

Subcellular Epithelial HMGB1 Expression Is Associated with Colorectal Neoplastic Progression, Male Sex, Mismatch Repair Protein Expression, Lymph Node Positivity, and an ‘Immune Cold’ Phenotype Associated with Poor Survival

 , ,
, ,  ,
,
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Tissue Specimens
2.2. Colorectal Cancer Tissue Microarray
2.3. Colonic Tissue Biopsies and Endoscopic Polypectomy Specimens
2.4. Immunohistochemistry
2.5. Scoring of Immunohistochemistry
2.6. Data Analysis and Statistics
3. Results
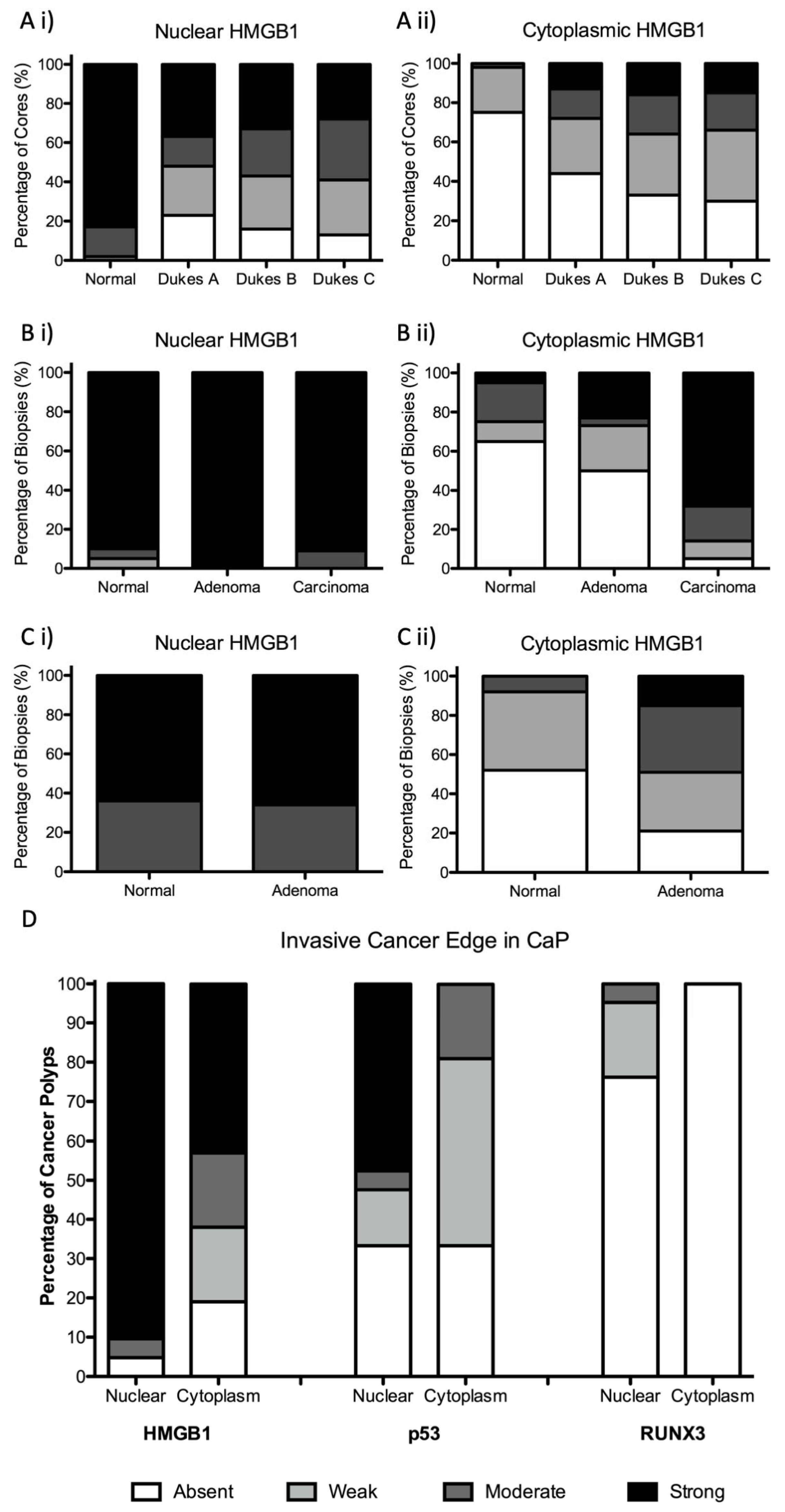
3.1. Subcellular Epithelial HMGB1 Expression Is Associated with Colonic Cancer, Lymph Node Positivity and Mismatch Repair Protein Expression
3.2. Epithelial Cytoplasmic HMGB1 Is Increased in Endoscopically Resected Colonic Adenomas
3.3. Epithelial HMGB1 Expression Is Prominent at the Leading Edge of Polyp Cancers (CaP)
3.4. Epithelial HMGB1 Is Not Associated with Epithelial p53 or RUNX3 Expression at the Invasive Cancer Margin or Molecular Phenotype
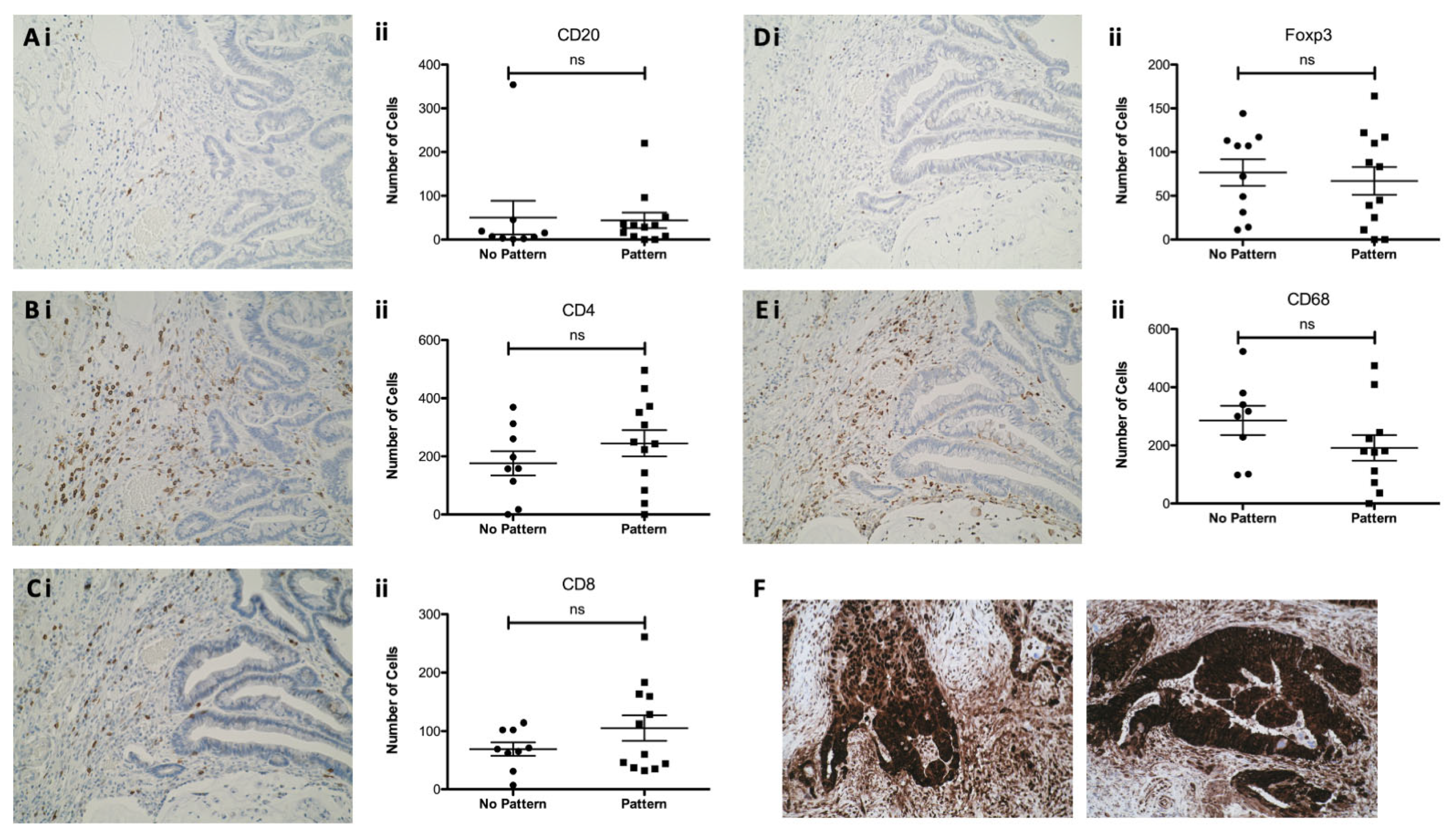
3.5. Epithelial HMGB1 at the Invasive Edge in CaP Is Not Associated with a Differential Adjacent Immune Cell Phenotype

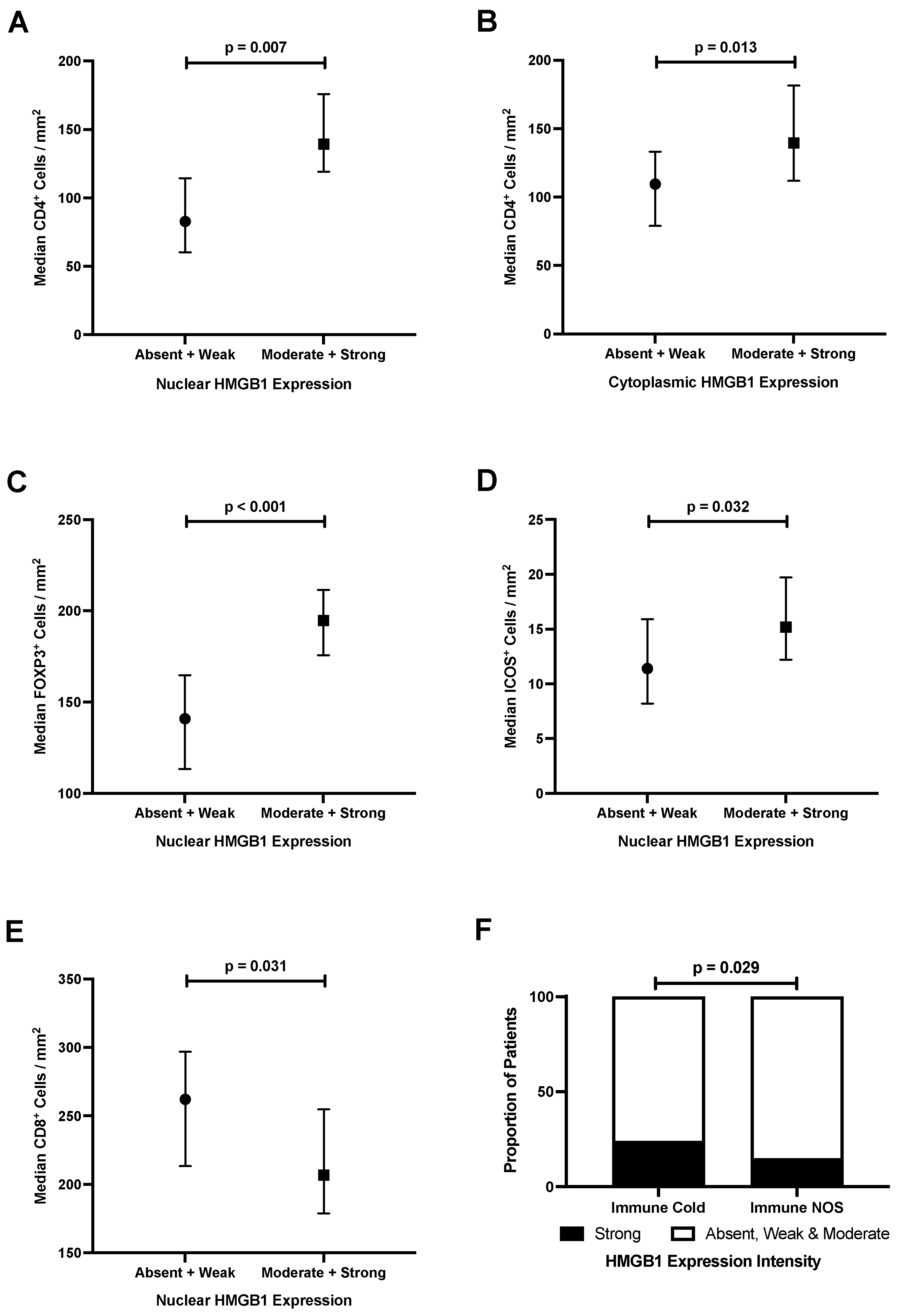
3.6. Epithelial HMGB1 Expression Is Associated with Increased Stromal CD4+, FOXP3+ and ICOS+ Cells, and Reduced CD8+ Cells in Colorectal Cancer
3.7. Strong Epithelial Cytoplasmic HMGB1 Is Associated with Immune Checkpoint Markers and an ‘Immune Cold’ Phenotype That Confers Poor Prognosis in Colorectal Cancer
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Morson, B.C. Evolution of cancer of the colon and rectum. Cancer 1974, 34, 845–849. [Google Scholar] [CrossRef]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Joanito, I.; Wirapati, P.; Zhao, N.; Nawaz, Z.; Yeo, G.; Lee, F.; Eng, C.L.P.; Macalinao, D.C.; Kahraman, M.; Srinivasan, H.; et al. Single-cell and bulk transcriptome sequencing identifies two epithelial tumor cell states and refines the consensus molecular classification of colorectal cancer. Nat. Genet. 2022, 54, 963–975. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Governa, V.; Trella, E.; Mele, V.; Tornillo, L.; Amicarella, F.; Cremonesi, E.; Muraro, M.G.; Xu, H.; Droeser, R.; Däster, S.R.; et al. The Interplay Between Neutrophils and CD8+ T Cells Improves Survival in Human Colorectal Cancer. Clin. Cancer Res. 2017, 23, 3847–3858. [Google Scholar] [CrossRef]
- Chen, R.; Kang, R.; Tang, D. The mechanism of HMGB1 secretion and release. Exp. Mol. Med. 2022, 54, 91–102. [Google Scholar] [CrossRef]
- Kang, R.; Chen, R.; Zhang, Q.; Hou, W.; Wu, S.; Cao, L.; Huang, J.; Yu, Y.; Fan, X.G.; Yan, Z.; et al. HMGB1 in health and disease. Mol. Asp. Med. 2014, 40, 1–116. [Google Scholar] [CrossRef]
- Bertheloot, D.; Latz, E. HMGB1, IL-1α, IL-33 and S100 proteins: Dual-function alarmins. Cell. Mol. Immunol. 2017, 14, 43–64. [Google Scholar] [CrossRef]
- Sinagra, T.; Merlo, S.; Spampinato, S.F.; Pasquale, R.D.; Sortino, M.A. High mobility group box 1 contributes to wound healing induced by inhibition of dipeptidylpeptidase 4 in cultured keratinocytes. Front. Pharmacol. 2015, 6, 126. [Google Scholar] [CrossRef]
- Wu, T.; Zhang, W.; Yang, G.; Li, H.; Chen, Q.; Song, R.; Zhao, L. HMGB1 overexpression as a prognostic factor for survival in cancer: A meta-analysis and systematic review. Oncotarget 2016, 7, 50417–50427. [Google Scholar] [CrossRef]
- Craig, S.G.; Humphries, M.P.; Alderdice, M.; Bingham, V.; Richman, S.D.; Loughrey, M.B.; Coleman, H.G.; Viratham-Pulsawatdi, A.; McCombe, K.; Murray, G.I.; et al. Immune status is prognostic for poor survival in colorectal cancer patients and is associated with tumour hypoxia. Br. J. Cancer 2020, 123, 1280–1288. [Google Scholar] [CrossRef] [PubMed]
- Porter, R.J.; Murray, G.I.; Alnabulsi, A.; Humphries, M.P.; James, J.A.; Salto-Tellez, M.; Craig, S.G.; Wang, J.M.; Yoshimura, T.; McLean, M.H. Colonic epithelial cathelicidin (LL-37) expression intensity is associated with progression of colorectal cancer and presence of CD8+ T cell infiltrate. J. Pathol. Clin. Res. 2021, 7, 495–506. [Google Scholar] [CrossRef]
- Alnabulsi, A.; Swan, R.; Cash, B.; Alnabulsi, A.; Murray, G.I. The differential expression of omega-3 and omega-6 fatty acid metabolising enzymes in colorectal cancer and its prognostic significance. Br. J. Cancer 2017, 116, 1612–1620. [Google Scholar] [CrossRef]
- Porter, R.J.; Murray, G.I.; Brice, D.P.; Petty, R.D.; McLean, M.H. Novel biomarkers for risk stratification of Barrett’s oesophagus associated neoplastic progression-epithelial HMGB1 expression and stromal lymphocytic phenotype. Br. J. Cancer 2020, 122, 545–554. [Google Scholar] [CrossRef]
- McLean, M.H.; Murray, G.I.; Stewart, K.N.; Norrie, G.; Mayer, C.; Hold, G.L.; Thomson, J.; Fyfe, N.; Hope, M.; Mowat, N.A.; et al. The inflammatory microenvironment in colorectal neoplasia. PLoS ONE 2011, 6, e15366. [Google Scholar] [CrossRef]
- Bankhead, P.; Loughrey, M.B.; Fernández, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef]
- Scottish Bowel Screening Programme Statistics: For the Period of Invitations from May 2019 to April 2021. National Statistics Publication. Public Health Scotland. 8 February 2022. Available online: https://publichealthscotland.scot/media/11605/2022-02-08-bowel-screening-publication-report.pdf (accessed on 8 February 2022).
- Bindea, G.; Mlecnik, B.; Galon, J. Immune sunrise: From the immunome to the cancer immune landscape. Oncoimmunology 2022, 11, 2019896. [Google Scholar] [CrossRef]
- Yalcin, S.; Philip, P.A.; Athanasiadis, I.; Bazarbashi, S.; Shamseddine, A. Classification of early-stage colon cancer with Immunoscore®: Clinical evidence and case studies. Future Oncol. 2022, 18, 613–623. [Google Scholar] [CrossRef]
- Li, Z.; Wang, H.; Song, B.; Sun, Y.; Han, J.; Xu, Z. Expression of high mobility group box-1 in colorectal cancer and its clinical significance. Zhonghua Wei Chang Wai Ke Za Zhi 2015, 18, 616–619. [Google Scholar] [PubMed]
- Peng, R.Q.; Wu, X.J.; Ding, Y.; Li, C.Y.; Yu, X.J.; Zhang, X.; Pan, Z.Z.; Wan, D.S.; Zheng, L.M.; Zeng, Y.X.; et al. Co-expression of nuclear and cytoplasmic HMGB1 is inversely associated with infiltration of CD45RO+ T cells and prognosis in patients with stage IIIB colon cancer. BMC Cancer 2010, 10, 496. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Q.; Huang, B.F.; Wang, Y.; Tang, C.H.; Jin, H.C.; Shao, F.; Shao, J.K.; Wang, Q.; Zeng, Y. Subcellular localization of HMGB1 in colorectal cancer impacts on tumor grade and survival prognosis. Sci. Rep. 2020, 10, 18587. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.C.; Bodmer, W.F. Genomic landscape of colorectal carcinogenesis. J. Cancer Res. Clin. Oncol. 2022, 148, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Rowell, J.P.; Simpson, K.L.; Stott, K.; Watson, M.; Thomas, J.O. HMGB1-facilitated p53 DNA binding occurs via HMG-Box/p53 transactivation domain interaction, regulated by the acidic tail. Structure 2012, 20, 2014–2024. [Google Scholar] [CrossRef]
- Yan, H.X.; Wu, H.P.; Zhang, H.L.; Ashton, C.; Tong, C.; Wu, H.; Qian, Q.J.; Wang, H.Y.; Ying, Q.L. p53 promotes inflammation-associated hepatocarcinogenesis by inducing HMGB1 release. J. Hepatol. 2013, 59, 762–768. [Google Scholar] [CrossRef]
- Su, Z.; Lu, H.; Jiang, H.; Zhu, H.; Li, Z.; Zhang, P.; Ni, P.; Shen, H.; Xu, W.; Xu, H. IFN-γ-producing Th17 cells bias by HMGB1-T-bet/RUNX3 axis might contribute to progression of coronary artery atherosclerosis. Atherosclerosis 2015, 243, 421–428. [Google Scholar] [CrossRef]
- Davalos, A.R.; Kawahara, M.; Malhotra, G.K.; Schaum, N.; Huang, J.; Ved, U.; Beausejour, C.M.; Coppe, J.P.; Rodier, F.; Campisi, J. p53-dependent release of Alarmin HMGB1 is a central mediator of senescent phenotypes. J. Cell Biol. 2013, 201, 613–629. [Google Scholar] [CrossRef]
- Taverna, S.; Tonacci, A.; Ferraro, M.; Cammarata, G.; Cuttitta, G.; Bucchieri, S.; Pace, E.; Gangemi, S. High Mobility Group Box 1: Biological Functions and Relevance in Oxidative Stress Related Chronic Diseases. Cells 2022, 11, 849. [Google Scholar] [CrossRef]
- Yuan, F.; Gu, L.; Guo, S.; Wang, C.; Li, G.M. Evidence for involvement of HMGB1 protein in human DNA mismatch repair. J. Biol. Chem. 2004, 279, 20935–20940. [Google Scholar] [CrossRef]
- Balana, A.T.; Mukherjee, A.; Nagpal, H.; Moon, S.P.; Fierz, B.; Vasquez, K.M.; Pratt, M.R. O-GlcNAcylation of High Mobility Group Box 1 (HMGB1) Alters Its DNA Binding and DNA Damage Processing Activities. J. Am. Chem. Soc. 2021, 143, 16030–16040. [Google Scholar] [CrossRef]
- Din, S.; Wong, K.; Mueller, M.F.; Oniscu, A.; Hewinson, J.; Black, C.J.; Miller, M.L.; Jiménez-Sánchez, A.; Rabbie, R.; Rashid, M.; et al. Mutational Analysis Identifies Therapeutic Biomarkers in Inflammatory Bowel Disease-Associated Colorectal Cancers. Clin. Cancer Res. 2018, 24, 5133–5142. [Google Scholar] [CrossRef]
- Idos, G.E.; Kwok, J.; Bonthala, N.; Kysh, L.; Gruber, S.B.; Qu, C. The Prognostic Implications of Tumor Infiltrating Lymphocytes in Colorectal Cancer: A Systematic Review and Meta-Analysis. Sci. Rep. 2020, 10, 3360. [Google Scholar] [CrossRef] [PubMed]
- Alexander, P.G.; McMillan, D.C.; Park, J.H. The local inflammatory response in colorectal cancer—Type, location or density? A systematic review and meta-analysis. Cancer Treat. Rev. 2020, 83, 101949. [Google Scholar] [CrossRef]
- Ning, T.; Li, J.; He, Y.; Zhang, H.; Wang, X.; Deng, T.; Liu, R.; Li, H.; Bai, M.; Fan, Q.; et al. Exosomal miR-208b related with oxaliplatin resistance promotes Treg expansion in colorectal cancer. Mol. Ther. 2021, 29, 2723–2736. [Google Scholar] [CrossRef]
- Fujimoto, H.; Saito, Y.; Ohuchida, K.; Kawakami, E.; Fujiki, S.; Watanabe, T.; Ono, R.; Kaneko, A.; Takagi, S.; Najima, Y.; et al. Deregulated Mucosal Immune Surveillance through Gut-Associated Regulatory T Cells and PD-1+ T Cells in Human Colorectal Cancer. J. Immunol. 2018, 200, 3291–3303. [Google Scholar] [CrossRef]
- Liu, Z.; Falo, L.D., Jr.; You, Z. Knockdown of HMGB1 in tumor cells attenuates their ability to induce regulatory T cells and uncovers naturally acquired CD8 T cell-dependent antitumor immunity. J. Immunol. 2011, 187, 118–125. [Google Scholar] [CrossRef]
- Li, D.Y.; Xiong, X.Z. ICOS+ Tregs: A Functional Subset of Tregs in Immune Diseases. Front. Immunol. 2020, 11, 2104. [Google Scholar] [CrossRef]
- Galaine, J.; Turco, C.; Vauchy, C.; Royer, B.; Mercier-Letondal, P.; Queiroz, L.; Loyon, R.; Mouget, V.; Boidot, R.; Laheurte, C.; et al. CD4 T cells target colorectal cancer antigens upregulated by oxaliplatin. Int. J. Cancer 2019, 145, 3112–3125. [Google Scholar] [CrossRef]
- DiToro, D.; Basu, R. Emerging Complexity in CD4+T Lineage Programming and Its Implications in Colorectal Cancer. Front. Immunol. 2021, 12, 694833. [Google Scholar] [CrossRef]
- Bromham, N.; Kallioinen, M.; Hoskin, P.; Davies, R.J. Colorectal cancer: Summary of NICE guidance. BMJ 2020, 268, m461. [Google Scholar] [CrossRef] [PubMed]
- André, T.; Shiu, K.K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. KEYNOTE-177 Investigators. Pembrolizumab in Microsatellite-Instability-High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef] [PubMed]
- Zemskova, M.; Kurdykov, S.; James, J.; McClain, N.; Rafikov, R.; Rafikov, O. Sex-specific stress response and HMGB1 release in pulmonary endothelial cells. PLoS ONE 2020, 15, e0231267. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, R.; Rafikova, O.; O’Connor, P.M.; Sullivan, J.C. Greater high-mobility group box 1 in male compared with female spontaneously hypertensive rats worsens renal ischemia-reperfusion injury. Clin. Sci. 2020, 134, 1751–1762. [Google Scholar] [CrossRef]
- White, A.; Ironmonger, L.; Steele, R.J.C.; Ormiston-Smith, N.; Crawford, C.; Seims, A. A review of sex-related differences in colorectal cancer incidence, screening uptake, routes to diagnosis, cancer stage and survival in the UK. BMC Cancer 2018, 18, 906. [Google Scholar] [CrossRef]
- Hubert, P.; Roncarati, P.; Demoulin, S.; Pilard, C.; Ancion, M.; Reynders, C.; Lerho, T.; Bruyere, D.; Lebeau, A.; Radermecker, C.; et al. Extracellular HMGB1 blockade inhibits tumor growth through profoundly remodeling immune microenvironment and enhances checkpoint inhibitor-based immunotherapy. J. Immunother. Cancer 2021, 9, e001966. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comparisons | Absent v Weak v Moderate v Strong | Absent v Weak, Moderate and Strong | Absent and Weak v Moderate and Strong | Strong v Absent, Weak and Moderate | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nucleus | Cytoplasm | Nucleus | Cytoplasm | Nucleus | Cytoplasm | Nucleus | Cytoplasm | |||||||||
| χ2 | p | χ2 | p | χ2 | p | χ2 | p | χ2 | p | χ2 | p | χ2 | p | χ2 | p | |
| Colorectal Cancer TMA | ||||||||||||||||
| Normal Colon vs. Colorectal Cancer | 54.143 | <0.001 | 36.266 | <0.001 | 9.073 | 0.003 | 32.695 | <0.001 | 30.747 | <0.001 | 20.772 | <0.001 | 51.827 | <0.001 | 8.529 | 0.003 |
| Cancer Polyps | ||||||||||||||||
| Normal vs. Adenoma | 2.310 | 0.315 | 5.837 | 0.120 | * | * | 0.963 | 0.327 | 1.127 | 0.288 | 0.028 | 0.867 | 2.310 | 0.129 | 2.689 | 0.101 |
| Normal vs. Carcinoma | 1.346 | 0.510 | 22.491 | <0.001 | * | * | 17.230 | <0.001 | 1.127 | 0.288 | 16.108 | <0.001 | 0.010 | 0.920 | 17.733 | <0.001 |
| Adenoma vs. Carcinoma | 2.095 | 0.148 | 16.419 | 0.001 | * | * | 11.458 | 0.001 | * | * | 15.655 | <0.001 | 2.095 | 0.148 | 9.167 | 0.002 |
| Non-Cancerous Polyps | ||||||||||||||||
| Normal vs. Adenoma | 0.280 | 0.868 | 13.484 | 0.004 | * | * | 7.086 | 0.008 | * | * | 12.066 | 0.001 | 0.028 | 0.868 | 4.124 | 0.042 |
| Comparison | Absent v Weak v Moderate v Strong | Absent v Weak, Moderate and Strong | Absent and Weak v Moderate and Strong ** | Strong v Absent, Weak and Moderate | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nucleus | Cytoplasm | Nucleus | Cytoplasm | Nucleus | Cytoplasm | Nucleus | Cytoplasm | |||||||||
| TS * | p | TS * | p | TS * | p | TS * | p | TS * | p | TS * | p | TS * | p | TS * | p | |
| Density (using continuous cell count data) | ||||||||||||||||
| CD4+ | 11.134 | 0.011 | 15.367 | 0.002 | 24,128.000 | 0.007 | 38,520.000 | <0.001 | 41,767.000 | 0.007 | 38,552.000 | 0.013 | 36,984.000 | 0.010 | 21,168.000 | 0.587 |
| CD3+ | 4.710 | 0.194 | 3.472 | 0.324 | 18,399.000 | 0.074 | 34,346.000 | 0.358 | 35,565.000 | 0.249 | 33,706.000 | 0.444 | 33,358.000 | 0.772 | 19,519.000 | 0.268 |
| CD8+ | 6.659 | 0.084 | 2.477 | 0.479 | 16,744.000 | 0.022 | 29,439.500 | 0.498 | 31,718.500 | 0.031 | 32,339.000 | 0.498 | 29,022.500 | 0.130 | 18,139.000 | 0.133 |
| FOXP3+ | 22.489 | <0.001 | 3.479 | 0.323 | 25,718.000 | 0.001 | 33,913.000 | 0.619 | 44,376.500 | <0.001 | 34,728.000 | 0.873 | 40,366.500 | <0.001 | 18,857.000 | 0.138 |
| IDO-1+ | 3.741 | 0.291 | 6.839 | 0.077 | 21,327.000 | 0.186 | 28,967.000 | 0.272 | 34,703 | 0.661 | 30,001.000 | 0.077 | 29,730.000 | 0.447 | 16,151.000 | 0.010 |
| ICOS+ | 10.026 | 0.018 | 10.393 | 0.016 | 23,326.500 | 0.013 | 30,417.500 | 0.981 | 39,203.500 | 0.032 | 29,905.000 | 0.050 | 35,980.500 | 0.009 | 16,078.000 | 0.002 |
| CD20+ | 2.515 | 0.472 | 0.288 | 0.962 | 14,849.5 | 0.224 | 25,674 | 0.817 | 26,485.5 | 0.296 | 23,336.5 | 0.757 | 23,420 | 0.977 | 13,035.5 | 0.802 |
| PDL1+ Stroma | 2.428 | 0.489 | 8.832 | 0.032 | 18,957.000 | 0.467 | 27,914.000 | 0.064 | 1804.757 | 0.141 | 32,329.000 | 0.528 | 30,554.000 | 0.644 | 17,125.000 | 0.027 |
| PDL1+ Tumour | 1.364 | 0.714 | 4.839 | 0.184 | 21,178 | 0.353 | 31,450 | 0.797 | 35,824 | 0.897 | 34,860 | 0.409 | 30,942 | 0.817 | 18,594 | 0.259 |
| Immune Cold vs. Immune NOS *** | ||||||||||||||||
| Immune Cold (low CD3/CD8) | 1.972 | 0.578 | 7.446 | 0.059 | 1.895 | 0.169 | 0.560 | 0.454 | 0.864 | 0.353 | 0.108 | 0.742 | 0.352 | 0.553 | 4.841 | 0.028 |
| Immune Cold (low CD3/CD8/CD4) | 1.722 | 0.632 | 7.230 | 0.065 | 1.491 | 0.222 | 0.626 | 0.429 | 0.641 | 0.423 | 0.239 | 0.625 | 0.685 | 0.408 | 4.777 | 0.029 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Porter, R.J.; Murray, G.I.; Hapca, S.; Hay, A.; Craig, S.G.; Humphries, M.P.; James, J.A.; Salto-Tellez, M.; Brice, D.P.; Berry, S.H.; et al. Subcellular Epithelial HMGB1 Expression Is Associated with Colorectal Neoplastic Progression, Male Sex, Mismatch Repair Protein Expression, Lymph Node Positivity, and an ‘Immune Cold’ Phenotype Associated with Poor Survival. Cancers 2023, 15, 1865. https://doi.org/10.3390/cancers15061865
Porter RJ, Murray GI, Hapca S, Hay A, Craig SG, Humphries MP, James JA, Salto-Tellez M, Brice DP, Berry SH, et al. Subcellular Epithelial HMGB1 Expression Is Associated with Colorectal Neoplastic Progression, Male Sex, Mismatch Repair Protein Expression, Lymph Node Positivity, and an ‘Immune Cold’ Phenotype Associated with Poor Survival. Cancers. 2023; 15(6):1865. https://doi.org/10.3390/cancers15061865
Chicago/Turabian StylePorter, Ross J., Graeme I. Murray, Sandra Hapca, Andrew Hay, Stephanie G. Craig, Matthew P. Humphries, Jacqueline A. James, Manuel Salto-Tellez, Daniel P. Brice, Susan H. Berry, and et al. 2023. "Subcellular Epithelial HMGB1 Expression Is Associated with Colorectal Neoplastic Progression, Male Sex, Mismatch Repair Protein Expression, Lymph Node Positivity, and an ‘Immune Cold’ Phenotype Associated with Poor Survival" Cancers 15, no. 6: 1865. https://doi.org/10.3390/cancers15061865
APA StylePorter, R. J., Murray, G. I., Hapca, S., Hay, A., Craig, S. G., Humphries, M. P., James, J. A., Salto-Tellez, M., Brice, D. P., Berry, S. H., & McLean, M. H. (2023). Subcellular Epithelial HMGB1 Expression Is Associated with Colorectal Neoplastic Progression, Male Sex, Mismatch Repair Protein Expression, Lymph Node Positivity, and an ‘Immune Cold’ Phenotype Associated with Poor Survival. Cancers, 15(6), 1865. https://doi.org/10.3390/cancers15061865