.; Hildreth, B.E., 3rd; Sharma, S.M.; Pitarresi, J.R.; Han, L.; Everett, C.; Koivisto, C.; Cuitino, M.C.; et al. Stat3 in tumor fibroblasts promotes an immunosuppressive microenvironment in pancreatic cancer. Life Sci. Alliance 2022, 5, e202201460. [Google Scholar] [CrossRef] [PubMed]
- Hegab, A.E.; Ozaki, M.; Kameyama, N.; Gao, J.; Kagawa, S.; Yasuda, H.; Soejima, K.; Yin, Y.; Guzy, R.D.; Nakamura, Y.; et al. Effect of fgf/fgfr pathway blocking on lung adenocarcinoma and its cancer-associated fibroblasts. J. Pathol. 2019, 249, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Henriksson, M.L.; Edin, S.; Dahlin, A.M.; Oldenborg, P.A.; Oberg, A.; Van Guelpen, B.; Rutegard, J.; Stenling, R.; Palmqvist, R. Colorectal cancer cells activate adjacent fibroblasts resulting in fgf1/fgfr3 signaling and increased invasion. Am. J. Pathol. 2011, 178, 1387–1394. [Google Scholar] [CrossRef]
- Zhang, A.; Qian, Y.; Ye, Z.; Chen, H.; Xie, H.; Zhou, L.; Shen, Y.; Zheng, S. Cancer-associated fibroblasts promote m2 polarization of macrophages in pancreatic ductal adenocarcinoma. Cancer Med. 2017, 6, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Syed, V. Tgf-beta signaling in cancer. J. Cell. Biochem. 2016, 117, 1279–1287. [Google Scholar] [CrossRef]
- Krishnamurty, A.T.; Shyer, J.A.; Thai, M.; Gandham, V.; Buechler, M.B.; Yang, Y.A.; Pradhan, R.N.; Wang, A.W.; Sanchez, P.L.; Qu, Y.; et al. Lrrc15(+) myofibroblasts dictate the stromal setpoint to suppress tumour immunity. Nature 2022, 611, 148–154. [Google Scholar] [CrossRef]
- Heichler, C.; Scheibe, K.; Schmied, A.; Geppert, C.I.; Schmid, B.; Wirtz, S.; Thoma, O.M.; Kramer, V.; Waldner, M.J.; Buttner, C.; et al. Stat3 activation through il-6/il-11 in cancer-associated fibroblasts promotes colorectal tumour development and correlates with poor prognosis. Gut 2020, 69, 1269–1282. [Google Scholar] [CrossRef]
- New, J.; Arnold, L.; Ananth, M.; Alvi, S.; Thornton, M.; Werner, L.; Tawfik, O.; Dai, H.; Shnayder, Y.; Kakarala, K.; et al. Secretory autophagy in cancer-associated fibroblasts promotes head and neck cancer progression and offers a novel therapeutic target. Cancer Res. 2017, 77, 6679–6691. [Google Scholar] [CrossRef]
- Rizwani, W.; Allen, A.E.; Trevino, J.G. Hepatocyte growth factor from a clinical perspective: A pancreatic cancer challenge. Cancers 2015, 7, 1785–1805. [Google Scholar] [CrossRef]
- Kumar; Kandl, C.; Hamilton, C.D.; Shnayder, Y.; Tsue, T.T.; Kakarala, K.; Ledgerwood, L.; Sun, X.S.; Huang, H.J.; Girod, D.; et al. Mitigation of Tumor-Associated Fibroblast-Facilitated Head and Neck Cancer Progression with Anti-Hepatocyte Growth Factor Antibody Ficlatuzumab. JAMA Otolaryngol. Head Neck Surg. 2015, 141, 1133–1139. [Google Scholar] [CrossRef]
- Teng, F.; Tian, W.Y.; Wang, Y.M.; Zhang, Y.F.; Guo, F.; Zhao, J.; Gao, C.; Xue, F.X. Cancer-Associated Fibroblasts Promote the Progression of Endometrial Cancer Via the Sdf-1/Cxcr4 Axis. J. Hematol. Oncol. 2016, 9, 8. [Google Scholar] [CrossRef]
- Zhu, X.; Han, S.; Wu, S.; Bai, Y.; Zhang, N.; Wei, L. Dual role of twist1 in cancer-associated fibroblasts and tumor cells promoted epithelial-mesenchymal transition of esophageal cancer. Exp. Cell Res. 2019, 375, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.Y.; Lee, J.J.; Yeh, C.Y.; Yang, C.J.; Kok, S.H.; Ko, J.Y.; Tsai, F.C.; Chia, J.S. Reciprocal activation of cancer-associated fibroblasts and oral squamous carcinoma cells through cxcl1. Oral Oncol. 2019, 88, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Bordignon, P.; Bottoni, G.; Xu, X.; Popescu, A.S.; Truan, Z.; Guenova, E.; Kofler, L.; Jafari, P.; Ostano, P.; Rocken, M.; et al. Dualism of fgf and tgf-beta signaling in heterogeneous cancer-associated fibroblast activation with etv1 as a critical determinant. Cell Rep. 2019, 28, 2358–2372.e2356. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Xu, Q.; Pan, D.; Xu, D.; Niu, B.; Hong, W.; Zhang, R.; Li, X.; Chen, S. Hic-5 in cancer-associated fibroblasts contributes to esophageal squamous cell carcinoma progression. Cell Death Dis. 2019, 10, 873. [Google Scholar] [CrossRef]
- Chen, B.; Sang, Y.; Song, X.; Zhang, D.; Wang, L.; Zhao, W.; Liang, Y.; Zhang, N.; Yang, Q. Exosomal mir-500a-5p derived from cancer-associated fibroblasts promotes breast cancer cell proliferation and metastasis through targeting usp28. Theranostics 2021, 11, 3932–3947. [Google Scholar] [CrossRef]
- Boelens, M.C.; Wu, T.J.; Nabet, B.Y.; Xu, B.; Qiu, Y.; Yoon, T.; Azzam, D.J.; Twyman-Saint Victor, C.; Wiemann, B.Z.; Ishwaran, H.; et al. Exosome transfer from stromal to breast cancer cells regulates therapy resistance pathways. Cell 2014, 159, 499–513. [Google Scholar] [CrossRef]
- Sansone, P.; Berishaj, M.; Rajasekhar, V.K.; Ceccarelli, C.; Chang, Q.; Strillacci, A.; Savini, C.; Shapiro, L.; Bowman, R.L.; Mastroleo, C.; et al. Evolution of cancer stem-like cells in endocrine-resistant metastatic breast cancers is mediated by stromal microvesicles. Cancer Res. 2017, 77, 1927–1941. [Google Scholar] [CrossRef]
- Zhang, H.; Deng, T.; Liu, R.; Ning, T.; Yang, H.; Liu, D.; Zhang, Q.; Lin, D.; Ge, S.; Bai, M.; et al. Caf secreted mir-522 suppresses ferroptosis and promotes acquired chemo-resistance in gastric cancer. Mol. Cancer 2020, 19, 43. [Google Scholar] [CrossRef]
- Giusti, I.; Di Francesco, M.; Poppa, G.; Esposito, L.; D’Ascenzo, S.; Dolo, V. Tumor-derived extracellular vesicles activate normal human fibroblasts to a cancer-associated fibroblast-like phenotype, sustaining a pro-tumorigenic microenvironment. Front. Oncol. 2022, 12, 839880. [Google Scholar] [CrossRef] [PubMed]
- Najafi, M.; Hashemi Goradel, N.; Farhood, B.; Salehi, E.; Nashtaei, M.S.; Khanlarkhani, N.; Khezri, Z.; Majidpoor, J.; Abouzaripour, M.; Habibi, M.; et al. Macrophage polarity in cancer: A review. J. Cell. Biochem. 2019, 120, 2756–2765. [Google Scholar] [CrossRef] [PubMed]
- Tang, P.C.; Chung, J.Y.; Xue, V.W.; Xiao, J.; Meng, X.M.; Huang, X.R.; Zhou, S.; Chan, A.S.; Tsang, A.C.; Cheng, A.S.; et al. Smad3 promotes cancer-associated fibroblasts generation via macrophage-myofibroblast transition. Adv. Sci. 2022, 9, e2101235. [Google Scholar] [CrossRef]
- Li, X.; Sun, Z.; Peng, G.; Xiao, Y.; Guo, J.; Wu, B.; Li, X.; Zhou, W.; Li, J.; Li, Z.; et al. Single-cell rna sequencing reveals a pro-invasive cancer-associated fibroblast subgroup associated with poor clinical outcomes in patients with gastric cancer. Theranostics 2022, 12, 620–638. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Kinoshita, J.; Yamaguchi, T.; Aoki, T.; Saito, H.; Hamabe-Horiike, T.; Harada, S.; Nomura, S.; Inaki, N.; Fushida, S. Crosstalk between cancer-associated fibroblasts and immune cells in peritoneal metastasis: Inhibition in the migration of m2 macrophages and mast cells by tranilast. Gastric Cancer 2022, 25, 515–526. [Google Scholar] [CrossRef]
- Huang, Q.; Huang, M.; Meng, F.; Sun, R. Activated pancreatic stellate cells inhibit nk cell function in the human pancreatic cancer microenvironment. Cell Mol. Immunol. 2019, 16, 87–89. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Li, H.; Deng, Y.; Tai, Y.; Zeng, K.; Zhang, Y.; Liu, W.; Zhang, Q.; Yang, Y. Cancer-associated fibroblasts induce pdl1+ neutrophils through the il6-stat3 pathway that foster immune suppression in hepatocellular carcinoma. Cell Death Dis. 2018, 9, 422. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Wang, Z.; Zhang, Y.; Pradhan, R.N.; Ganguly, D.; Chandra, R.; Murimwa, G.; Wright, S.; Gu, X.; Maddipati, R.; et al. Mesothelial cell-derived antigen-presenting cancer-associated fibroblasts induce expansion of regulatory t cells in pancreatic cancer. Cancer Cell. 2022, 40, 656–673.e657. [Google Scholar] [CrossRef]
- Yang, Y.; Ma, B.; Han, L.; Xu, W.; Du, X.; Wei, W.; Liao, T.; Ji, Q.; Qu, N.; Wang, Y. Integrated single-cell and bulk rna sequencing analyses reveal a prognostic signature of cancer-associated fibroblasts in head and neck squamous cell carcinoma. Front. Genet. 2022, 13, 1028469. [Google Scholar] [CrossRef]
- Mak, T.K.; Li, X.; Huang, H.; Wu, K.; Huang, Z.; He, Y.; Zhang, C. The cancer-associated fibroblast-related signature predicts prognosis and indicates immune microenvironment infiltration in gastric cancer. Front. Immunol. 2022, 13, 951214. [Google Scholar] [CrossRef]
- Kato, T.; Noma, K.; Ohara, T.; Kashima, H.; Katsura, Y.; Sato, H.; Komoto, S.; Katsube, R.; Ninomiya, T.; Tazawa, H.; et al. Cancer-associated fibroblasts affect intratumoral cd8(+) and foxp3(+) t cells via il6 in the tumor microenvironment. Clin. Cancer Res. 2018, 24, 4820–4833. [Google Scholar] [CrossRef]

| Factor | Receptor | Reference |
|---|---|---|
| TGF-β1 and TGF-β2 | TGF-βR type I and II | [61,62,63,64,65,66,67] |
| IL-6 | IL-6R | [68,69] |
| IL-8 | CXCR1/2 | [70,71] |
| MCP1 (CCL2) | CCR-2 | [72,73,74] |
| HGF (SF) | c-Met | [75,76] |
| LIF | LIFR | [77,78] |
| SDF-1 (CXCL12) | CXCR4 | [8,79] |
| CXCL1 | CXCR2 | [48,80,81] |
| FGF1, FGF2, and FGF9 | FGFR, FGFR2, and FGFR9 | [82,83] |
| CCL-5 | CCR1, CCR3, and CCR5 | [84] |
| IL-11 | IL-11R | [84] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wright, K.; Ly, T.; Kriet, M.; Czirok, A.; Thomas, S.M. Cancer-Associated Fibroblasts: Master Tumor Microenvironment Modifiers. Cancers 2023, 15, 1899. https://doi.org/10.3390/cancers15061899
Wright K, Ly T, Kriet M, Czirok A, Thomas SM. Cancer-Associated Fibroblasts: Master Tumor Microenvironment Modifiers. Cancers. 2023; 15(6):1899. https://doi.org/10.3390/cancers15061899
Chicago/Turabian StyleWright, Kellen, Thuc Ly, Matthew Kriet, Andras Czirok, and Sufi Mary Thomas. 2023. "Cancer-Associated Fibroblasts: Master Tumor Microenvironment Modifiers" Cancers 15, no. 6: 1899. https://doi.org/10.3390/cancers15061899
APA StyleWright, K., Ly, T., Kriet, M., Czirok, A., & Thomas, S. M. (2023). Cancer-Associated Fibroblasts: Master Tumor Microenvironment Modifiers. Cancers, 15(6), 1899. https://doi.org/10.3390/cancers15061899