
Non-Oncogene Addiction of KRAS-Mutant Cancers to IL-1β via Versican and Mononuclear IKKβ

 , , ,
, , ,  , ,
, ,  , , , add
Show full author list
, , , add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Murine and Human Study Approval
2.2. Reagents
2.3. Cells
2.4. Experimental Mice
2.5. Mouse Tumor Models
2.6. Bioluminescence and Biofluorescence Imaging
2.7. Sequencing
2.8. Constructs and Transfections
2.9. Intrapleural Catheter
2.10. Cytology
2.11. Flow Cytometry
2.12. Immunohistochemistry
2.13. Bone Marrow Transfer (BMT) and Liposomal Clodronate
2.14. Bone Marrow Derived Macrophages (BMDM)
2.15. Immunoblotting
2.16. qPCR and Microarrays
2.17. Shotgun Proteomics
2.18. Cellular Treatments
2.19. Mouse Treatments
2.20. Data Availability
2.21. Transcription Factor Binding Site Analyses
2.22. Statistics
3. Results
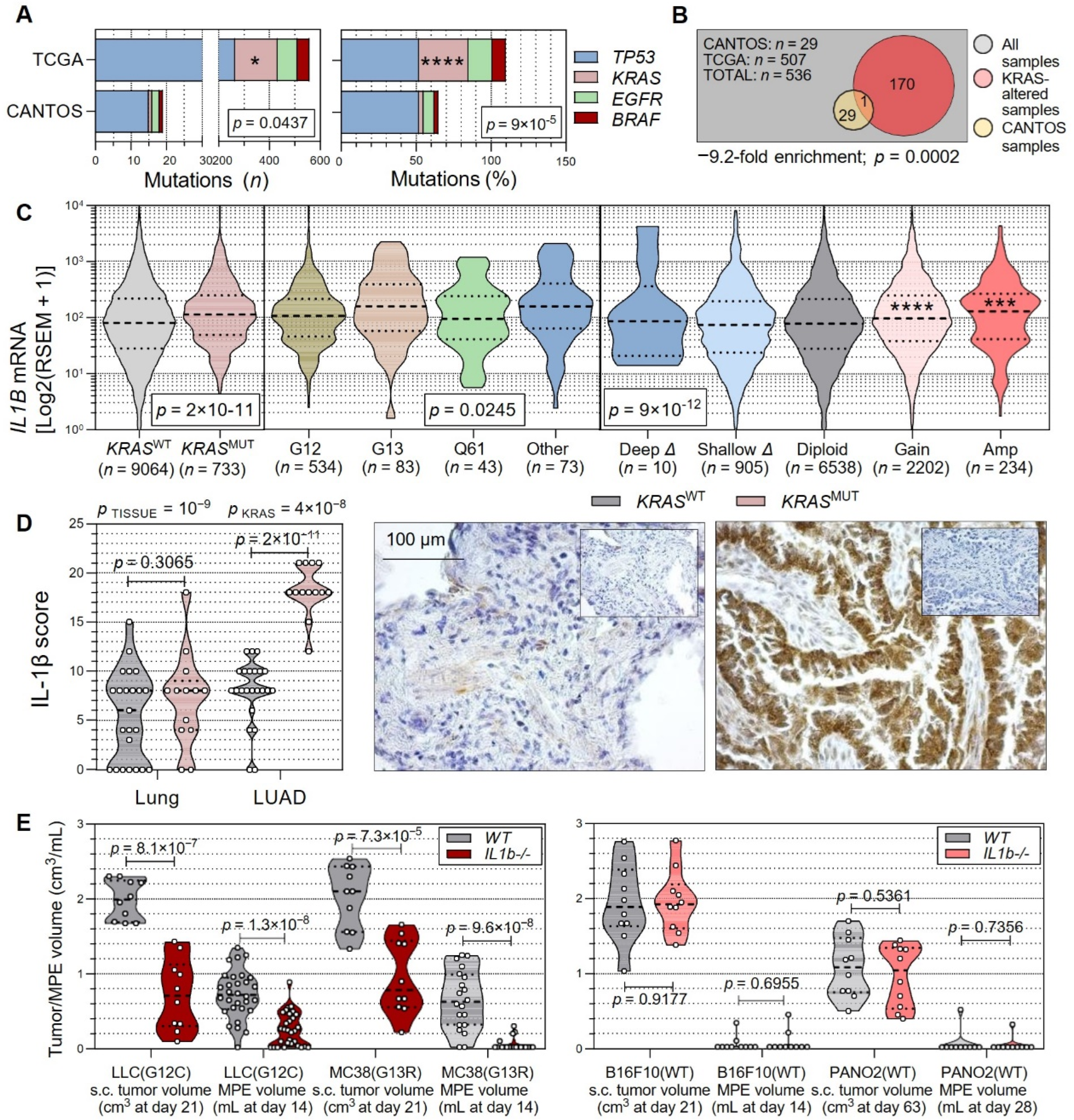
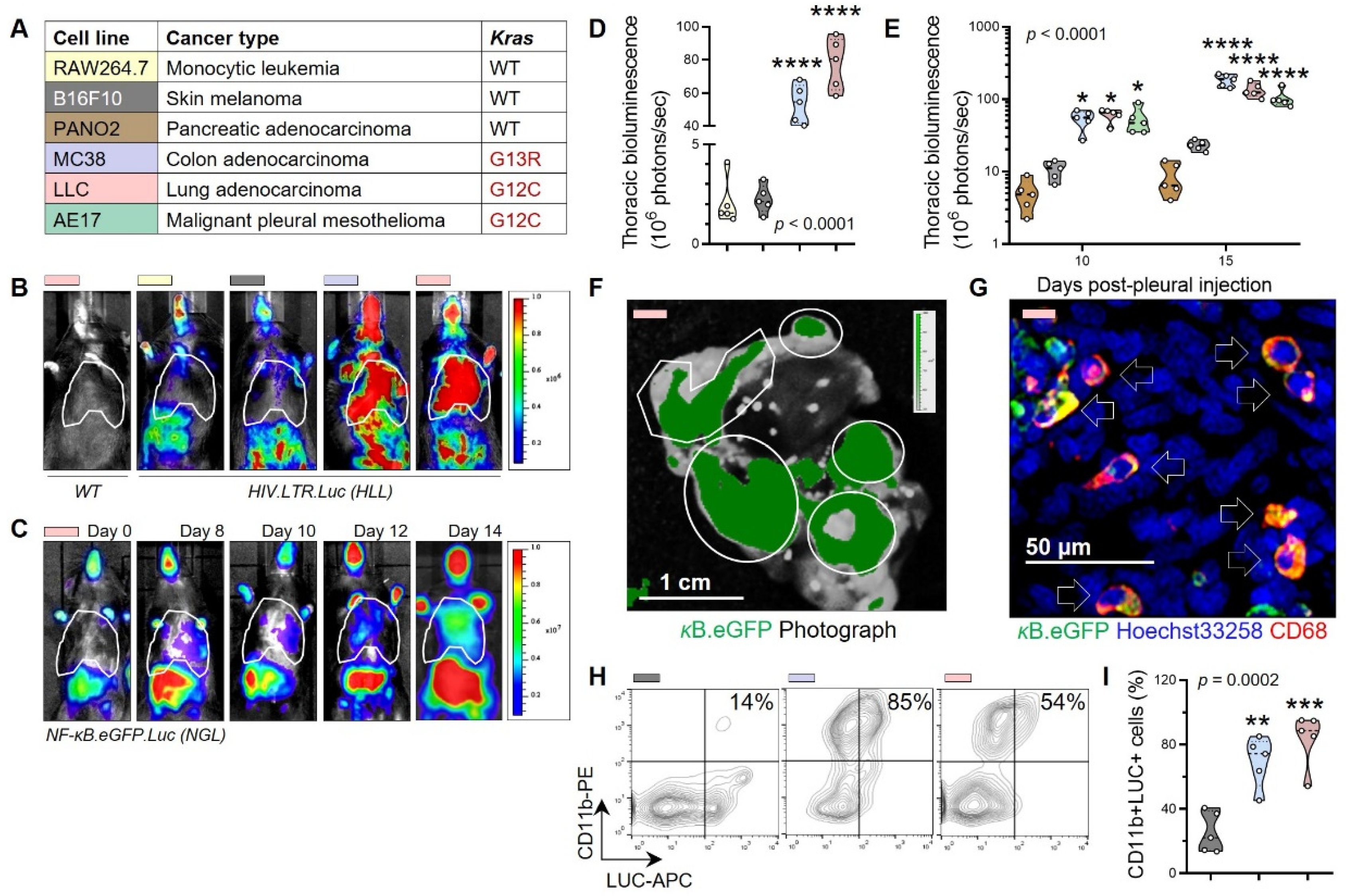
3.1. Non-Oncogene Addiction of KRAS-Mutant Human and Murine Cancers to IL-1β
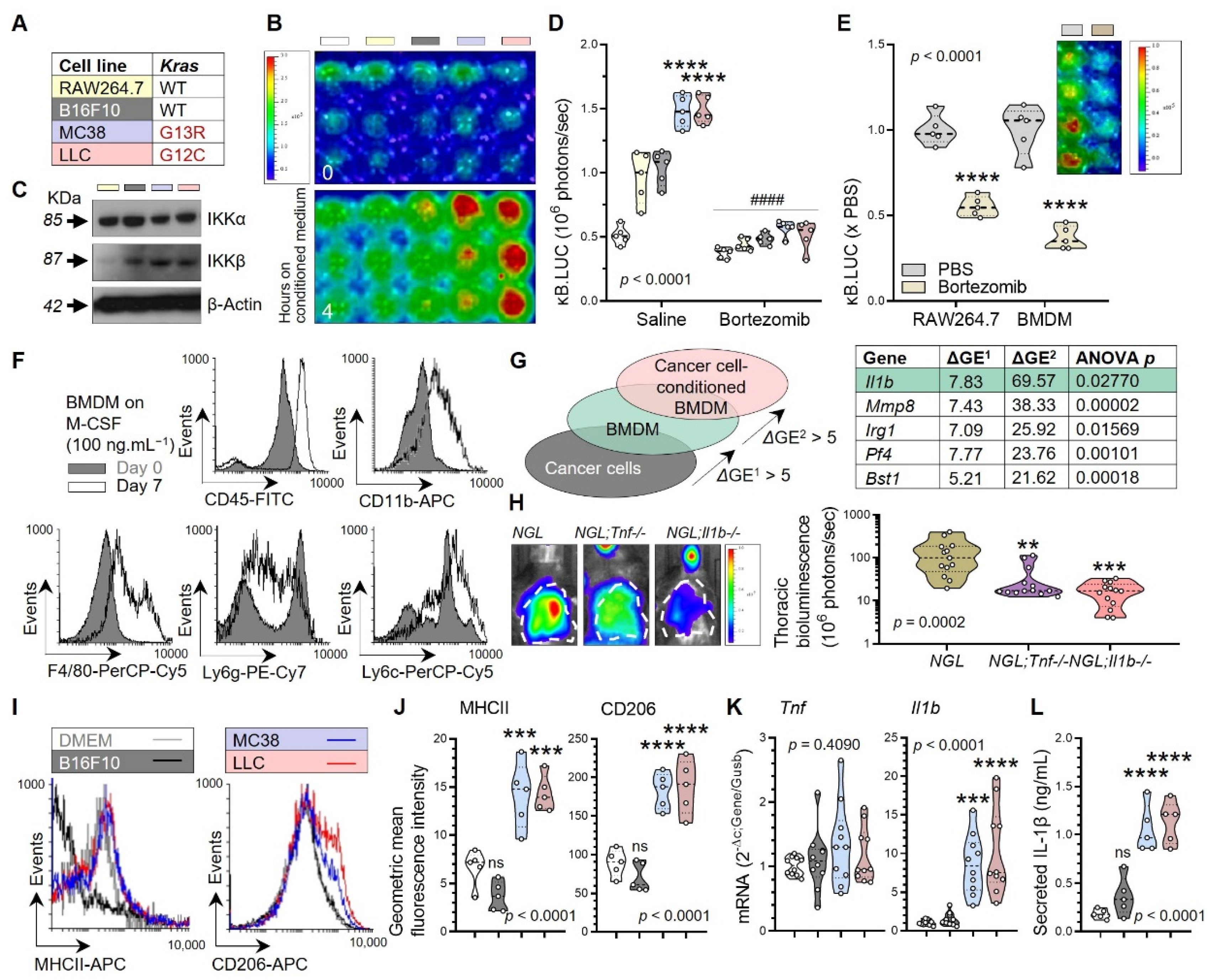
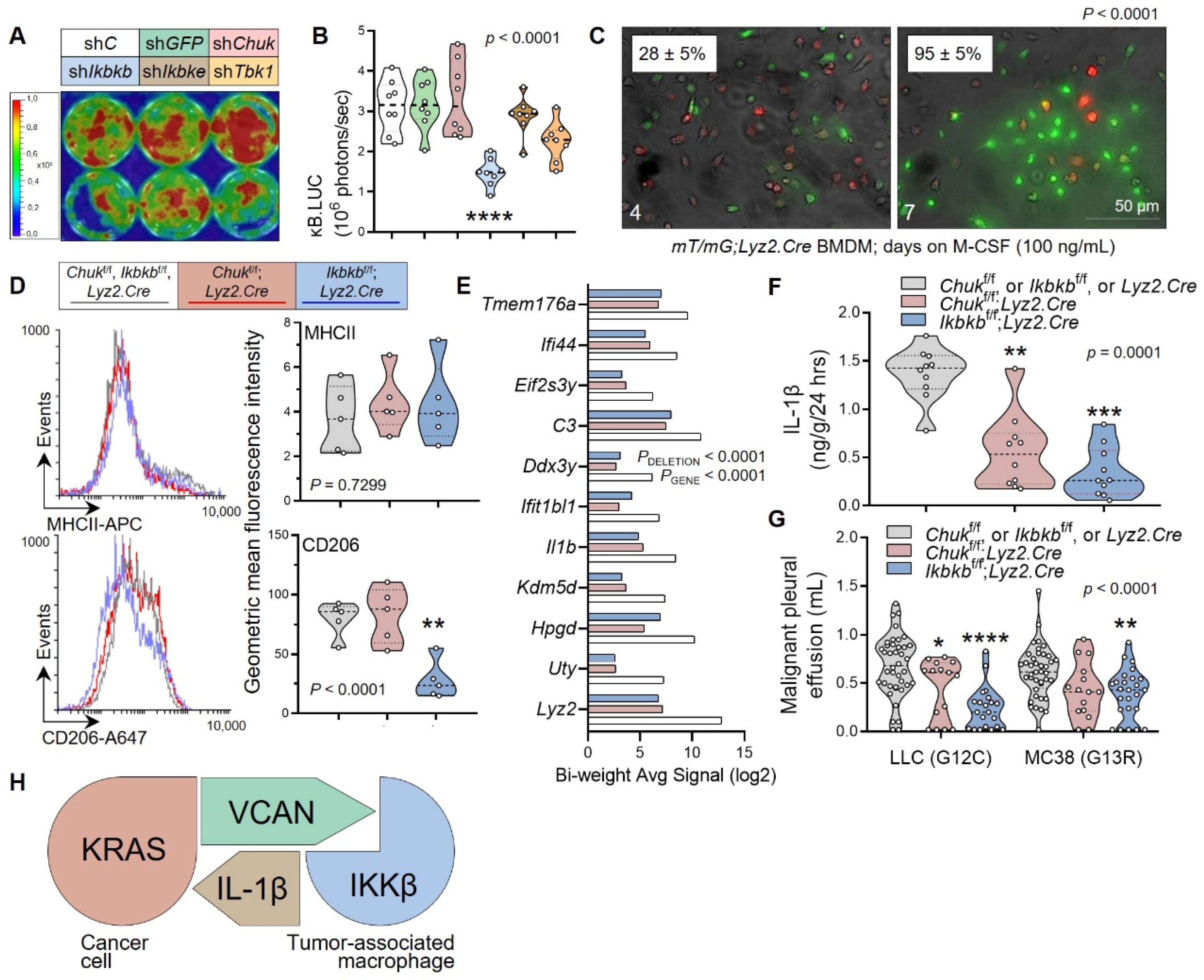
3.2. Tumor-Associated Macrophages as a Source of Tumorigenic IL-1β
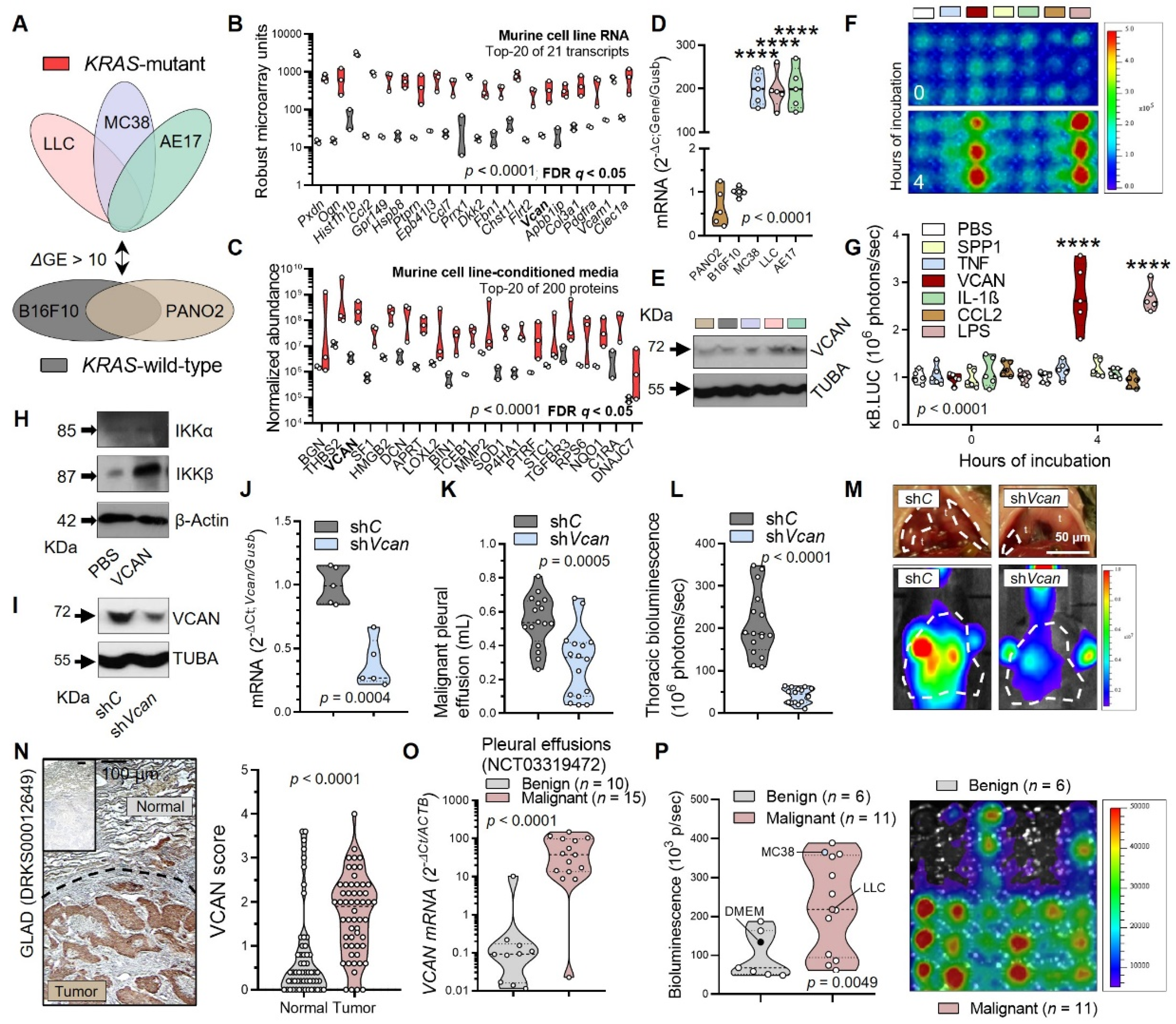
3.3. Tumor-Secreted Versican as a Key Macrophage Effector
3.4. Myeloid IKKB as the VCAN Accessory
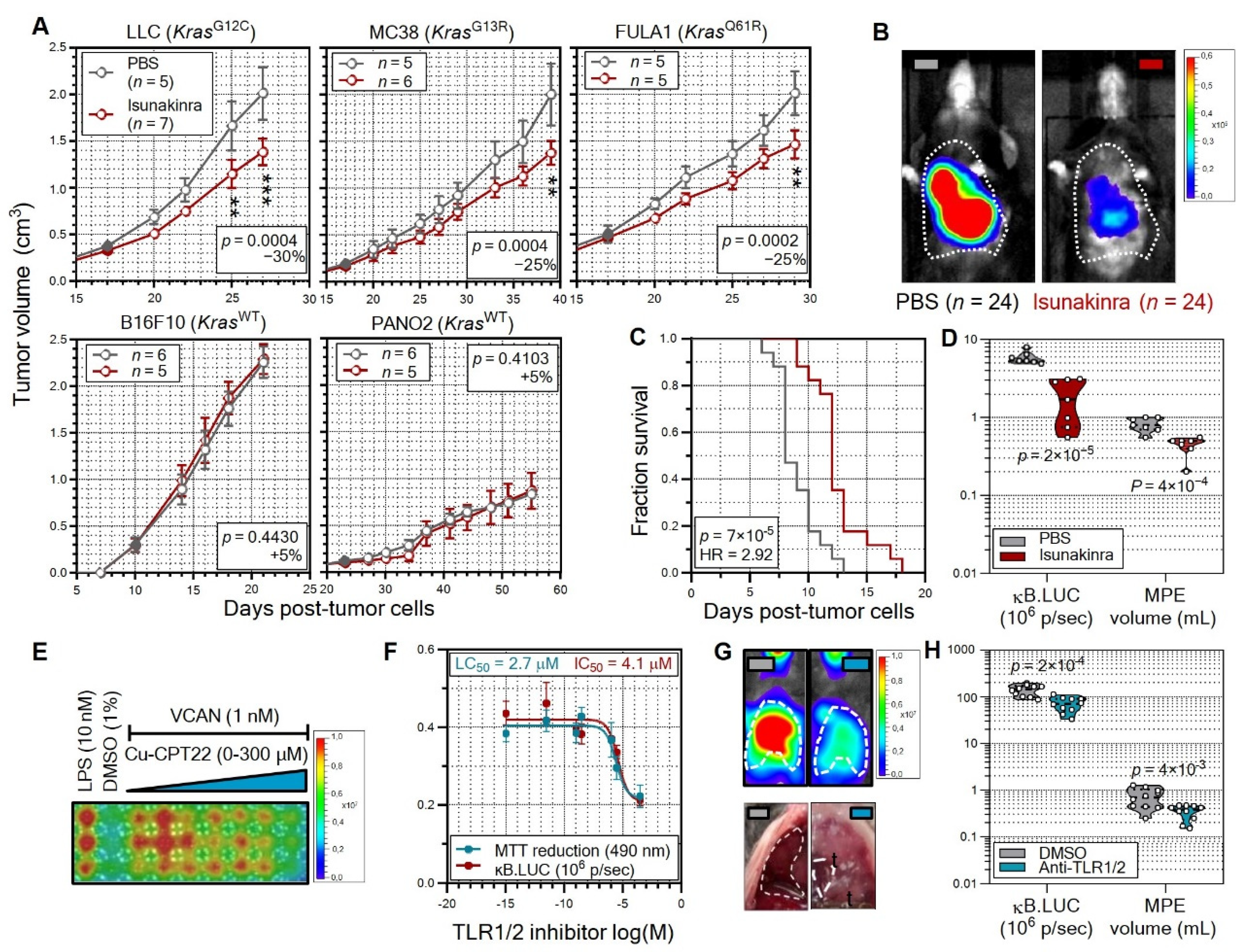
3.5. Non-oncogene Addiction of KRAS-Mutant Tumors to IL-1β Is Actionable
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Diakos, C.I.; Charles, K.A.; McMillan, D.C.; Clarke, S.J. Cancer-related inflammation and treatment effectiveness. Lancet Oncol. 2014, 15, e493–e503. [Google Scholar] [CrossRef]
- Ridker, P.M.; MacFadyen, J.G.; Thuren, T.; Everett, B.M.; Libby, P.; Glynn, R.J. Effect of interleukin-1beta inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: Exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 2017, 390, 1833–1842. [Google Scholar] [CrossRef]
- Available online: https://www.novartis.com/news/media-releases/novartis-provides-update-phase-iii-study-evaluating-canakinumab-acz885-second-or-third-line-treatment-combination-chemotherapy-non-small-cell-lung-cancer (accessed on 7 July 2021).
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef]
- Seguin, L.; Kato, S.; Franovic, A.; Camargo, M.F.; Lesperance, J.; Elliott, K.C.; Yebra, M.; Mielgo, A.; Lowy, A.M.; Husain, H.; et al. An integrin beta(3)-KRAS-RalB complex drives tumour stemness and resistance to EGFR inhibition. Nat. Cell Biol. 2014, 16, 457–468. [Google Scholar] [CrossRef]
- Soucek, L.; Lawlor, E.R.; Soto, D.; Shchors, K.; Swigart, L.B.; Evan, G.I. Mast cells are required for angiogenesis and macroscopic expansion of Myc-induced pancreatic islet tumors. Nat. Med. 2007, 13, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Bang, D.; Wilson, W.; Ryan, M.; Yeh, J.J.; Baldwin, A.S. GSK-3alpha promotes oncogenic KRAS function in pancreatic cancer via TAK1-TAB stabilization and regulation of noncanonical NF-kappaB. Cancer Discov. 2013, 3, 690–703. [Google Scholar] [CrossRef] [PubMed]
- Ling, J.; Kang, Y.; Zhao, R.; Xia, Q.; Lee, D.F.; Chang, Z.; Li, J.; Peng, B.; Fleming, J.B.; Wang, H.; et al. KrasG12D-induced IKK2/beta/NF-kappaB activation by IL-1alpha and p62 feedforward loops is required for development of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 105–120. [Google Scholar] [CrossRef]
- Agalioti, T.; Giannou, A.D.; Krontira, A.C.; Kanellakis, N.I.; Kati, D.; Vreka, M.; Pepe, M.; Spella, M.; Lilis, I.; Zazara, D.E.; et al. Mutant KRAS promotes malignant pleural effusion formation. Nat. Commun. 2017, 8, 15205. [Google Scholar] [CrossRef] [PubMed]
- Marazioti, A.; Lilis, I.; Vreka, M.; Apostolopoulou, H.; Kalogeropoulou, A.; Giopanou, I.; Giotopoulou, G.A.; Krontira, A.C.; Iliopoulou, M.; Kanellakis, N.I.; et al. Myeloid-derived interleukin-1beta drives oncogenic KRAS-NF-kappaBeta addiction in malignant pleural effusion. Nat. Commun. 2018, 9, 672. [Google Scholar] [CrossRef]
- Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=celex%3A32010L0063 (accessed on 17 January 2013).
- Klotz, L.V.; Courty, Y.; Lindner, M.; Petit-Courty, A.; Stowasser, A.; Koch, I.; Eichhorn, M.E.; Lilis, I.; Morresi-Hauf, A.; Arendt, K.A.M.; et al. Comprehensive clinical profiling of the Gauting locoregional lung adenocarcinoma donors. Cancer Med. 2019, 8, 1486–1499. [Google Scholar] [CrossRef]
- Marazioti, A.; Voulgaridis, A.; Psallidas, I.; Lamort, A.S.; Iliopoulou, M.; Krontira, A.C.; Lilis, I.; Asciak, R.; Kanellakis, N.I.; Rahman, N.M.; et al. Clinical identification of malignant pleural effusions. medRxiv 2021. [Google Scholar] [CrossRef]
- Available online: https://www.wma.net/policies-post/wma-declaration-of-helsinki-ethical-principles-for-medical-research-involving-human-subjects/ (accessed on 19 March 2016).
- Available online: https://www.drks.de/drks_web/navigate.do?navigationId=trial.HTML&TRIAL_ID=DRKS00012649 (accessed on 23 April 2017).
- Available online: https://clinicaltrials.gov/ct2/show/NCT03319472?term=NCT03319472&rank=1 (accessed on 24 October 2017).
- Hou, J.; Townson, S.A.; Kovalchin, J.T.; Masci, A.; Kiner, O.; Shu, Y.; King, B.M.; Schirmer, E.; Golden, K.; Thomas, C.; et al. Design of a superior cytokine antagonist for topical ophthalmic use. Proc. Natl. Acad. Sci. USA 2013, 110, 3913–3918. [Google Scholar] [CrossRef]
- Cheng, K.; Wang, X.; Zhang, S.; Yin, H. Discovery of small-molecule inhibitors of the TLR1/TLR2 complex. Angew. Chem. Int. Ed. Engl. 2012, 51, 12246–12249. [Google Scholar] [CrossRef] [PubMed]
- Giannou, A.D.; Marazioti, A.; Spella, M.; Kanellakis, N.I.; Apostolopoulou, H.; Psallidas, I.; Prijovich, Z.M.; Vreka, M.; Zazara, D.E.; Lilis, I.; et al. Mast cells mediate malignant pleural effusion formation. J. Clin. Investig. 2015, 125, 2317–2334. [Google Scholar] [CrossRef] [PubMed]
- Spella, M.; Lilis, I.; Pepe, M.A.; Chen, Y.; Armaka, M.; Lamort, A.S.; Zazara, D.E.; Roumelioti, F.; Vreka, M.; Kanellakis, N.I.; et al. Club cells form lung adenocarcinomas and maintain the alveoli of adult mice. eLife 2019, 8, e45571. [Google Scholar] [CrossRef] [PubMed]
- Vreka, M.; Lilis, I.; Papageorgopoulou, M.; Giotopoulou, G.A.; Lianou, M.; Giopanou, I.; Kanellakis, N.I.; Spella, M.; Agalioti, T.; Armenis, V.; et al. IkappaB Kinase alpha Is Required for Development and Progression of KRAS-Mutant Lung Adenocarcinoma. Cancer Res. 2018, 78, 2939–2951. [Google Scholar] [CrossRef]
- Kanellakis, N.I.; Giannou, A.D.; Pepe, M.A.A.; Agalioti, T.; Zazara, D.E.; Giopanou, I.; Psallidas, I.; Spella, M.; Marazioti, A.; Arendt, K.A.M.; et al. Tobacco chemical-induced mouse lung adenocarcinoma cell lines pin the prolactin orthologue proliferin as a lung tumour promoter. Carcinogenesis 2019, 40, 1352–1362. [Google Scholar] [CrossRef]
- Blackwell, T.S.; Yull, F.E.; Chen, C.L.; Venkatakrishnan, A.; Blackwell, T.R.; Hicks, D.J.; Lancaster, L.H.; Christman, J.W.; Kerr, L.D. Multiorgan nuclear factor kappa B activation in a transgenic mouse model of systemic inflammation. Am. J. Respir. Crit. Care Med. 2000, 162, 1095–1101. [Google Scholar] [CrossRef]
- Everhart, M.B.; Han, W.; Sherrill, T.P.; Arutiunov, M.; Polosukhin, V.V.; Burke, J.R.; Sadikot, R.T.; Christman, J.W.; Yull, F.E.; Blackwell, T.S. Duration and intensity of NF-kappaB activity determine the severity of endotoxin-induced acute lung injury. J. Immunol. 2006, 176, 4995–5005. [Google Scholar] [CrossRef]
- Muzumdar, M.D.; Tasic, B.; Miyamichi, K.; Li, L.; Luo, L. A global double-fluorescent Cre reporter mouse. Genesis 2007, 45, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Clausen, B.E.; Burkhardt, C.; Reith, W.; Renkawitz, R.; Forster, I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999, 8, 265–277. [Google Scholar] [CrossRef]
- Voehringer, D.; Liang, H.E.; Locksley, R.M. Homeostasis and effector function of lymphopenia-induced “memory-like” T cells in constitutively T cell-depleted mice. J. Immunol. 2008, 180, 4742–4753. [Google Scholar] [CrossRef] [PubMed]
- Pasparakis, M.; Alexopoulou, L.; Episkopou, V.; Kollias, G. Immune and inflammatory responses in TNF alpha-deficient mice: A critical requirement for TNF alpha in the formation of primary B cell follicles, follicular dendritic cell networks and germinal centers, and in the maturation of the humoral immune response. J. Exp. Med. 1996, 184, 1397–1411. [Google Scholar] [CrossRef]
- Gareus, R.; Huth, M.; Breiden, B.; Nenci, A.; Rosch, N.; Haase, I.; Bloch, W.; Sandhoff, K.; Pasparakis, M. Normal epidermal differentiation but impaired skin-barrier formation upon keratinocyte-restricted IKK1 ablation. Nat. Cell Biol. 2007, 9, 461–469. [Google Scholar] [CrossRef]
- Pasparakis, M.; Courtois, G.; Hafner, M.; Schmidt-Supprian, M.; Nenci, A.; Toksoy, A.; Krampert, M.; Goebeler, M.; Gillitzer, R.; Israel, A.; et al. TNF-mediated inflammatory skin disease in mice with epidermis-specific deletion of IKK2. Nature 2002, 417, 861–866. [Google Scholar] [CrossRef]
- Horai, R.; Asano, M.; Sudo, K.; Kanuka, H.; Suzuki, M.; Nishihara, M.; Takahashi, M.; Iwakura, Y. Production of mice deficient in genes for interleukin (IL)-1alpha, IL-1beta, IL-1alpha/beta, and IL-1 receptor antagonist shows that IL-1beta is crucial in turpentine-induced fever development and glucocorticoid secretion. J. Exp. Med. 1998, 187, 1463–1475. [Google Scholar] [CrossRef] [PubMed]
- Feyerabend, T.B.; Weiser, A.; Tietz, A.; Stassen, M.; Harris, N.; Kopf, M.; Radermacher, P.; Moller, P.; Benoist, C.; Mathis, D.; et al. Cre-mediated cell ablation contests mast cell contribution in models of antibody- and T cell-mediated autoimmunity. Immunity 2011, 35, 832–844. [Google Scholar] [CrossRef] [PubMed]
- Stathopoulos, G.T.; Sherrill, T.P.; Cheng, D.S.; Scoggins, R.M.; Han, W.; Polosukhin, V.V.; Connelly, L.; Yull, F.E.; Fingleton, B.; Blackwell, T.S. Epithelial NF-kappaB activation promotes urethane-induced lung carcinogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 18514–18519. [Google Scholar] [CrossRef]
- Merrick, C.; Sherrill, T.; Kanellakis, N.I.; Asciak, R.; Stathopoulos, G.T.; Maldonado, F.; Rahman, N.M.; Blackwell, T.; Psallidas, I. Novel mouse model of indwelling pleural catheter in mice with malignant pleural effusion. ERJ Open Res. 2019, 5, 00226-2018. [Google Scholar] [CrossRef] [PubMed]
- Available online: http://flowingsoftware.btk.fi/ (accessed on 17 May 2018).
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Everhart, M.B.; Han, W.; Parman, K.S.; Polosukhin, V.V.; Zeng, H.; Sadikot, R.T.; Li, B.; Yull, F.E.; Christman, J.W.; Blackwell, T.S. Intratracheal administration of liposomal clodronate accelerates alveolar macrophage reconstitution following fetal liver transplantation. J. Leukoc. Biol. 2005, 77, 173–180. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Grosche, A.; Hauser, A.; Lepper, M.F.; Mayo, R.; von Toerne, C.; Merl-Pham, J.; Hauck, S.M. The Proteome of Native Adult Muller Glial Cells From Murine Retina. Mol. Cell. Proteom. 2016, 15, 462–480. [Google Scholar] [CrossRef]
- Kall, L.; Canterbury, J.D.; Weston, J.; Noble, W.S.; MacCoss, M.J. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. Methods 2007, 4, 923–925. [Google Scholar] [CrossRef]
- Available online: https://www.encodeproject.org/ (accessed on 23 January 2020).
- ENCODE Project Consortium. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science 2004, 306, 636–640. [Google Scholar] [CrossRef]
- Lachmann, A.; Xu, H.; Krishnan, J.; Berger, S.I.; Mazloom, A.R.; Ma’ayan, A. ChEA: Transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics 2010, 26, 2438–2444. [Google Scholar] [CrossRef]
- Available online: https://maayanlab.cloud/Harmonizome/dataset/CHEA+Transcription+Factor+Targets (accessed on 26 January 2020).
- Faul, F.; Erdfelder, E.; Lang, A.G.; Buchner, A. G*Power 3: A flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav. Res. Methods 2007, 39, 175–191. [Google Scholar] [CrossRef]
- Available online: https://systems.crump.ucla.edu/hypergeometric/index.php (accessed on 18 February 2020).
- Wong, C.C.; Baum, J.; Silvestro, A.; Beste, M.T.; Bharani-Dharan, B.; Xu, S.; Wang, Y.A.; Wang, X.; Prescott, M.F.; Krajkovich, L.; et al. Inhibition of IL1beta by Canakinumab May Be Effective against Diverse Molecular Subtypes of Lung Cancer: An Exploratory Analysis of the CANTOS Trial. Cancer Res. 2020, 80, 5597–5605. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.D.; Alexandrov, A.; Kim, J.; Wala, J.; Berger, A.H.; Pedamallu, C.S.; Shukla, S.A.; Guo, G.; Brooks, A.N.; Murray, B.A.; et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet. 2016, 48, 607–616. [Google Scholar] [CrossRef]
- Available online: https://www.cbioportal.org/ (accessed on 22 November 2018).
- Available online: https://bit.ly/3uSO0D4 (accessed on 23 October 2018).
- McLoed, A.G.; Sherrill, T.P.; Cheng, D.S.; Han, W.; Saxon, J.A.; Gleaves, L.A.; Wu, P.; Polosukhin, V.V.; Karin, M.; Yull, F.E.; et al. Neutrophil-Derived IL-1beta Impairs the Efficacy of NF-kappaB Inhibitors against Lung Cancer. Cell Rep. 2016, 16, 120–132. [Google Scholar] [CrossRef]
- Voigt, C.; May, P.; Gottschlich, A.; Markota, A.; Wenk, D.; Gerlach, I.; Voigt, S.; Stathopoulos, G.T.; Arendt, K.A.M.; Heise, C.; et al. Cancer cells induce interleukin-22 production from memory CD4(+) T cells via interleukin-1 to promote tumor growth. Proc. Natl. Acad. Sci. USA 2017, 114, 12994–12999. [Google Scholar] [CrossRef]
- Hiscott, J.; Marois, J.; Garoufalis, J.; D’Addario, M.; Roulston, A.; Kwan, I.; Pepin, N.; Lacoste, J.; Nguyen, H.; Bensi, G.; et al. Characterization of a functional NF-kappa B site in the human interleukin 1 beta promoter: Evidence for a positive autoregulatory loop. Mol. Cell. Biol. 1993, 13, 6231–6240. [Google Scholar] [CrossRef] [PubMed]
- Manthiram, K.; Zhou, Q.; Aksentijevich, I.; Kastner, D.L. The monogenic autoinflammatory diseases define new pathways in human innate immunity and inflammation. Nat. Immunol. 2017, 18, 832–842. [Google Scholar] [CrossRef]
- Kim, S.; Takahashi, H.; Lin, W.W.; Descargues, P.; Grivennikov, S.; Kim, Y.; Luo, J.L.; Karin, M. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature 2009, 457, 102–106. [Google Scholar] [CrossRef]
- Kabbout, M.; Garcia, M.M.; Fujimoto, J.; Liu, D.D.; Woods, D.; Chow, C.W.; Mendoza, G.; Momin, A.A.; James, B.P.; Solis, L.; et al. ETS2 mediated tumor suppressive function and MET oncogene inhibition in human non-small cell lung cancer. Clin. Cancer Res. 2013, 19, 3383–3395. [Google Scholar] [CrossRef] [PubMed]
- Brouwer-Visser, J.; Cheng, W.Y.; Bauer-Mehren, A.; Maisel, D.; Lechner, K.; Andersson, E.; Dudley, J.T.; Milletti, F. Regulatory T-cell Genes Drive Altered Immune Microenvironment in Adult Solid Cancers and Allow for Immune Contextual Patient Subtyping. Cancer Epidemiol. Biomark. Prev. 2018, 27, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.A.; Beare, D.; Bindal, N.; Bamford, S.; Ward, S.; Cole, C.G.; Jia, M.; Kok, C.; Boutselakis, H.; De, T.; et al. COSMIC: High-Resolution Cancer Genetics Using the Catalogue of Somatic Mutations in Cancer. Curr. Protoc. Hum. Genet. 2016, 91, 10–11. [Google Scholar] [CrossRef]
- Nagy, A.; Lanczky, A.; Menyhart, O.; Gyorffy, B. Validation of miRNA prognostic power in hepatocellular carcinoma using expression data of independent datasets. Sci. Rep. 2018, 8, 9227. [Google Scholar] [CrossRef]
- Sotorasib Edges Closer to Approval. Cancer Discov. 2021, 11, OF2. [CrossRef] [PubMed]
- Stephen, A.G.; Esposito, D.; Bagni, R.K.; McCormick, F. Dragging ras back in the ring. Cancer Cell 2014, 25, 272–281. [Google Scholar] [CrossRef]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRAS(G12C) Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Greten, F.R.; Eckmann, L.; Greten, T.F.; Park, J.M.; Li, Z.W.; Egan, L.J.; Kagnoff, M.F.; Karin, M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 2004, 118, 285–296. [Google Scholar] [CrossRef]
- Yang, J.; Hawkins, O.E.; Barham, W.; Gilchuk, P.; Boothby, M.; Ayers, G.D.; Joyce, S.; Karin, M.; Yull, F.E.; Richmond, A. Myeloid IKKbeta promotes antitumor immunity by modulating CCL11 and the innate immune response. Cancer Res. 2014, 74, 7274–7284. [Google Scholar] [CrossRef]
- Miyakoshi, S.; Kami, M.; Yuji, K.; Matsumura, T.; Takatoku, M.; Sasaki, M.; Narimatsu, H.; Fujii, T.; Kawabata, M.; Taniguchi, S.; et al. Severe pulmonary complications in Japanese patients after bortezomib treatment for refractory multiple myeloma. Blood 2006, 107, 3492–3494. [Google Scholar] [CrossRef] [PubMed]
- Moreau, P.; Mateos, M.V.; Berenson, J.R.; Weisel, K.; Lazzaro, A.; Song, K.; Dimopoulos, M.A.; Huang, M.; Zahlten-Kumeli, A.; Stewart, A.K. Once weekly versus twice weekly carfilzomib dosing in patients with relapsed and refractory multiple myeloma (A.R.R.O.W.): Interim analysis results of a randomised, phase 3 study. Lancet Oncol. 2018, 19, 953–964. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spella, M.; Ntaliarda, G.; Skiadas, G.; Lamort, A.-S.; Vreka, M.; Marazioti, A.; Lilis, I.; Bouloukou, E.; Giotopoulou, G.A.; Pepe, M.A.A.; et al. Non-Oncogene Addiction of KRAS-Mutant Cancers to IL-1β via Versican and Mononuclear IKKβ. Cancers 2023, 15, 1866. https://doi.org/10.3390/cancers15061866
Spella M, Ntaliarda G, Skiadas G, Lamort A-S, Vreka M, Marazioti A, Lilis I, Bouloukou E, Giotopoulou GA, Pepe MAA, et al. Non-Oncogene Addiction of KRAS-Mutant Cancers to IL-1β via Versican and Mononuclear IKKβ. Cancers. 2023; 15(6):1866. https://doi.org/10.3390/cancers15061866
Chicago/Turabian StyleSpella, Magda, Giannoula Ntaliarda, Georgios Skiadas, Anne-Sophie Lamort, Malamati Vreka, Antonia Marazioti, Ioannis Lilis, Eleni Bouloukou, Georgia A. Giotopoulou, Mario A. A. Pepe, and et al. 2023. "Non-Oncogene Addiction of KRAS-Mutant Cancers to IL-1β via Versican and Mononuclear IKKβ" Cancers 15, no. 6: 1866. https://doi.org/10.3390/cancers15061866
APA StyleSpella, M., Ntaliarda, G., Skiadas, G., Lamort, A.-S., Vreka, M., Marazioti, A., Lilis, I., Bouloukou, E., Giotopoulou, G. A., Pepe, M. A. A., Weiss, S. A. I., Petrera, A., Hauck, S. M., Koch, I., Lindner, M., Hatz, R. A., Behr, J., Arendt, K. A. M., Giopanou, I., ... Stathopoulos, G. T. (2023). Non-Oncogene Addiction of KRAS-Mutant Cancers to IL-1β via Versican and Mononuclear IKKβ. Cancers, 15(6), 1866. https://doi.org/10.3390/cancers15061866