

Drugs That Mimic Hypoxia Selectively Target EBV-Positive Gastric Cancer Cells

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Cells
2.3. Chemical Mimics of Hypoxia and Other Drugs
2.4. Cell Viability Assays
2.5. Western Blot Analysis
2.6. Immunofluorescence Assays
2.7. Generation of EBV-Positive Xenograft Tumors
3. Results
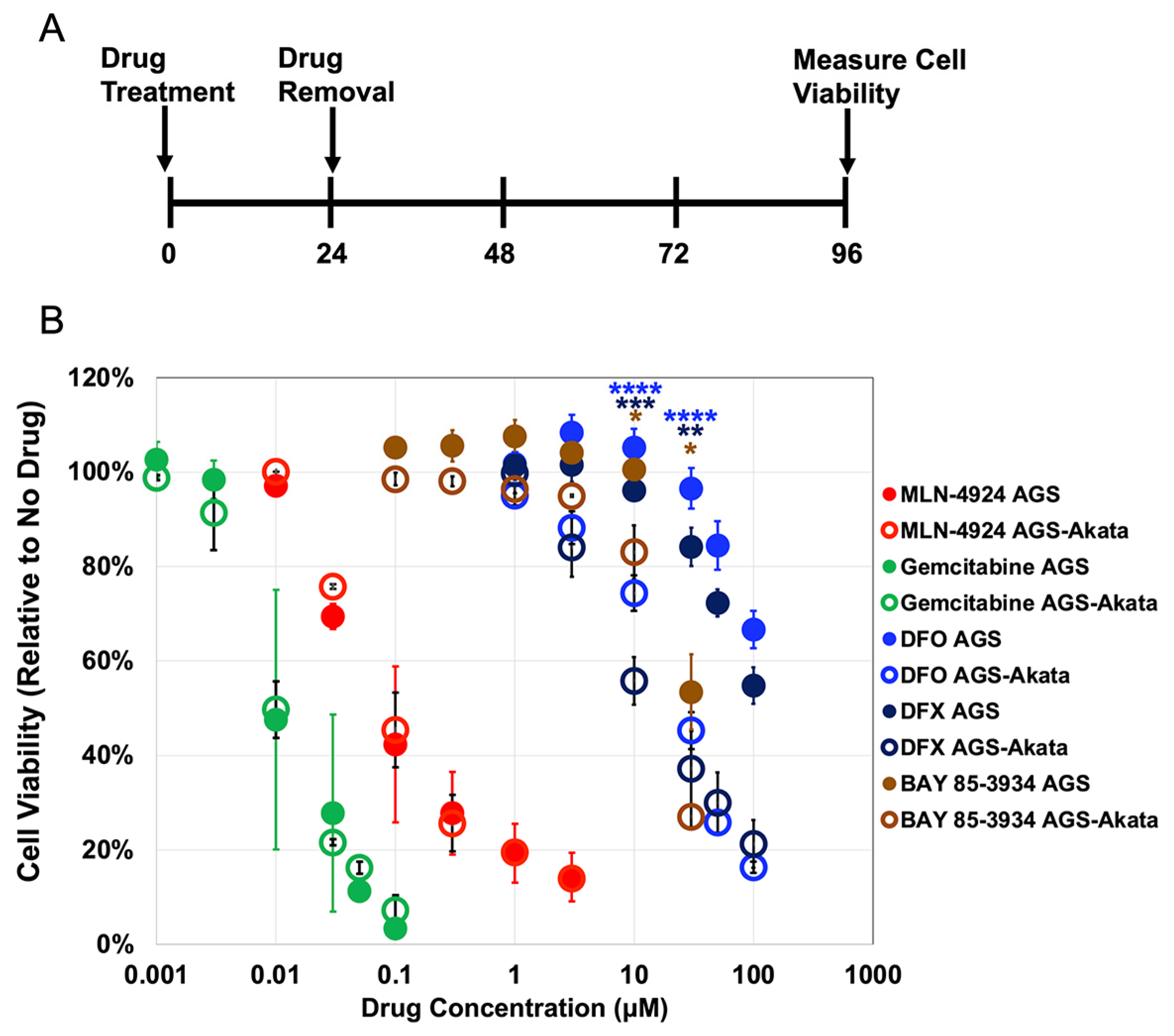
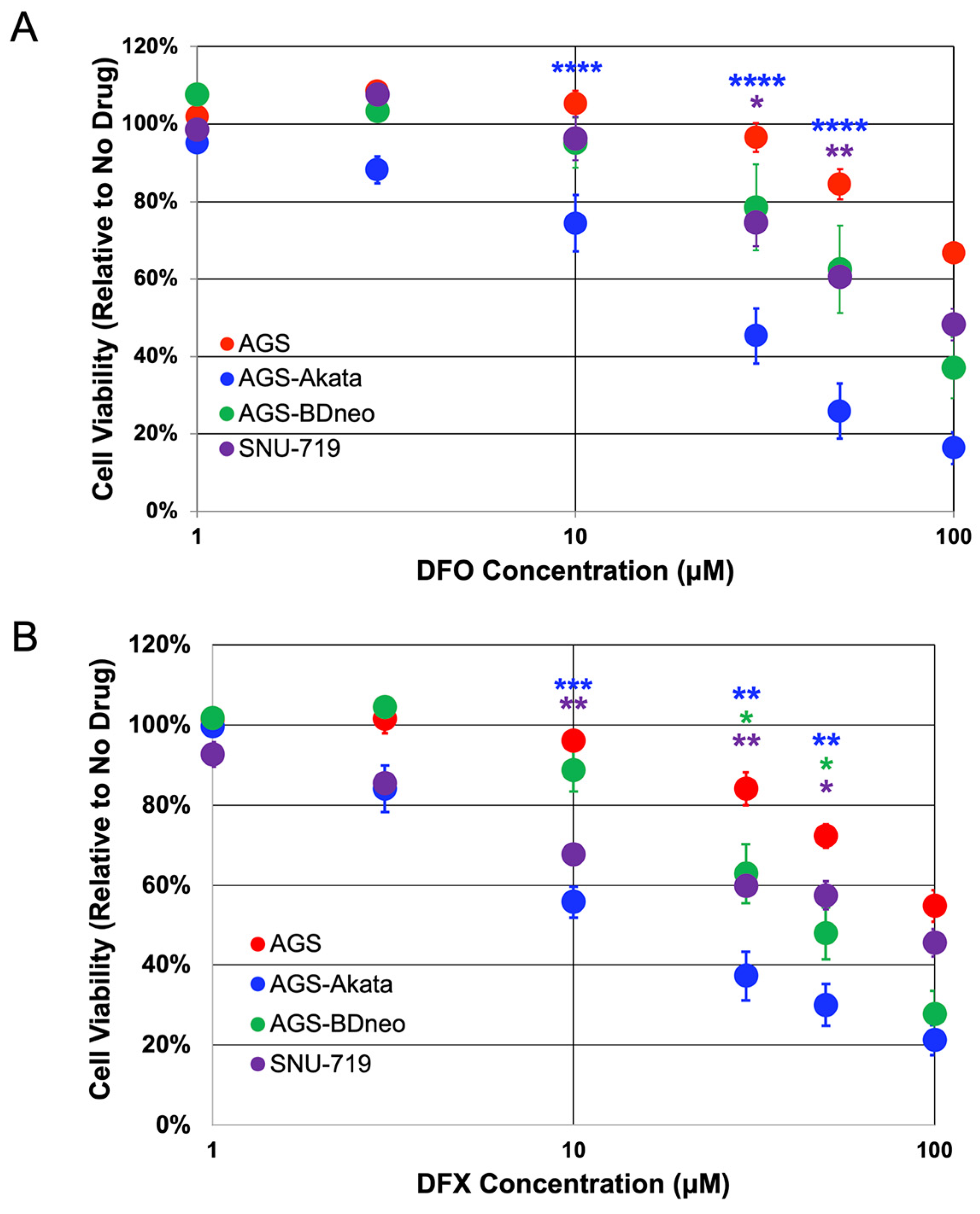
3.1. EBV-positive Gastric Cancer Cells Are Preferentially Killed by Iron Chelators
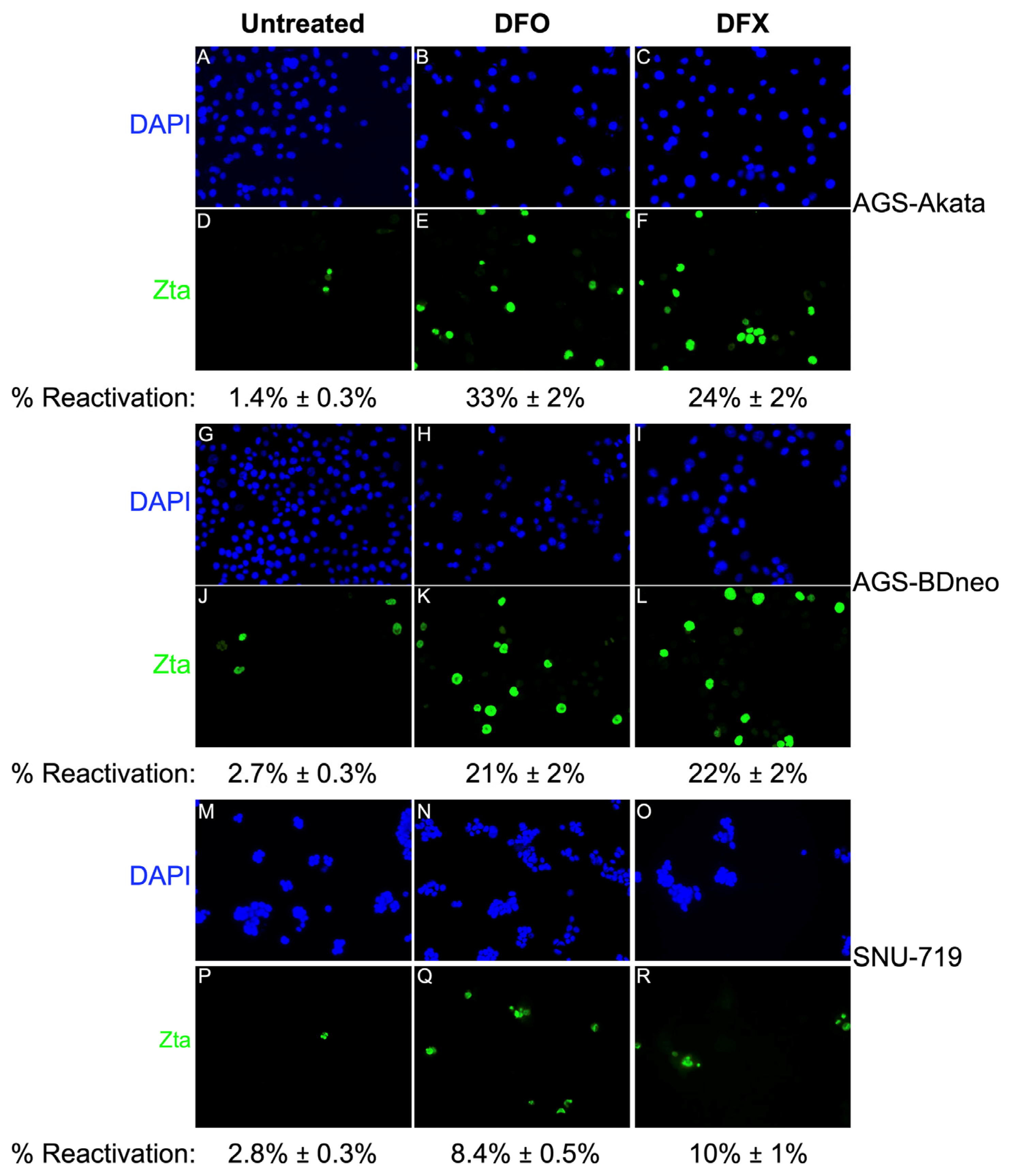
3.2. DFO and DFX Induce Synthesis of EBV Lytic Antigens in EBV-positive Gastric Cancer Cell Lines
3.3. DFO and DFX Fail to Significantly Induce EBV Reactivation In Vivo in a Xenograft Tumor Model in Young Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Longnecker, R.M.; Kieff, E.; Cohen, J.I. Epstein-Barr Virus. In Fields Virology; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013. [Google Scholar]
- Kutok, J.; Wang, F. Spectrum of epstein-barr virus–associated diseases. Annu. Rev. Pathol. Mech. Dis. 2006, 1, 375–404. [Google Scholar] [CrossRef] [PubMed]
- Khan, G.; Hashim, M.J. Global burden of deaths from Epstein-Barr virus attributable malignancies 1990-2010. Infect. Agents Cancer 2014, 9, 38. [Google Scholar] [CrossRef] [PubMed]
- Iizasa, H.; Nanbo, A.; Nishikawa, J.; Jinushi, M.; Yoshiyama, H. Epstein-Barr Virus (EBV)-associated Gastric Carcinoma. Viruses 2012, 4, 3420–3439. [Google Scholar] [CrossRef] [PubMed]
- van Beek, J.; zur Hausen, A.; Kranenbarg, E.M.-K.; van de Velde, C.J.; Middeldorp, J.M.; van den Brule, A.J.C.; Meijer, C.J.L.M.; Bloemena, E. EBV-Positive Gastric Adenocarcinomas: A Distinct Clinicopathologic Entity With a Low Frequency of Lymph Node Involvement. J. Clin. Oncol. 2004, 22, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, J.; Iizasa, H.; Yoshiyama, H.; Shimokuri, K.; Kobayashi, Y.; Sasaki, S.; Nakamura, M.; Yanai, H.; Sakai, K.; Suehiro, Y.; et al. Clinical Importance of Epstein–Barr Virus-Associated Gastric Cancer. Cancers 2018, 10, 167. [Google Scholar] [CrossRef]
- Kenney, S.C.; Mertz, J.E. Regulation of the latent-lytic switch in Epstein–Barr virus. Semin. Cancer Biol. 2014, 26, 60–68. [Google Scholar] [CrossRef]
- Hau, P.M.; Lung, H.L.; Wu, M.; Tsang, C.M.; Wong, K.-L.; Mak, N.K.; Lo, K.W. Targeting Epstein-Barr Virus in Nasopharyngeal Carcinoma. Front. Oncol. 2020, 10, 600. [Google Scholar] [CrossRef]
- Yiu, S.P.T.; Dorothea, M.; Hui, K.F.; Chiang, A.K.S. Lytic Induction Therapy against Epstein–Barr Virus-Associated Malignancies: Past, Present, and Future. Cancers 2020, 12, 2142. [Google Scholar] [CrossRef]
- Murata, T.; Sugimoto, A.; Inagaki, T.; Yanagi, Y.; Watanabe, T.; Sato, Y.; Kimura, H. Molecular Basis of Epstein–Barr Virus Latency Establishment and Lytic Reactivation. Viruses 2021, 13, 2344. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.; Hagemeier, S.R.; Fingeroth, J.D.; Gershburg, E.; Pagano, J.S.; Kenney, S.C. The Epstein-Barr Virus (EBV)-Encoded Protein Kinase, EBV-PK, but Not the Thymidine Kinase (EBV-TK), Is Required for Ganciclovir and Acyclovir Inhibition of Lytic Viral Production. J. Virol. 2010, 84, 4534–4542. [Google Scholar] [CrossRef]
- Freeman, S.M.; Abboud, C.N.; A Whartenby, K.; Packman, C.H.; Koeplin, D.S.; Moolten, F.L.; Abraham, G.N. The “bystander effect”: Tumor regression when a fraction of the tumor mass is genetically modified. Cancer Res 1993, 53, 5274–5283. [Google Scholar] [PubMed]
- Feng, W.-H.; Hong, G.; Delecluse, H.-J.; Kenney, S.C. Lytic Induction Therapy for Epstein-Barr Virus-Positive B-Cell Lymphomas. J. Virol. 2004, 78, 1893–1902. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.G.; Kim, H.; Kim, E.J.; Park, P.-G.; Dong, S.M.; Choi, T.H.; Kim, H.; Chong, C.R.; Liu, J.O.; Chen, J.; et al. Targeted therapy for Epstein-Barr virus-associated gastric carcinoma using low-dose gemcitabine-induced lytic activation. Oncotarget 2015, 6, 31018–31029. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.-H.; Kenney, S.C. Valproic Acid Enhances the Efficacy of Chemotherapy in EBV-Positive Tumors by Increasing Lytic Viral Gene Expression. Cancer Res. 2006, 66, 8762–8769. [Google Scholar] [CrossRef]
- Hui, K.F.; Cheung, A.K.L.; Choi, C.K.; Yeung, P.L.; Middeldorp, J.M.; Lung, M.L.; Tsao, S.W.; Chiang, A.K.S. Inhibition of class I histone deacetylases by romidepsin potently induces Epstein-Barr virus lytic cycle and mediates enhanced cell death with ganciclovir. Int. J. Cancer 2015, 138, 125–136. [Google Scholar] [CrossRef]
- Mentzer, S.J.; Fingeroth, J.; Reilly, J.J.; Perrine, S.P.; Faller, D.V. Arginine Butyrate-Induced Susceptibility to Ganciclovir in an Epstein–Barr-Virus-Associated Lymphoma. Blood Cells Mol. Dis. 1998, 24, 114–123. [Google Scholar] [CrossRef]
- Ghosh, S.K.; Forman, L.W.; Akinsheye, I.; Perrine, S.P.; Faller, D.V. Short, discontinuous exposure to butyrate effectively sensitizes latently EBV-infected lymphoma cells to nucleoside analogue antiviral agents. Blood Cells Mol. Dis. 2006, 38, 57–65. [Google Scholar] [CrossRef]
- Perrine, S.P.; Hermine, O.; Small, T.; Suarez, F.; O’Reilly, R.; Boulad, F.; Fingeroth, J.; Askin, M.; Levy, A.; Mentzer, S.J.; et al. A phase 1/2 trial of arginine butyrate and ganciclovir in patients with Epstein-Barr virus–associated lymphoid malignancies. Blood 2006, 109, 2571–2578. [Google Scholar] [CrossRef]
- Haverkos, B.M.; Alpdogan, O.; Baiocchi, R.; E Brammer, J.; Feldman, T.A.; Capra, M.; A Brem, E.; Nair, S.M.; Scheinberg, P.; Pereira, J.; et al. Nanatinostat (Nstat) and Valganciclovir (VGCV) in Relapsed/Refractory (R/R) Epstein-Barr Virus-Positive (EBV +) Lymphomas: Final Results from the Phase 1b/2 VT3996-201 Study. Blood 2021, 138, 623. [Google Scholar] [CrossRef]
- Wildeman, M.A.; Novalić, Z.; Verkuijlen, S.A.; Juwana, H.; Huitema, A.D.; Tan, I.B.; Middeldorp, J.M.; De Boer, J.P.; Greijer, A.E. Cytolytic Virus Activation Therapy for Epstein-Barr Virus–Driven TumorsNew Therapy for Ep-stein-Barr Virus–Driven Tumors. Clin. Cancer Res. 2012, 18, 5061–5070. [Google Scholar] [CrossRef]
- Hsu, C.-L.; Kuo, Y.-C.; Huang, Y.; Huang, Y.-C.; Lui, K.-W.; Chang, K.-P.; Lin, T.-L.; Fan, H.-C.; Lin, A.-C.; Hsieh, C.-H.; et al. Application of a patient-derived xenograft model in cytolytic viral activation therapy for nasopharyngeal carcinoma. Oncotarget 2015, 6, 31323–31334. [Google Scholar] [CrossRef]
- Stoker, S.D.; Novalić, Z.; Wildeman, M.A.; Huitema, A.D.R.; Verkuijlen, S.A.W.M.; Juwana, H.; Greijer, A.E.; Tan, I.B.; Middeldorp, J.; De Boer, J.P. Epstein–Barr virus-targeted therapy in nasopharyngeal carcinoma. J. Cancer Res. Clin. Oncol. 2015, 141, 1845–1857. [Google Scholar] [CrossRef]
- Kraus, R.J.; Yu, X.; Cordes, B.-L.A.; Sathiamoorthi, S.; Iempridee, T.; Nawandar, D.M.; Ma, S.; Romero-Masters, J.C.; McChesney, K.G.; Lin, Z.; et al. Hypoxia-inducible factor-1α plays roles in Epstein-Barr virus’s natural life cycle and tumorigenesis by inducing lytic infection through direct binding to the immediate-early BZLF1 gene promoter. PLOS Pathog. 2017, 13, e1006404. [Google Scholar] [CrossRef] [PubMed]
- Yiu, S.P.T.; Hui, K.F.; Choi, C.K.; Kao, R.Y.T.; Ma, C.W.; Yang, D.; Chiang, A.K.S. Intracellular Iron Chelation by a Novel Compound, C7, Reactivates Epstein–Barr Virus (EBV) Lytic Cycle via the ERK-Autophagy Axis in EBV-Positive Epithelial Cancers. Cancers 2018, 10, 505. [Google Scholar] [CrossRef] [PubMed]
- Kraus, R.J.; Cordes, B.-L.A.; Sathiamoorthi, S.; Patel, P.; Yuan, X.; Iempridee, T.; Yu, X.; Lee, D.L.; Lambert, P.F.; Mertz, J.E. Reactivation of Epstein-Barr Virus by HIF-1α Requires p53. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-Inducible Factors and the Response to Hypoxic Stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef] [PubMed]
- Fong, G.-H.; Takeda, K. Role and regulation of prolyl hydroxylase domain proteins. Cell Death Differ. 2008, 15, 635–641. [Google Scholar] [CrossRef]
- Lee, J.-W.; Bae, S.-H.; Jeong, J.-W.; Kim, S.-H.; Kim, K.-W. Hypoxia-inducible factor (HIF-1)α: Its protein stability and biological functions. Exp. Mol. Med. 2004, 36, 1–12. [Google Scholar] [CrossRef]
- Kontoghiorghes, G.J.; Kontoghiorghe, C.N. Efficacy and safety of iron-chelation therapy with deferoxamine, deferiprone, and deferasirox for the treatment of iron-loaded patients with non-transfusion-dependent thalassemia syndromes. Drug Des. Dev. Ther. 2016, ume 10, 465–481. [Google Scholar] [CrossRef]
- Beck, H.; Jeske, M.; Thede, K.; Stoll, F.; Flamme, I.; Akbaba, M.; Ergüden, J.-K.; Karig, G.; Keldenich, J.; Oehme, F.; et al. Discovery of Molidustat (BAY 85-3934): A Small-Molecule Oral HIF-Prolyl Hydroxylase (HIF-PH) Inhibitor for the Treatment of Renal Anemia. Chemmedchem 2018, 13, 988–1003. [Google Scholar] [CrossRef]
- Zhou, L.; Zhang, W.; Sun, Y.; Jia, L. Protein neddylation and its alterations in human cancers for targeted therapy. Cell. Signal. 2018, 44, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Molesworth, S.J.; Lake, C.M.; Borza, C.M.; Turk, S.M.; Hutt-Fletcher, L.M. Epstein-Barr Virus gH Is Essential for Penetration of B Cells but Also Plays a Role in Attachment of Virus to Epithelial Cells. J. Virol. 2000, 74, 6324–6332. [Google Scholar] [CrossRef] [PubMed]
- Borza, C.M.; Hutt-Fletcher, L.M. Epstein-Barr Virus Recombinant Lacking Expression of Glycoprotein gp150 Infects B Cells Normally but Is Enhanced for Infection of Epithelial Cells. J. Virol. 1998, 72, 7577–7582. [Google Scholar] [CrossRef]
- Park, J.G.; Yang, H.K.; Kim, W.H.; Chung, J.K.; Kang, M.S.; Lee, J.H.; Oh, J.H.; Park, H.S.; Yeo, K.S.; Kang, S.H.; et al. Establishment and Characterization of Human Gastric Carcinoma Cell Lines. Int. J. Cancer J. Int. Cancer 1997, 70, 443–449. [Google Scholar] [CrossRef]
- Adamson, A.L.; Kenney, S.; Manninen, A.; Huotari, P.; Hiipakka, M.; Renkema, G.H.; Saksela, K. Epstein-Barr Virus Immediate-Early Protein BZLF1 Is SUMO-1 Modified and Disrupts Promyelocytic Leukemia Bodies. J. Virol. 2001, 75, 3034–3037. [Google Scholar] [CrossRef]
- Jiang, B.-H.; Zheng, J.Z.; Leung, S.W.; Roe, R.; Semenza, G.L. Transactivation and Inhibitory Domains of Hypox-ia-Inducible Factor 1α: Modulation of Transcriptional Activity by Oxygen Tension. J. Biol. Chem. 1997, 272, 19253–19260. [Google Scholar] [CrossRef]
- Zhao, Y.; Xiong, X.; Jia, L.; Sun, Y. Targeting Cullin-RING ligases by MLN4924 induces autophagy via modulating the HIF1-REDD1-TSC1-mTORC1-DEPTOR axis. Cell Death Dis. 2012, 3, e386. [Google Scholar] [CrossRef] [PubMed]
- Flamme, I.; Oehme, F.; Ellinghaus, P.; Jeske, M.; Keldenich, J.; Thuss, U. Mimicking Hypoxia to Treat Anemia: HIF-Stabilizer BAY 85-3934 (Molidustat) Stimulates Erythropoietin Production without Hypertensive Effects. PLOS ONE 2014, 9, e111838. [Google Scholar] [CrossRef]
- Lan, H.; Tang, Z.; Jin, H.; Sun, Y. Neddylation inhibitor MLN4924 suppresses growth and migration of human gastric cancer cells. Sci. Rep. 2016, 6, 24218. [Google Scholar] [CrossRef]
- Kim, J.L.; Lee, D.-H.; Na, Y.J.; Kim, B.R.; Jeong, Y.A.; Lee, S.I.; Kang, S.; Joung, S.Y.; Lee, S.-Y.; Oh, S.C.; et al. Iron chelator-induced apoptosis via the ER stress pathway in gastric cancer cells. Tumor Biol. 2016, 37, 9709–9719. [Google Scholar] [CrossRef]
- Min, K.; Lee, S.K. EBV miR-BART10-3p Promotes Cell Proliferation and Migration by Targeting DKK1. Int. J. Biol. Sci. 2019, 15, 657–667. [Google Scholar] [CrossRef]
- Israel, B.F.; Kenney, S.C. Virally targeted therapies for EBV-associated malignancies. Oncogene 2003, 22, 5122–5130. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, V.; Talarico, C.L.; Stanat, S.C.; Davis, M.; Coen, D.M.; Biron, K.K. A protein kinase homologue controls phosphorylation of ganciclovir in human cytomegalovirus-infected cells. Nature 1992, 358, 162–164. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.M.; Cannon, J.S.; Tanhehco, Y.C.; Hamzeh, F.M.; Ambinder, R.F. Induction of Epstein-Barr Virus Kinases To Sensitize Tumor Cells to Nucleoside Analogues. Antimicrob. Agents Chemother. 2001, 45, 2082–2091. [Google Scholar] [CrossRef] [PubMed]
- Hider, R.C.; Hoffbrand, A.V. The Role of Deferiprone in Iron Chelation. N. Engl. J. Med. 2018, 379, 2140–2150. [Google Scholar] [CrossRef]
- Poggiali, E.; Cassinerio, E. An update on iron chelation therapy. Blood Transfus. 2012, 10, 411. [Google Scholar] [CrossRef] [PubMed]
- Gai, W.; Peng, Z.; Liu, C.H.; Zhang, L.; Jiang, H. Advances in Cancer Treatment by Targeting the Neddylation Path-way. Front. Cell Dev. Biol. 2021, 9, 653882. [Google Scholar] [CrossRef]
- Akizawa, T.; MacDougall, I.C.; Berns, J.S.; Bernhardt, T.; Staedtler, G.; Taguchi, M.; Iekushi, K.; Krueger, T. Long-Term Efficacy and Safety of Molidustat for Anemia in Chronic Kidney Disease: DIALOGUE Extension Studies. Am. J. Nephrol. 2019, 49, 271–280. [Google Scholar] [CrossRef]
- Macdougall, I.C.; Akizawa, T.; Berns, J.S.; Bernhardt, T.; Krueger, T. Effects of Molidustat in the Treatment of Ane-mia in CKD. Clin. J. Am. Soc. Nephrol. 2019, 14, 28–39. [Google Scholar] [CrossRef]
- Moysan, E.; Bastiat, G.; Benoit, J.-P. Gemcitabine versus Modified Gemcitabine: A Review of Several Promising Chemical Modifications. Mol. Pharm. 2012, 10, 430–444. [Google Scholar] [CrossRef]
- Rensvold, J.W.; Ong, S.-E.; Jeevananthan, A.; Carr, S.A.; Mootha, V.K.; Pagliarini, D.J. Complementary RNA and Protein Profiling Identifies Iron as a Key Regulator of Mitochondrial Biogenesis. Cell Rep. 2013, 3, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Yiu, S.P.T.; Hui, K.F.; Münz, C.; Lo, K.-W.; Tsao, S.W.; Kao, R.Y.T.; Yang, D.; Chiang, A.K.S. Autophagy-Dependent Reactivation of Epstein-Barr Virus Lytic Cycle and Combinatorial Effects of Autophagy-Dependent and Independent Lytic Inducers in Nasopharyngeal Carcinoma. Cancers 2019, 11, 1871. [Google Scholar] [CrossRef]
- Herr, C.Q.; Hausinger, R.P. Amazing Diversity in Biochemical Roles of Fe(II)/2-Oxoglutarate Oxygenases. Trends Biochem. Sci. 2018, 43, 517–532. [Google Scholar] [CrossRef] [PubMed]
- Woellmer, A.; Hammerschmidt, W. Epstein–Barr virus and host cell methylation: Regulation of latency, replication and virus reactivation. Curr. Opin. Virol. 2013, 3, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Badal, S.; Her, Y.F.; Maher, L.J. Nonantibiotic Effects of Fluoroquinolones in Mammalian Cells. J. Biol. Chem. 2015, 290, 22287–22297. [Google Scholar] [CrossRef]
- Namba-Fukuyo, H.; Funata, S.; Matsusaka, K.; Fukuyo, M.; Rahmutulla, B.; Mano, Y.; Fukayama, M.; Aburatani, H.; Kaneda, A. TET2 functions as a resistance factor against DNA methylation acquisition during Epstein-Barr virus infection. Oncotarget 2016, 7, 81512–81526. [Google Scholar] [CrossRef]
- Wille, C.K.; Nawandar, D.M.; Henning, A.N.; Ma, S.; Oetting, K.M.; Lee, D.; Lambert, P.; Johannsen, E.C.; Kenney, S.C. 5-hydroxymethylation of the EBV genome regulates the latent to lytic switch. Proc. Natl. Acad. Sci. USA 2015, 112, E7257–E7265. [Google Scholar] [CrossRef]
- Sun, K.; Jia, K.; Lv, H.; Wang, S.-Q.; Wu, Y.; Lei, H.; Chen, X. EBV-Positive Gastric Cancer: Current Knowledge and Future Perspectives. Front. Oncol. 2020, 10, 583463. [Google Scholar] [CrossRef]
- Hagiwara, A.; Takahashi, T.; Kitamura, K.; Sakakura, C.; Shirasu, M.; Ohgaki, M.; Imanishi, T.; Yamasaki, J. Endoscopic local injection of a new drug delivery formulation, anticancer drug bound to carbon particles, for digestive cancers: Pilot study. J. Gastroenterol. 1997, 32, 141–147. [Google Scholar] [CrossRef]
- Monga, S.P.; Wadleigh, R.; Adib, H.; Harmon, J.W.; Berlin, M.; Mishra, L. Endoscopic treatment of gastric cancer with intratumoral cisplatin/epinephrine injectable gel: A case report. Gastrointest. Endosc. 1998, 48, 415–417. [Google Scholar] [CrossRef]
- Lang, J.; Zhao, X.; Wang, X.; Zhao, Y.; Li, Y.; Zhao, R.; Cheng, K.; Li, Y.; Han, X.; Zheng, X.; et al. Targeted Co-delivery of the Iron Chelator Deferoxamine and a HIF1α Inhibitor Impairs Pancreatic Tumor Growth. ACS Nano 2019. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cordes, B.-l.A.; Bilger, A.; Kraus, R.J.; Ward-Shaw, E.T.; Labott, M.R.; Lee, S.; Lambert, P.F.; Mertz, J.E. Drugs That Mimic Hypoxia Selectively Target EBV-Positive Gastric Cancer Cells. Cancers 2023, 15, 1846. https://doi.org/10.3390/cancers15061846
Cordes B-lA, Bilger A, Kraus RJ, Ward-Shaw ET, Labott MR, Lee S, Lambert PF, Mertz JE. Drugs That Mimic Hypoxia Selectively Target EBV-Positive Gastric Cancer Cells. Cancers. 2023; 15(6):1846. https://doi.org/10.3390/cancers15061846
Chicago/Turabian StyleCordes, Blue-leaf A., Andrea Bilger, Richard J. Kraus, Ella T. Ward-Shaw, Madeline R. Labott, Shinhyo Lee, Paul F. Lambert, and Janet E. Mertz. 2023. "Drugs That Mimic Hypoxia Selectively Target EBV-Positive Gastric Cancer Cells" Cancers 15, no. 6: 1846. https://doi.org/10.3390/cancers15061846
APA StyleCordes, B.-l. A., Bilger, A., Kraus, R. J., Ward-Shaw, E. T., Labott, M. R., Lee, S., Lambert, P. F., & Mertz, J. E. (2023). Drugs That Mimic Hypoxia Selectively Target EBV-Positive Gastric Cancer Cells. Cancers, 15(6), 1846. https://doi.org/10.3390/cancers15061846