
Target Antigen Attributes and Their Contributions to Clinically Approved Antibody-Drug Conjugates (ADCs) in Haematopoietic and Solid Cancers

 , ,
, ,  and
and 
Abstract
Simple Summary
Abstract
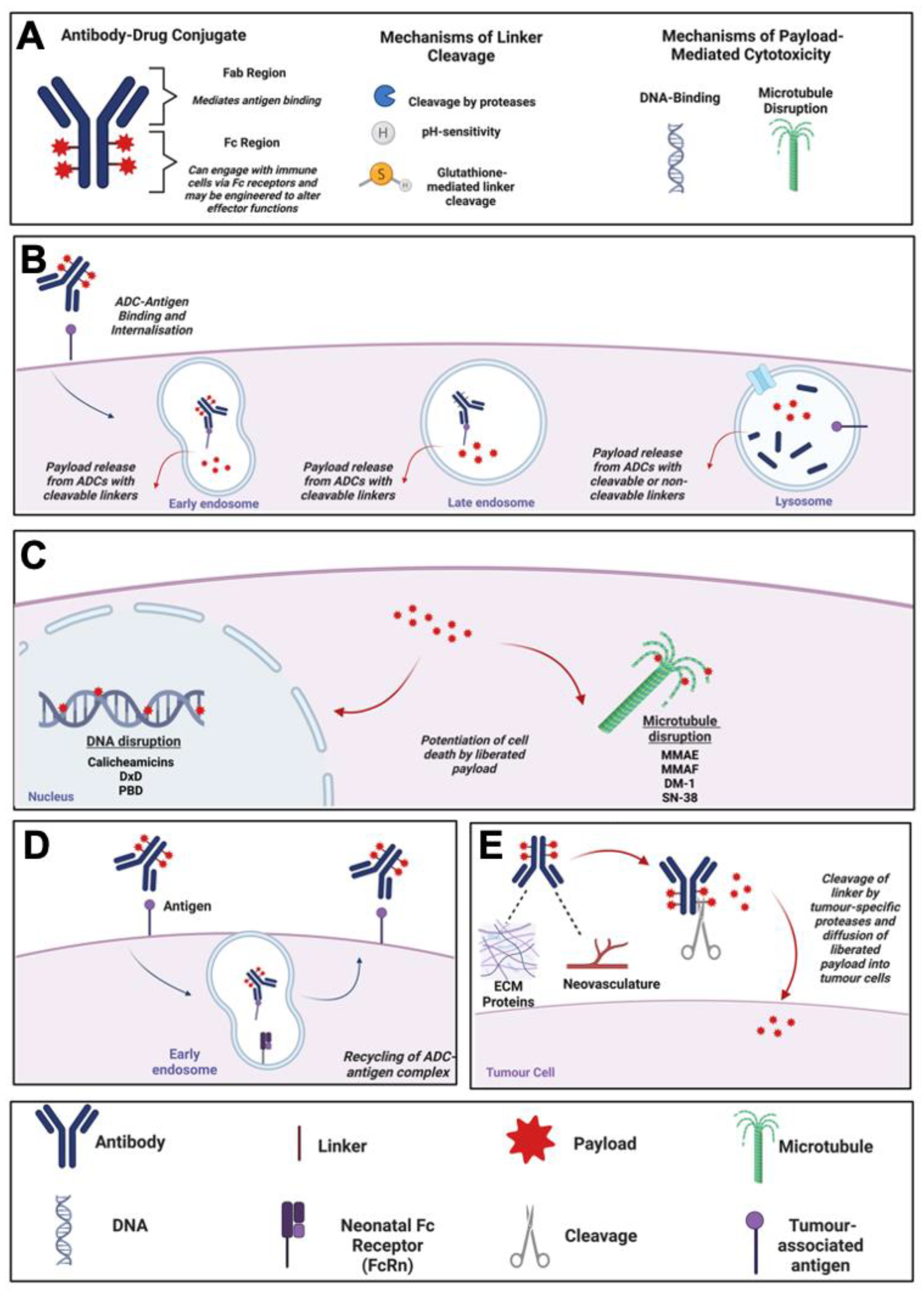
1. Key Features of ADCs
1.1. Introduction to Antibody Therapeutics in Oncology
1.2. Antibody-Drug Conjugates (ADCs)
1.3. Mechanism of Action of ADCs
1.4. Classes of ADC Payloads
1.4.1. Tubulin Inhibitors
1.4.2. Topoisomerase Inhibitors
1.4.3. DNA Cross-Linking Agents
1.4.4. DNA-Cleaving Agents
2. Features of Antigens for Antibody Target Selection in ADC Design
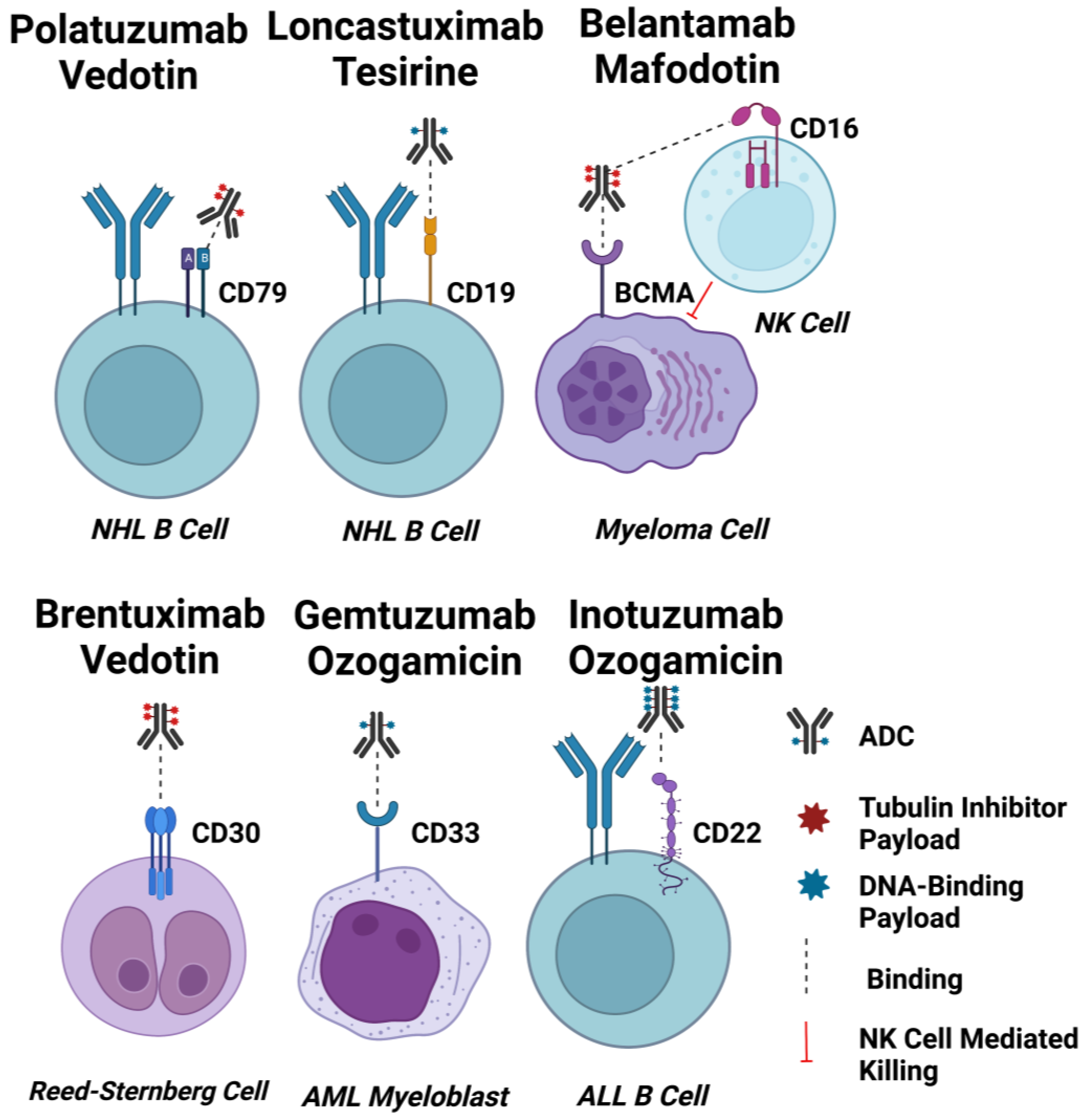
2.1. ADC Targets for Haematological Cancers
2.1.1. CD33
2.1.2. CD22
2.1.3. CD19
2.1.4. CD79b
2.1.5. BCMA
2.1.6. CD30
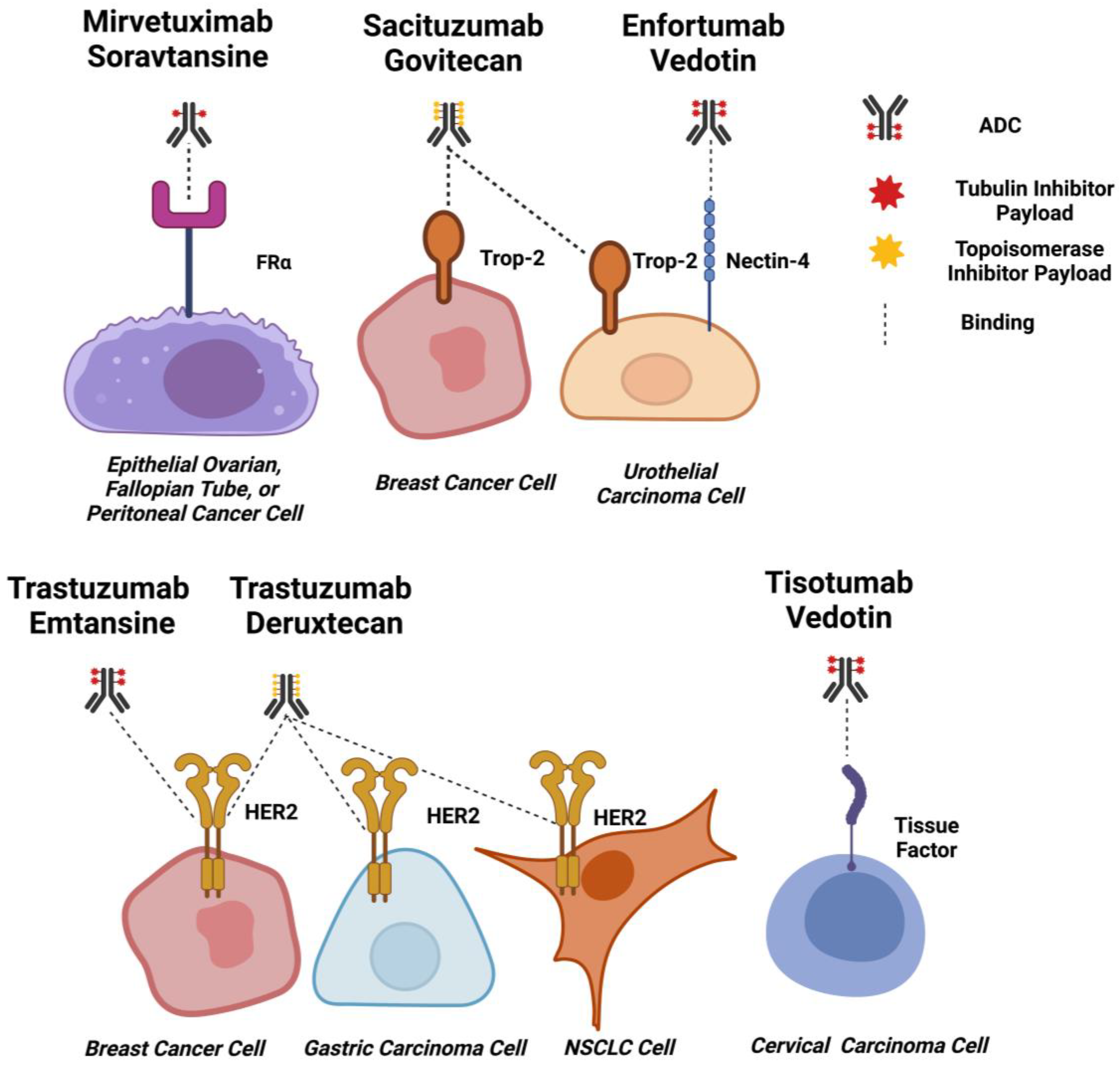
2.2. ADC Targets for Solid Tumours
2.2.1. HER2
2.2.2. TROP-2
2.2.3. Nectin-4
2.2.4. Tissue Factor
2.2.5. FRα
3. Target Selection in the Design of ADCs
3.1. Target Expression Levels
3.2. Toxicities Associated with Target Expression on Non-Malignant Tissues
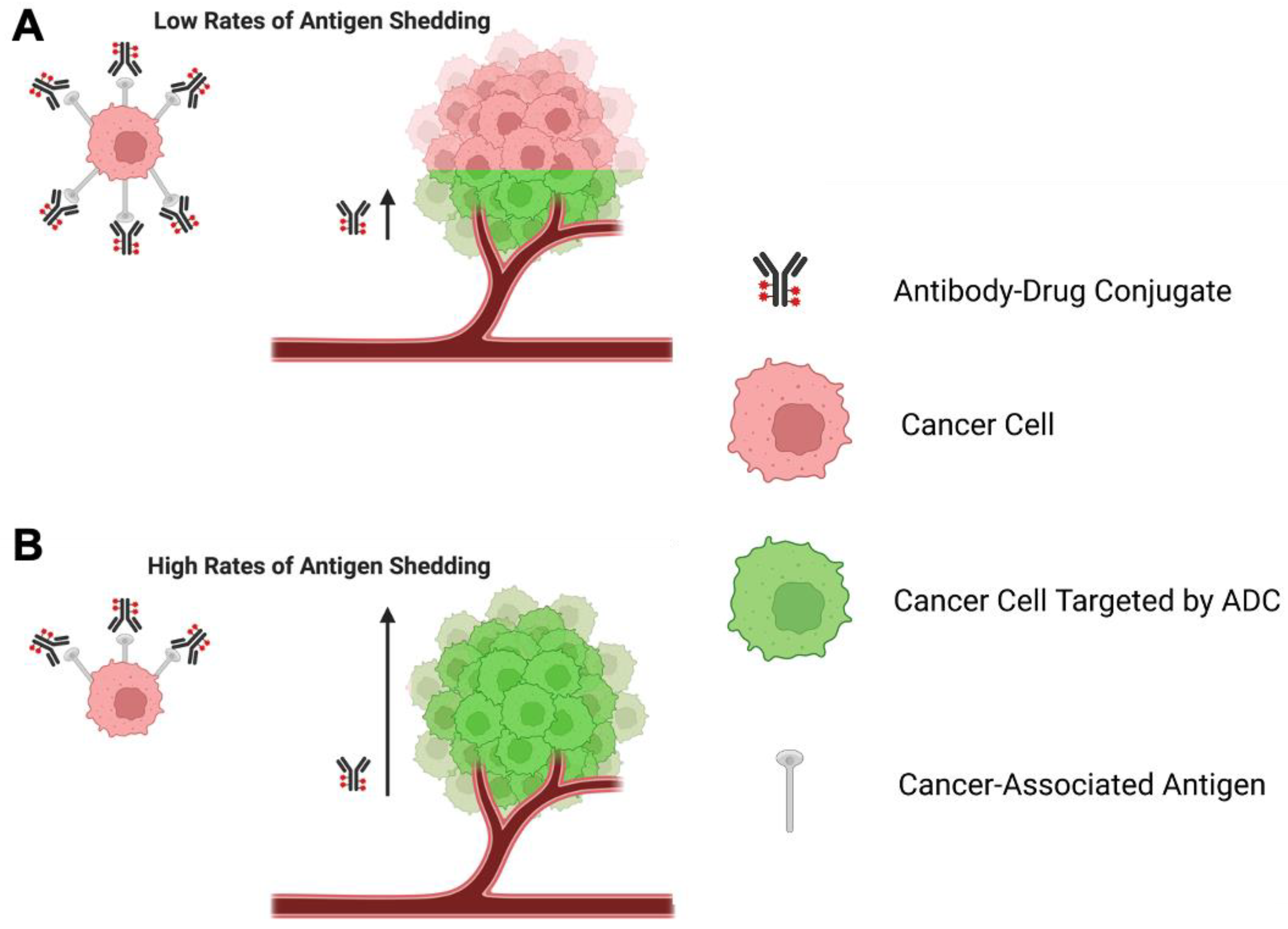
3.3. Significance of Antigen Shedding on ADC Function
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Goydel, R.S.; Rader, C. Antibody-based cancer therapy. Oncogene 2021, 40, 3655–3664. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, R.M.; Coumbe, B.G.T.; Josephs, D.H.; Mele, S.; Ilieva, K.M.; Cheung, A.; Tutt, A.N.; Spicer, J.F.; Thurston, D.E.; Crescioli, S.; et al. Antibody structure and engineering considerations for the design and function of Antibody Drug Conjugates (ADCs). Oncoimmunology 2018, 7, e1395127. [Google Scholar] [CrossRef] [PubMed]
- Pysz, I.; Jackson, P.J.M.; Thurston, D.E. Chapter 1: Introduction to Antibody–Drug Conjugates (ADCs). In Cytotoxic Payloads for Antibody–Drug Conjugates; The Royal Society of Chemistry: London, UK, 2019; pp. 1–30. [Google Scholar]
- Chau, C.H.; Steeg, P.S.; Figg, W.D. Antibody-drug conjugates for cancer. Lancet 2019, 394, 793–804. [Google Scholar] [CrossRef] [PubMed]
- Yaghoubi, S.; Karimi, M.H.; Lotfinia, M.; Gharibi, T.; Mahi-Birjand, M.; Kavi, E.; Hosseini, F.; Sineh Sepehr, K.; Khatami, M.; Bagheri, N.; et al. Potential drugs used in the antibody-drug conjugate (ADC) architecture for cancer therapy. J. Cell. Physiol. 2020, 235, 31–64. [Google Scholar] [CrossRef]
- James, J.S.; Dubs, G. FDA approves new kind of lymphoma treatment. Food and Drug Administration. AIDS Treat. News. 1997, 284, 2–3. [Google Scholar]
- Goldenberg, M.M. Trastuzumab, a recombinant DNA-derived humanized monoclonal antibody, a novel agent for the treatment of metastatic breast cancer. Clin. Ther. 1999, 21, 309–318. [Google Scholar] [CrossRef]
- Gogesch, P.; Dudek, S.; van Zandbergen, G.; Waibler, Z.; Anzaghe, M. The Role of Fc Receptors on the Effectiveness of Therapeutic Monoclonal Antibodies. Int. J. Mol. Sci. 2021, 22, 8947. [Google Scholar] [CrossRef]
- Yu, J.; Song, Y.; Tian, W. How to select IgG subclasses in developing anti-tumor therapeutic antibodies. J. Hematol. Oncol. 2020, 13, 45. [Google Scholar] [CrossRef]
- Vukovic, N.; van Elsas, A.; Verbeek, J.S.; Zaiss, D.M.W. Isotype selection for antibody-based cancer therapy. Clin. Exp. Immunol. 2021, 203, 351–365. [Google Scholar] [CrossRef]
- Abdollahpour-Alitappeh, M.; Lotfinia, M.; Gharibi, T.; Mardaneh, J.; Farhadihosseinabadi, B.; Larki, P.; Faghfourian, B.; Sepehr, K.S.; Abbaszadeh-Goudarzi, K.; Abbaszadeh-Goudarzi, G.; et al. Antibody-drug conjugates (ADCs) for cancer therapy: Strategies, challenges, and successes. J. Cell. Physiol. 2019, 234, 5628–5642. [Google Scholar] [CrossRef]
- Criscitiello, C.; Morganti, S.; Curigliano, G. Antibody-drug conjugates in solid tumors: A look into novel targets. J. Hematol. Oncol. 2021, 14, 20. [Google Scholar] [CrossRef]
- Diamantis, N.; Banerji, U. Antibody-drug conjugates—An emerging class of cancer treatment. Br. J. Cancer 2016, 114, 362–367. [Google Scholar] [CrossRef]
- Joubert, N.; Beck, A.; Dumontet, C.; Denevault-Sabourin, C. Antibody-Drug Conjugates: The Last Decade. Pharmaceuticals 2020, 13, 245. [Google Scholar] [CrossRef]
- Khongorzul, P.; Ling, C.J.; Khan, F.U.; Ihsan, A.U.; Zhang, J. Antibody-Drug Conjugates: A Comprehensive Review. Mol. Cancer Res. 2020, 18, 3–19. [Google Scholar] [CrossRef]
- Janthur, W.D.; Cantoni, N.; Mamot, C. Drug conjugates such as Antibody Drug Conjugates (ADCs), immunotoxins and immunoliposomes challenge daily clinical practice. Int. J. Mol. Sci. 2012, 13, 16020–16045. [Google Scholar] [CrossRef]
- Bross, P.F.; Beitz, J.; Chen, G.; Chen, X.H.; Duffy, E.; Kieffer, L.; Roy, S.; Sridhara, R.; Rahman, A.; Williams, G.; et al. Approval summary: Gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin. Cancer Res. 2001, 7, 1490–1496. [Google Scholar]
- Kang, M.S.; Kong, T.W.S.; Khoo, J.Y.X.; Loh, T.P. Recent developments in chemical conjugation strategies targeting native amino acids in proteins and their applications in antibody-drug conjugates. Chem. Sci. 2021, 12, 13613–13647. [Google Scholar] [CrossRef]
- Fu, Z.; Li, S.; Han, S.; Shi, C.; Zhang, Y. Antibody drug conjugate: The “biological missile” for targeted cancer therapy. Signal Transduct. Target. Ther. 2022, 7, 93. [Google Scholar] [CrossRef]
- Jabbour, E.; Paul, S.; Kantarjian, H. The clinical development of antibody-drug conjugates—Lessons from leukaemia. Nat. Rev. Clin. Oncol. 2021, 18, 418–433. [Google Scholar] [CrossRef]
- McCombs, J.R.; Owen, S.C. Antibody drug conjugates: Design and selection of linker, payload and conjugation chemistry. AAPS J. 2015, 17, 339–351. [Google Scholar] [CrossRef]
- Ritchie, M.; Tchistiakova, L.; Scott, N. Implications of receptor-mediated endocytosis and intracellular trafficking dynamics in the development of antibody drug conjugates. MAbs 2013, 5, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Birrer, M.J.; Moore, K.N.; Betella, I.; Bates, R.C. Antibody-Drug Conjugate-Based Therapeutics: State of the Science. J. Natl. Cancer Inst. 2019, 111, 538–549. [Google Scholar] [CrossRef]
- Ashman, N.; Bargh, J.D.; Spring, D.R. Non-internalising antibody–drug conjugates. Chem. Soc. Rev. 2022, 51, 9182–9202. [Google Scholar] [CrossRef] [PubMed]
- Gébleux, R.; Stringhini, M.; Casanova, R.; Soltermann, A.; Neri, D. Non-internalizing antibody-drug conjugates display potent anti-cancer activity upon proteolytic release of monomethyl auristatin E in the subendothelial extracellular matrix. Int. J. Cancer 2017, 140, 1670–1679. [Google Scholar] [CrossRef] [PubMed]
- Dal Corso, A.; Gebleux, R.; Murer, P.; Soltermann, A.; Neri, D. A non-internalizing antibody-drug conjugate based on an anthracycline payload displays potent therapeutic activity in vivo. J. Control. Release 2017, 264, 211–218. [Google Scholar] [CrossRef]
- Nittoli, T.; Kelly, M.P.; Delfino, F.; Rudge, J.; Kunz, A.; Markotan, T.; Spink, J.; Chen, Z.; Shan, J.; Navarro, E.; et al. Antibody drug conjugates of cleavable amino-alkyl and aryl maytansinoids. Bioorg. Med. Chem. 2018, 26, 2271–2279. [Google Scholar] [CrossRef]
- Oroudjev, E.; Lopus, M.; Wilson, L.; Audette, C.; Provenzano, C.; Erickson, H.; Kovtun, Y.; Chari, R.; Jordan, M.A. Maytansinoid-antibody conjugates induce mitotic arrest by suppressing microtubule dynamic instability. Mol. Cancer Ther. 2010, 9, 2700–2713. [Google Scholar] [CrossRef]
- Akaiwa, M.; Dugal-Tessier, J.; Mendelsohn, B.A. Antibody-Drug Conjugate Payloads; Study of Auristatin Derivatives. Chem. Pharm. Bull. 2020, 68, 201–211. [Google Scholar] [CrossRef]
- Doronina, S.O.; Toki, B.E.; Torgov, M.Y.; Mendelsohn, B.A.; Cerveny, C.G.; Chace, D.F.; DeBlanc, R.L.; Gearing, R.P.; Bovee, T.D.; Siegall, C.B.; et al. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat. Biotechnol. 2003, 21, 778–784. [Google Scholar] [CrossRef]
- Li, F.; Emmerton, K.K.; Jonas, M.; Zhang, X.; Miyamoto, J.B.; Setter, J.R.; Nicholas, N.D.; Okeley, N.M.; Lyon, R.P.; Benjamin, D.R.; et al. Intracellular Released Payload Influences Potency and Bystander-Killing Effects of Antibody-Drug Conjugates in Preclinical Models. Cancer Res. 2016, 76, 2710–2719. [Google Scholar] [CrossRef]
- Alley, S.C.; Zhang, X.; Okeley, N.M.; Anderson, M.; Law, C.L.; Senter, P.D.; Benjamin, D.R. The pharmacologic basis for antibody-auristatin conjugate activity. J. Pharmacol. Exp. Ther. 2009, 330, 932–938. [Google Scholar] [CrossRef]
- Johansson, M.P.; Maaheimo, H.; Ekholm, F.S. New insight on the structural features of the cytotoxic auristatins MMAE and MMAF revealed by combined NMR spectroscopy and quantum chemical modelling. Sci. Rep. 2017, 7, 15920. [Google Scholar] [CrossRef]
- Lonial, S.; Lee, H.C.; Badros, A.; Trudel, S.; Nooka, A.K.; Chari, A.; Abdallah, A.O.; Callander, N.; Lendvai, N.; Sborov, D.; et al. Belantamab mafodotin for relapsed or refractory multiple myeloma (DREAMM-2): A two-arm, randomised, open-label, phase 2 study. Lancet Oncol. 2020, 21, 207–221. [Google Scholar] [CrossRef]
- Bailly, C. Irinotecan: 25 years of cancer treatment. Pharmacol. Res. 2019, 148, 104398. [Google Scholar] [CrossRef]
- Thomas, A.; Pommier, Y. Targeting Topoisomerase I in the Era of Precision Medicine. Clin. Cancer Res. 2019, 25, 6581–6589. [Google Scholar] [CrossRef]
- Yamato, M.; Hasegawa, J.; Maejima, T.; Hattori, C.; Kumagai, K.; Watanabe, A.; Nishiya, Y.; Shibutani, T.; Aida, T.; Hayakawa, I.; et al. DS-7300a, a DNA Topoisomerase I Inhibitor, DXd-Based Antibody–Drug Conjugate Targeting B7-H3, Exerts Potent Antitumor Activities in Preclinical Models. Mol. Cancer Ther. 2022, 21, 635–646. [Google Scholar] [CrossRef]
- Ogitani, Y.; Hagihara, K.; Oitate, M.; Naito, H.; Agatsuma, T. Bystander killing effect of DS-8201a, a novel anti-human epidermal growth factor receptor 2 antibody-drug conjugate, in tumors with human epidermal growth factor receptor 2 heterogeneity. Cancer Sci. 2016, 107, 1039–1046. [Google Scholar] [CrossRef]
- Mantaj, J.; Jackson, P.J.M.; Rahman, K.M.; Thurston, D.E. From Anthramycin to Pyrrolobenzodiazepine (PBD)-Containing Antibody–Drug Conjugates (ADCs). Angew. Chem. Int. Ed. 2017, 56, 462–488. [Google Scholar] [CrossRef]
- Lee, A. Loncastuximab Tesirine: First Approval. Drugs 2021, 81, 1229–1233. [Google Scholar] [CrossRef]
- Zammarchi, F.; Corbett, S.; Adams, L.; Tyrer, P.C.; Kiakos, K.; Janghra, N.; Marafioti, T.; Britten, C.E.; Havenith, C.E.G.; Chivers, S.; et al. ADCT-402, a PBD dimer-containing antibody drug conjugate targeting CD19-expressing malignancies. Blood 2018, 131, 1094–1105. [Google Scholar] [CrossRef]
- Shor, B.; Kahler, J.; Dougher, M.; Xu, J.; Mack, M.; Rosfjord, E.; Wang, F.; Melamud, E.; Sapra, P. Enhanced Antitumor Activity of an Anti-5T4 Antibody-Drug Conjugate in Combination with PI3K/mTOR inhibitors or Taxanes. Clin. Cancer Res. 2016, 22, 383–394. [Google Scholar] [CrossRef] [PubMed]
- FDA Approves Inotuzumab Ozogamicin for Relapsed or Refractory B-Cell Precursor ALL. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-inotuzumab-ozogamicin-relapsed-or-refractory-b-cell-precursor-all (accessed on 9 February 2023).
- Harper, J.; Hollingsworth, R. Selecting Optimal Antibody–Drug Conjugate Targets Using Indication-Dependent or Indication-Independent Approaches. In Antibody Drug Conjugates: Fundamentals, Drug Development, and Clinical Outcomes to Target Cancer; Wiley: Hoboken, NJ, USA, 2016; pp. 33–58. [Google Scholar]
- Drago, J.Z.; Modi, S.; Chandarlapaty, S. Unlocking the potential of antibody-drug conjugates for cancer therapy. Nat. Rev. Clin. Oncol. 2021, 18, 327–344. [Google Scholar] [CrossRef] [PubMed]
- Bander, N.H. Antibody–Drug Conjugate Target Selection: Critical Factors. In Antibody-Drug Conjugates; Ducry, L., Ed.; Humana Press: Totowa, NJ, USA, 2013; pp. 29–40. [Google Scholar]
- Panowski, S.; Bhakta, S.; Raab, H.; Polakis, P.; Junutula, J.R. Site-specific antibody drug conjugates for cancer therapy. MAbs 2014, 6, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Thurber, G.M.; Schmidt, M.M.; Wittrup, K.D. Antibody tumor penetration: Transport opposed by systemic and antigen-mediated clearance. Adv. Drug Deliv. Rev. 2008, 60, 1421–1434. [Google Scholar] [CrossRef]
- Hammood, M.; Craig, A.W.; Leyton, J.V. Impact of Endocytosis Mechanisms for the Receptors Targeted by the Currently Approved Antibody-Drug Conjugates (ADCs)-A Necessity for Future ADC Research and Development. Pharmaceuticals 2021, 14, 674. [Google Scholar] [CrossRef]
- Zhang, Y.; Pastan, I. High shed antigen levels within tumors: An additional barrier to immunoconjugate therapy. Clin. Cancer Res. 2008, 14, 7981–7986. [Google Scholar] [CrossRef]
- Kim, E.P.Y. A Perspective on the Effects of Antigen Shedding on Targeted Delivery of Immunotoxins in Solid Tumors: A Mathematical Model Study. Bull. Korean Chem. Soc. 2016, 37, 1977–1984. [Google Scholar] [CrossRef]
- Pak, Y.; Pastan, I.; Kreitman, R.J.; Lee, B. Effect of antigen shedding on targeted delivery of immunotoxins in solid tumors from a mathematical model. PLoS ONE 2014, 9, e110716. [Google Scholar] [CrossRef]
- Pak, Y.; Zhang, Y.; Pastan, I.; Lee, B. Antigen shedding may improve efficiencies for delivery of antibody-based anticancer agents in solid tumors. Cancer Res. 2012, 72, 3143–3152. [Google Scholar] [CrossRef]
- Tong, J.T.W.; Harris, P.W.R.; Brimble, M.A.; Kavianinia, I. An Insight into FDA Approved Antibody-Drug Conjugates for Cancer Therapy. Molecules 2021, 26, 5847. [Google Scholar] [CrossRef]
- Damelin, M.; Zhong, W.; Myers, J.; Sapra, P. Evolving Strategies for Target Selection for Antibody-Drug Conjugates. Pharm. Res. 2015, 32, 3494–3507. [Google Scholar] [CrossRef]
- Chu, Y.; Zhou, X.; Wang, X. Antibody-drug conjugates for the treatment of lymphoma: Clinical advances and latest progress. J. Hematol. Oncol. 2021, 14, 88. [Google Scholar] [CrossRef]
- Andrews, R.G.; Singer, J.W.; Bernstein, I.D. Precursors of colony-forming cells in humans can be distinguished from colony-forming cells by expression of the CD33 and CD34 antigens and light scatter properties. J. Exp. Med. 1989, 169, 1721–1731. [Google Scholar] [CrossRef]
- Laszlo, G.S.; Estey, E.H.; Walter, R.B. The past and future of CD33 as therapeutic target in acute myeloid leukemia. Blood Rev. 2014, 28, 143–153. [Google Scholar] [CrossRef]
- Jilani, I.; Estey, E.; Huh, Y.; Joe, Y.; Manshouri, T.; Yared, M.; Giles, F.; Kantarjian, H.; Cortes, J.; Thomas, D.; et al. Differences in CD33 intensity between various myeloid neoplasms. Am. J. Clin. Pathol. 2002, 118, 560–566. [Google Scholar] [CrossRef]
- Molica, M.; Perrone, S.; Mazzone, C.; Niscola, P.; Cesini, L.; Abruzzese, E.; de Fabritiis, P. CD33 Expression and Gentuzumab Ozogamicin in Acute Myeloid Leukemia: Two Sides of the Same Coin. Cancers 2021, 13, 3214. [Google Scholar] [CrossRef]
- Petersdorf, S.H.; Kopecky, K.J.; Slovak, M.; Willman, C.; Nevill, T.; Brandwein, J.; Larson, R.A.; Erba, H.P.; Stiff, P.J.; Stuart, R.K.; et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood 2013, 121, 4854–4860. [Google Scholar] [CrossRef]
- Giles, F.J.; Kantarjian, H.M.; Kornblau, S.M.; Thomas, D.A.; Garcia-Manero, G.; Waddelow, T.A.; David, C.L.; Phan, A.T.; Colburn, D.E.; Rashid, A.; et al. Mylotarg™ (gemtuzumab ozogamicin) therapy is associated with hepatic venoocclusive disease in patients who have not received stem cell transplantation. Cancer Interdiscip. Int. J. Am. Cancer Soc. 2001, 92, 406–413. [Google Scholar] [CrossRef]
- Rajvanshi, P.; Shulman, H.M.; Sievers, E.L.; McDonald, G.B. Hepatic sinusoidal obstruction after gemtuzumab ozogamicin (Mylotarg) therapy. Blood 2002, 99, 2310–2314. [Google Scholar] [CrossRef]
- Van Der Velden, V.H.; te Marvelde, J.G.; Hoogeveen, P.G.; Bernstein, I.D.; Houtsmuller, A.B.; Berger, M.S.; van Dongen, J.J. Targeting of the CD33-calicheamicin immunoconjugate Mylotarg (CMA-676) in acute myeloid leukemia: In vivo and in vitro saturation and internalization by leukemic and normal myeloid cells. Blood 2001, 97, 3197–3204. [Google Scholar] [CrossRef]
- Taksin, A.L.; Legrand, O.; Raffoux, E.; de Revel, T.; Thomas, X.; Contentin, N.; Bouabdallah, R.; Pautas, C.; Turlure, P.; Reman, O.; et al. High efficacy and safety profile of fractionated doses of Mylotarg as induction therapy in patients with relapsed acute myeloblastic leukemia: A prospective study of the alfa group. Leukemia 2007, 21, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Norsworthy, K.J.; Ko, C.W.; Lee, J.E.; Liu, J.; John, C.S.; Przepiorka, D.; Farrell, A.T.; Pazdur, R. FDA Approval Summary: Mylotarg for Treatment of Patients with Relapsed or Refractory CD33-Positive Acute Myeloid Leukemia. Oncologist 2018, 23, 1103–1108. [Google Scholar] [CrossRef] [PubMed]
- Wyeth Pharmaceuticals. Mylotarg (Gemtuzumab Ozogamicin) [Package Insert]. US Food and Drug Administration Website. Revised June 2020. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761060s004lbl.pdf. (accessed on 23 June 2022).
- Pollard, J.A.; Loken, M.; Gerbing, R.B.; Raimondi, S.C.; Hirsch, B.A.; Aplenc, R.; Bernstein, I.D.; Gamis, A.S.; Alonzo, T.A.; Meshinchi, S. CD33 Expression and Its Association With Gemtuzumab Ozogamicin Response: Results From the Randomized Phase III Children’s Oncology Group Trial AAML0531. J. Clin. Oncol. 2016, 34, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Van der Velden, V.H.; Boeckx, N.; Jedema, I.; te Marvelde, J.G.; Hoogeveen, P.G.; Boogaerts, M.; van Dongen, J.J. High CD33-antigen loads in peripheral blood limit the efficacy of gemtuzumab ozogamicin (Mylotarg) treatment in acute myeloid leukemia patients. Leukemia 2004, 18, 983–988. [Google Scholar] [CrossRef] [PubMed]
- Walter, R.B.; Gooley, T.A.; van der Velden, V.H.; Loken, M.R.; van Dongen, J.J.; Flowers, D.A.; Bernstein, I.D.; Appelbaum, F.R. CD33 expression and P-glycoprotein-mediated drug efflux inversely correlate and predict clinical outcome in patients with acute myeloid leukemia treated with gemtuzumab ozogamicin monotherapy. Blood 2007, 109, 4168–4170. [Google Scholar] [CrossRef]
- Doody, G.M.; Justement, L.B.; Delibrias, C.C.; Matthews, R.J.; Lin, J.; Thomas, M.L.; Fearon, D.T. A role in B cell activation for CD22 and the protein tyrosine phosphatase SHP. Science 1995, 269, 242–244. [Google Scholar] [CrossRef]
- Tedder, T.F.; Tuscano, J.; Sato, S.; Kehrl, J.H. CD22, a B lymphocyte-specific adhesion molecule that regulates antigen receptor signaling. Annu. Rev. Immunol. 1997, 15, 481–504. [Google Scholar] [CrossRef]
- O’Reilly, M.K.; Tian, H.; Paulson, J.C. CD22 is a recycling receptor that can shuttle cargo between the cell surface and endosomal compartments of B cells. J. Immunol. 2011, 186, 1554–1563. [Google Scholar] [CrossRef]
- DiJoseph, J.F.; Armellino, D.C.; Boghaert, E.R.; Khandke, K.; Dougher, M.M.; Sridharan, L.; Kunz, A.; Hamann, P.R.; Gorovits, B.; Udata, C.; et al. Antibody-targeted chemotherapy with CMC-544: A CD22-targeted immunoconjugate of calicheamicin for the treatment of B-lymphoid malignancies. Blood 2004, 103, 1807–1814. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; DeAngelo, D.J.; Stelljes, M.; Liedtke, M.; Stock, W.; Gökbuget, N.; O’Brien, S.M.; Jabbour, E.; Wang, T.; Liang White, J.; et al. Inotuzumab ozogamicin versus standard of care in relapsed or refractory acute lymphoblastic leukemia: Final report and long-term survival follow-up from the randomized, phase 3 INO-VATE study. Cancer 2019, 125, 2474–2487. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; DeAngelo, D.J.; Stelljes, M.; Martinelli, G.; Liedtke, M.; Stock, W.; Gökbuget, N.; O’Brien, S.; Wang, K.; Wang, T.; et al. Inotuzumab Ozogamicin versus Standard Therapy for Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2016, 375, 740–753. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; Stock, W.; Cassaday, R.D.; DeAngelo, D.J.; Jabbour, E.; O’Brien, S.M.; Stelljes, M.; Wang, T.; Paccagnella, M.L.; Nguyen, K.; et al. Efficacy and Safety Outcomes in the Phase 3 INO-Vate Trial By Baseline CD22 Positivity Assessed By Local Laboratories. Blood 2019, 134 (Suppl. S1), 1344. [Google Scholar] [CrossRef]
- Wang, K.; Wei, G.; Liu, D. CD19: A biomarker for B cell development, lymphoma diagnosis and therapy. Exp. Hematol. Oncol. 2012, 1, 36. [Google Scholar] [CrossRef]
- Ingle, G.S.; Chan, P.; Elliott, J.M.; Chang, W.S.; Koeppen, H.; Stephan, J.P.; Scales, S.J. High CD21 expression inhibits internalization of anti-CD19 antibodies and cytotoxicity of an anti-CD19-drug conjugate. Br. J. Haematol. 2008, 140, 46–58. [Google Scholar] [CrossRef]
- Scheuermann, R.H.; Racila, E. CD19 antigen in leukemia and lymphoma diagnosis and immunotherapy. Leuk. Lymphoma 1995, 18, 385–397. [Google Scholar] [CrossRef]
- Makita, S.; Tobinai, K. Antibody therapy targeting CD19 for B-cell non-Hodgkin’s lymphoma. Ann. Oncol. 2018, 29, 1086–1089. [Google Scholar] [CrossRef]
- Caimi, P.F.; Ai, W.; Alderuccio, J.P.; Ardeshna, K.M.; Hamadani, M.; Hess, B.; Kahl, B.S.; Radford, J.; Solh, M.; Stathis, A.; et al. Loncastuximab tesirine in relapsed or refractory diffuse large B-cell lymphoma (LOTIS-2): A multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2021, 22, 790–800. [Google Scholar] [CrossRef]
- Dornan, D.; Bennett, F.; Chen, Y.; Dennis, M.; Eaton, D.; Elkins, K.; French, D.; Go, M.A.; Jack, A.; Junutula, J.R.; et al. Therapeutic potential of an anti-CD79b antibody-drug conjugate, anti-CD79b-vc-MMAE, for the treatment of non-Hodgkin lymphoma. Blood 2009, 114, 2721–2729. [Google Scholar] [CrossRef]
- Polson, A.G.; Yu, S.F.; Elkins, K.; Zheng, B.; Clark, S.; Ingle, G.S.; Slaga, D.S.; Giere, L.; Du, C.; Tan, C.; et al. Antibody-drug conjugates targeted to CD79 for the treatment of non-Hodgkin lymphoma. Blood 2007, 110, 616–623. [Google Scholar] [CrossRef]
- Teicher, B.A. Antibody-drug conjugate targets. Curr. Cancer Drug Targets 2009, 9, 982–1004. [Google Scholar] [CrossRef]
- Deeks, E.D. Polatuzumab Vedotin: First Global Approval. Drugs 2019, 79, 1467–1475. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, M.; Zheng, B.; Erdmann, T.; Koeppen, H.; McCord, R.; Grau, M.; Staiger, A.; Chai, A.; Sandmann, T.; Madle, H.; et al. Anti-CD22 and anti-CD79B antibody drug conjugates are active in different molecular diffuse large B-cell lymphoma subtypes. Leukemia 2015, 29, 1578–1586. [Google Scholar] [CrossRef] [PubMed]
- Coiffier, B.; Lepage, E.; Briere, J.; Herbrecht, R.; Tilly, H.; Bouabdallah, R.; Morel, P.; Van Den Neste, E.; Salles, G.; Gaulard, P.; et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N. Engl. J. Med. 2002, 346, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Sehn, L.H.; Herrera, A.F.; Flowers, C.R.; Kamdar, M.K.; McMillan, A.; Hertzberg, M.; Assouline, S.; Kim, T.M.; Kim, W.S.; Ozcan, M.; et al. Polatuzumab Vedotin in Relapsed or Refractory Diffuse Large B-Cell Lymphoma. J. Clin. Oncol. 2020, 38, 155–165. [Google Scholar] [CrossRef]
- FDA Approves Polatuzumab Vedotin-Piiq for Diffuse Large B-Cell Lymphoma. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-polatuzumab-vedotin-piiq-diffuse-large-b-cell-lymphoma (accessed on 9 February 2023).
- Bourbon, E.; Salles, G. Polatuzumab vedotin: An investigational anti-CD79b antibody drug conjugate for the treatment of diffuse large B-cell lymphoma. Expert Opin. Investig. Drugs 2020, 29, 1079–1088. [Google Scholar] [CrossRef]
- Herrera, A.F.; Tracy, S.; Croft, B.; Opat, S.; Ray, J.; Lovejoy, A.F.; Musick, L.; Paulson, J.N.; Sehn, L.H.; Jiang, Y. Risk profiling of patients with relapsed/refractory diffuse large B-cell lymphoma by measuring circulating tumor DNA. Blood Adv. 2022, 6, 1651–1660. [Google Scholar] [CrossRef]
- Tai, Y.-T.; Mayes, P.A.; Acharya, C.; Zhong, M.Y.; Cea, M.; Cagnetta, A.; Craigen, J.; Yates, J.; Gliddon, L.; Fieles, W.; et al. Novel anti–B-cell maturation antigen antibody-drug conjugate (GSK2857916) selectively induces killing of multiple myeloma. Blood 2014, 123, 3128–3138. [Google Scholar] [CrossRef]
- Cho, S.F.; Anderson, K.C.; Tai, Y.T. Targeting B Cell Maturation Antigen (BCMA) in Multiple Myeloma: Potential Uses of BCMA-Based Immunotherapy. Front. Immunol. 2018, 9, 1821. [Google Scholar] [CrossRef]
- Markham, A. Belantamab Mafodotin: First Approval. Drugs. 2020, 80, 1607–1613. [Google Scholar] [CrossRef]
- Wahab, A.; Rafae, A.; Mushtaq, K.; Masood, A.; Ehsan, H.; Khakwani, M.; Khan, A. Ocular Toxicity of Belantamab Mafodotin, an Oncological Perspective of Management in Relapsed and Refractory Multiple Myeloma. Front. Oncol. 2021, 11, 678634. [Google Scholar] [CrossRef]
- Aschauer, J.; Donner, R.; Lammer, J.; Roberts, P.; Funk, M.; Agis, H.; Schmidinger, G. Corneal Toxicity Associated With Belantamab Mafodotin Is Not Restricted to the Epithelium: Neuropathy Studied With Confocal Microscopy. Am. J. Ophthalmol. 2022, 242, 116–124. [Google Scholar] [CrossRef]
- Abeykoon, J.P.; Vaxman, J.; Patel, S.V.; Kumar, S.; Malave, G.C.; Young, K.S.; Ailawadhi, S.; Larsen, J.T.; Dispenzieri, A.; Muchtar, E.; et al. Impact of belantamab mafodotin-induced ocular toxicity on outcomes of patients with advanced multiple myeloma. Br. J. Haematol. 2022, 199, 95–99. [Google Scholar] [CrossRef]
- GSK Provides Update on DREAMM-3 Phase III Trial for Blenrep in Relapsed/Refractory Multiple Myeloma: GSK. Available online: https://www.gsk.com/en-gb/media/press-releases/gsk-provides-update-on-dreamm-3-phase-iii-trial-for-blenrep/ (accessed on 25 January 2023).
- Van der Weyden, C.A.; Pileri, S.A.; Feldman, A.L.; Whisstock, J.; Prince, H.M. Understanding CD30 biology and therapeutic targeting: A historical perspective providing insight into future directions. Blood Cancer J. 2017, 7, e603. [Google Scholar] [CrossRef]
- Deutsch, Y.E.; Tadmor, T.; Podack, E.R.; Rosenblatt, J.D. CD30: An important new target in hematologic malignancies. Leuk. Lymphoma 2011, 52, 1641–1654. [Google Scholar] [CrossRef]
- Senter, P.D.; Sievers, E.L. The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat. Biotechnol. 2012, 30, 631–637. [Google Scholar] [CrossRef]
- Younes, A.; Yasothan, U.; Kirkpatrick, P. Brentuximab vedotin. Nat. Rev. Drug Discov. 2012, 11, 19–20. [Google Scholar] [CrossRef]
- Straus, D.J.; Długosz-Danecka, M.; Alekseev, S.; Illés, Á.; Picardi, M.; Lech-Maranda, E.; Feldman, T.; Smolewski, P.; Savage, K.J.; Bartlett, N.L.; et al. Brentuximab vedotin with chemotherapy for stage III/IV classical Hodgkin lymphoma: 3-year update of the ECHELON-1 study. Blood 2020, 135, 735–742. [Google Scholar] [CrossRef]
- Okeley, N.M.; Miyamoto, J.B.; Zhang, X.; Sanderson, R.J.; Benjamin, D.R.; Sievers, E.L.; Senter, P.D.; Alley, S.C. Intracellular activation of SGN-35, a potent anti-CD30 antibody-drug conjugate. Clin. Cancer Res. 2010, 16, 888–897. [Google Scholar] [CrossRef]
- Velasco, R.; Domingo-Domenech, E.; Sureda, A. Brentuximab-Induced Peripheral Neurotoxicity: A Multidisciplinary Approach to Manage an Emerging Challenge in Hodgkin Lymphoma Therapy. Cancers 2021, 13, 6125. [Google Scholar] [CrossRef]
- Oh, D.Y.; Bang, Y.J. HER2-targeted therapies—A role beyond breast cancer. Nat. Rev. Clin. Oncol. 2020, 17, 33–48. [Google Scholar] [CrossRef]
- Schettini, F.; Prat, A. Dissecting the biological heterogeneity of HER2-positive breast cancer. Breast 2021, 59, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Houghton, S.C.; Hankinson, S.E. Cancer Progress and Priorities: Breast Cancer. Cancer Epidemiol. Biomark. Prev. 2021, 30, 822–844. [Google Scholar] [CrossRef] [PubMed]
- Leone, F.; Perissinotto, E.; Cavalloni, G.; Fonsato, V.; Bruno, S.; Surrenti, N.; Hong, D.; Capaldi, A.; Geuna, M.; Piacibello, W.; et al. Expression of the c-ErbB-2/HER2 proto-oncogene in normal hematopoietic cells. J. Leukoc. Biol. 2003, 74, 593–601. [Google Scholar] [CrossRef] [PubMed]
- You, F.; Roberts, L.A.; Kang, S.P.; Nunes, R.A.; Dias, C.; Iglehart, J.D.; Solomon, N.A.; Friedman, P.N.; Harris, L.N. Low-level expression of HER2 and CK19 in normal peripheral blood mononuclear cells: Relevance for detection of circulating tumor cells. J. Hematol. Oncol. 2008, 1, 2. [Google Scholar] [CrossRef] [PubMed]
- Casalini, P.; Iorio, M.V.; Galmozzi, E.; Menard, S. Role of HER receptors family in development and differentiation. J. Cell. Physiol. 2004, 200, 343–350. [Google Scholar] [CrossRef]
- Scaltriti, M.; Rojo, F.; Ocana, A.; Anido, J.; Guzman, M.; Cortes, J.; Di Cosimo, S.; Matias-Guiu, X.; Ramon y Cajal, S.; Arribas, J.; et al. Expression of p95HER2, a truncated form of the HER2 receptor, and response to anti-HER2 therapies in breast cancer. J. Natl. Cancer Inst. 2007, 99, 628–638. [Google Scholar] [CrossRef]
- Tse, C.; Gauchez, A.S.; Jacot, W.; Lamy, P.J. HER2 shedding and serum HER2 extracellular domain: Biology and clinical utility in breast cancer. Cancer Treat. Rev. 2012, 38, 133–142. [Google Scholar] [CrossRef]
- Albanell, J.; Baselga, J. Trastuzumab, a humanized anti-HER2 monoclonal antibody, for the treatment of breast cancer. Drugs Today 1999, 35, 931–946. [Google Scholar]
- Austin, C.D.; De Maziere, A.M.; Pisacane, P.I.; van Dijk, S.M.; Eigenbrot, C.; Sliwkowski, M.X.; Klumperman, J.; Scheller, R.H. Endocytosis and sorting of ErbB2 and the site of action of cancer therapeutics trastuzumab and geldanamycin. Mol. Biol. Cell 2004, 15, 5268–5282. [Google Scholar] [CrossRef]
- Barok, M.; Joensuu, H.; Isola, J. Trastuzumab emtansine: Mechanisms of action and drug resistance. Breast Cancer Res. 2014, 16, 209. [Google Scholar] [CrossRef]
- Hunter, F.W.; Barker, H.R.; Lipert, B.; Rothe, F.; Gebhart, G.; Piccart-Gebhart, M.J.; Sotiriou, C.; Jamieson, S.M.F. Mechanisms of resistance to trastuzumab emtansine (T-DM1) in HER2-positive breast cancer. Br. J. Cancer 2020, 122, 603–612. [Google Scholar] [CrossRef]
- Seidman, A.; Hudis, C.; Pierri, M.K.; Shak, S.; Paton, V.; Ashby, M.; Murphy, M.; Stewart, S.J.; Keefe, D. Cardiac dysfunction in the trastuzumab clinical trials experience. J. Clin. Oncol. 2002, 20, 1215–1221. [Google Scholar] [CrossRef]
- Dempsey, N.; Rosenthal, A.; Dabas, N.; Kropotova, Y.; Lippman, M.; Bishopric, N.H. Trastuzumab-induced cardiotoxicity: A review of clinical risk factors, pharmacologic prevention, and cardiotoxicity of other HER2-directed therapies. Breast Cancer Res. Treat. 2021, 188, 21–36. [Google Scholar] [CrossRef]
- Onitilo, A.A.; Engel, J.M.; Stankowski, R.V. Cardiovascular toxicity associated with adjuvant trastuzumab therapy: Prevalence, patient characteristics, and risk factors. Ther. Adv. Drug Saf. 2014, 5, 154–166. [Google Scholar] [CrossRef]
- Mohan, N.; Jiang, J.; Dokmanovic, M.; Wu, W.J. Trastuzumab-mediated cardiotoxicity: Current understanding, challenges, and frontiers. Antib. Ther. 2018, 1, 13–17. [Google Scholar] [CrossRef]
- Krop, I.E.; Suter, T.M.; Dang, C.T.; Dirix, L.; Romieu, G.; Zamagni, C.; Citron, M.L.; Campone, M.; Xu, N.; Smitt, M.; et al. Feasibility and cardiac safety of trastuzumab emtansine after anthracycline-based chemotherapy as (neo)adjuvant therapy for human epidermal growth factor receptor 2-positive early-stage breast cancer. J. Clin. Oncol. 2015, 33, 1136–1142. [Google Scholar] [CrossRef]
- Ponde, N.; Ameye, L.; Lambertini, M.; Paesmans, M.; Piccart, M.; de Azambuja, E. Trastuzumab emtansine (T-DM1)-associated cardiotoxicity: Pooled analysis in advanced HER2-positive breast cancer. Eur. J. Cancer 2020, 126, 65–73. [Google Scholar] [CrossRef]
- Dieras, V.; Miles, D.; Verma, S.; Pegram, M.; Welslau, M.; Baselga, J.; Krop, I.E.; Blackwell, K.; Hoersch, S.; Xu, J.; et al. Trastuzumab emtansine versus capecitabine plus lapatinib in patients with previously treated HER2-positive advanced breast cancer (EMILIA): A descriptive analysis of final overall survival results from a randomised, open-label, phase 3 trial. Lancet Oncol. 2017, 18, 732–742. [Google Scholar] [CrossRef]
- Verma, S.; Miles, D.; Gianni, L.; Krop, I.E.; Welslau, M.; Baselga, J.; Pegram, M.; Oh, D.Y.; Dieras, V.; Guardino, E.; et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N. Engl. J. Med. 2012, 367, 1783–1791. [Google Scholar] [CrossRef]
- Perez, E.A.; Barrios, C.; Eiermann, W.; Toi, M.; Im, Y.H.; Conte, P.; Martin, M.; Pienkowski, T.; Pivot, X.; Burris, H., III; et al. Trastuzumab Emtansine With or Without Pertuzumab Versus Trastuzumab Plus Taxane for Human Epidermal Growth Factor Receptor 2-Positive, Advanced Breast Cancer: Primary Results From the Phase III MARIANNE Study. J. Clin. Oncol. 2017, 35, 141–148. [Google Scholar] [CrossRef]
- Modi, S.; Saura, C.; Yamashita, T.; Park, Y.H.; Kim, S.B.; Tamura, K.; Andre, F.; Iwata, H.; Ito, Y.; Tsurutani, J.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer. N. Engl. J. Med. 2020, 382, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.; Park, H.; Murthy, R.K.; Iwata, H.; Tamura, K.; Tsurutani, J.; Moreno-Aspitia, A.; Doi, T.; Sagara, Y.; Redfern, C.; et al. Antitumor Activity and Safety of Trastuzumab Deruxtecan in Patients With HER2-Low-Expressing Advanced Breast Cancer: Results From a Phase Ib Study. J. Clin. Oncol. 2020, 38, 1887–1896. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.; Jacot, W.; Yamashita, T.; Sohn, J.; Vidal, M.; Tokunaga, E.; Tsurutani, J.; Ueno, N.T.; Prat, A.; Chae, Y.S.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Low Advanced Breast Cancer. N. Engl. J. Med. 2022, 387, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Indini, A.; Rijavec, E.; Grossi, F. Trastuzumab Deruxtecan: Changing the Destiny of HER2 Expressing Solid Tumors. Int. J. Mol. Sci. 2021, 22, 4774. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.; Kim, S.B.; Chung, W.P.; Im, S.A.; Park, Y.H.; Hegg, R.; Kim, M.H.; Tseng, L.M.; Petry, V.; Chung, C.F.; et al. Trastuzumab Deruxtecan versus Trastuzumab Emtansine for Breast Cancer. N. Engl. J. Med. 2022, 386, 1143–1154. [Google Scholar] [CrossRef]
- FDA, D.I.S.C.O. Burst Edition: FDA Approval of Enhertu (Fam-Trastuzumab Deruxtecan-Nxki) for Adult Patients with Unresectable or Metastatic HER2-Positive Breast Cancer. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-disco-burst-edition-fda-approval-enhertu-fam-trastuzumab-deruxtecan-nxki-adult-patients (accessed on 9 February 2023).
- FDA Grants Accelerated Approval to Fam-Trastuzumab Deruxtecan-Nxki for HER2-Mutant Non-Small Cell Lung Cancer. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-fam-trastuzumab-deruxtecan-nxki-her2-mutant-non-small-cell-lung (accessed on 9 February 2023).
- Goto, K.; Sang-We, K.; Kubo, T.; Goto, Y.; Ahn, M.J.; Planchard, D.; Kim, D.W.; Yang, J.C.H.; Yang, T.Y.; Pereira, K.M.C.; et al. LBA55 Trastuzumab deruxtecan (T-DXd) in patients (Pts) with HER2-mutant metastatic non-small cell lung cancer (NSCLC): Interim results from the phase 2 DESTINY-Lung02 trial. Ann. Oncol. 2022, 33, S1422. [Google Scholar] [CrossRef]
- FDA Approves Fam-Trastuzumab Deruxtecan-Nxki for HER2-Positive Gastric Adenocarcinomas. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-fam-trastuzumab-deruxtecan-nxki-her2-positive-gastric-adenocarcinomas (accessed on 9 February 2023).
- Stepan, L.P.; Trueblood, E.S.; Hale, K.; Babcook, J.; Borges, L.; Sutherland, C.L. Expression of Trop2 cell surface glycoprotein in normal and tumor tissues: Potential implications as a cancer therapeutic target. J. Histochem. Cytochem. 2011, 59, 701–710. [Google Scholar] [CrossRef]
- Dum, D.; Taherpour, N.; Menz, A.; Höflmayer, D.; Völkel, C.; Hinsch, A.; Gorbokon, N.; Lennartz, M.; Hube-Magg, C.; Fraune, C.; et al. Trophoblast Cell Surface Antigen 2 Expression in Human Tumors: A Tissue Microarray Study on 18,563 Tumors. Pathobiology 2022, 89, 245–258. [Google Scholar] [CrossRef]
- Lenart, S.; Lenart, P.; Smarda, J.; Remsik, J.; Soucek, K.; Benes, P. Trop2: Jack of All Trades, Master of None. Cancers 2020, 12, 3328. [Google Scholar] [CrossRef]
- Shvartsur, A.; Bonavida, B. Trop2 and its overexpression in cancers: Regulation and clinical/therapeutic implications. Genes Cancer 2015, 6, 84–105. [Google Scholar] [CrossRef]
- Trerotola, M.; Guerra, E.; Ali, Z.; Aloisi, A.L.; Ceci, M.; Simeone, P.; Acciarito, A.; Zanna, P.; Vacca, G.; D’Amore, A.; et al. Trop-2 cleavage by ADAM10 is an activator switch for cancer growth and metastasis. Neoplasia 2021, 23, 415–428. [Google Scholar] [CrossRef]
- Ambrogi, F.; Fornili, M.; Boracchi, P.; Trerotola, M.; Relli, V.; Simeone, P.; La Sorda, R.; Lattanzio, R.; Querzoli, P.; Pedriali, M.; et al. Trop-2 is a determinant of breast cancer survival. PLoS ONE 2014, 9, e96993. [Google Scholar] [CrossRef]
- Jeon, Y.; Jo, U.; Hong, J.; Gong, G.; Lee, H.J. Trophoblast cell-surface antigen 2 (TROP2) expression in triple-negative breast cancer. BMC Cancer 2022, 22, 1014. [Google Scholar] [CrossRef]
- FDA Grants Regular Approval to Sacituzumab Govitecan for Triple-Negative Breast Cancer. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-regular-approval-sacituzumab-govitecan-triple-negative-breast-cancer (accessed on 9 February 2023).
- Bardia, A.; Mayer, I.A.; Vahdat, L.T.; Tolaney, S.M.; Isakoff, S.J.; Diamond, J.R.; O’Shaughnessy, J.; Moroose, R.L.; Santin, A.D.; Abramson, V.G.; et al. Sacituzumab Govitecan-hziy in Refractory Metastatic Triple-Negative Breast Cancer. N. Engl. J. Med. 2019, 380, 741–751. [Google Scholar] [CrossRef]
- FDA Grants Accelerated Approval to Sacituzumab Govitecan-Hziy for Metastatic Triple Negative Breast Cancer. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-sacituzumab-govitecan-hziy-metastatic-triple-negative-breast-cancer (accessed on 9 February 2023).
- Bardia, A.; Hurvitz, S.A.; Tolaney, S.M.; Loirat, D.; Punie, K.; Oliveira, M.; Brufsky, A.; Sardesai, S.D.; Kalinsky, K.; Zelnak, A.B.; et al. Sacituzumab Govitecan in Metastatic Triple-Negative Breast Cancer. N. Engl. J. Med. 2021, 384, 1529–1541. [Google Scholar] [CrossRef]
- Olivier, T.; Prasad, V. Sacituzumab govitecan in metastatic triple negative breast cancer (TNBC): Four design features in the ASCENT trial potentially favored the experimental arm. Transl. Oncol. 2022, 15, 101248. [Google Scholar] [CrossRef]
- Bardia, A.; Tolaney, S.M.; Punie, K.; Loirat, D.; Oliveira, M.; Kalinsky, K.; Zelnak, A.; Aftimos, P.; Dalenc, F.; Sardesai, S.; et al. Biomarker analyses in the phase III ASCENT study of sacituzumab govitecan versus chemotherapy in patients with metastatic triple-negative breast cancer. Ann. Oncol. 2021, 32, 1148–1156. [Google Scholar] [CrossRef]
- FDA Approves Sacituzumab Govitecan-Hziy for HR-Positive Breast Cancer. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-sacituzumab-govitecan-hziy-hr-positive-breast-cancer (accessed on 9 February 2023).
- Rugo, H.S.; Bardia, A.; Marmé, F.; Cortes, J.; Schmid, P.; Loirat, D.; Tredan, O.; Ciruelos, E.; Dalenc, F.; Gómez Pardo, P.; et al. Primary results from TROPiCS-02: A randomized phase 3 study of sacituzumab govitecan (SG) versus treatment of physician’s choice (TPC) in patients (Pts) with hormone receptor–positive/HER2-negative (HR+/HER2−) advanced breast cancer. J. Clin. Oncol. 2022, 40 (Suppl. S17), LBA1001. [Google Scholar] [CrossRef]
- Tagawa, S.T.; Balar, A.V.; Petrylak, D.P.; Kalebasty, A.R.; Loriot, Y.; Flechon, A.; Jain, R.K.; Agarwal, N.; Bupathi, M.; Barthelemy, P.; et al. TROPHY-U-01: A Phase II Open-Label Study of Sacituzumab Govitecan in Patients With Metastatic Urothelial Carcinoma Progressing After Platinum-Based Chemotherapy and Checkpoint Inhibitors. J. Clin. Oncol. 2021, 39, 2474–2485. [Google Scholar] [CrossRef]
- Rikitake, Y.; Mandai, K.; Takai, Y. The role of nectins in different types of cell-cell adhesion. J. Cell Sci. 2012, 125 Pt 16, 3713–3722. [Google Scholar] [CrossRef]
- Nishiwada, S.; Sho, M.; Yasuda, S.; Shimada, K.; Yamato, I.; Akahori, T.; Kinoshita, S.; Nagai, M.; Konishi, N.; Nakajima, Y. Nectin-4 expression contributes to tumor proliferation, angiogenesis and patient prognosis in human pancreatic cancer. J. Exp. Clin. Cancer Res. 2015, 34, 30. [Google Scholar] [CrossRef] [PubMed]
- Challita-Eid, P.M.; Satpayev, D.; Yang, P.; An, Z.; Morrison, K.; Shostak, Y.; Raitano, A.; Nadell, R.; Liu, W.; Lortie, D.R.; et al. Enfortumab Vedotin Antibody-Drug Conjugate Targeting Nectin-4 Is a Highly Potent Therapeutic Agent in Multiple Preclinical Cancer Models. Cancer Res. 2016, 76, 3003–3013. [Google Scholar] [CrossRef] [PubMed]
- Fabre-Lafay, S.; Garrido-Urbani, S.; Reymond, N.; Gonçalves, A.; Dubreuil, P.; Lopez, M. Nectin-4, a new serological breast cancer marker, is a substrate for tumor necrosis factor-alpha-converting enzyme (TACE)/ADAM-17. J. Biol. Chem. 2005, 280, 19543–19550. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, P.C.; Boylan, K.L.M.; Walcheck, B.; Heinze, R.; Geller, M.A.; Argenta, P.A.; Skubitz, A.P.N. Ectodomain shedding of the cell adhesion molecule Nectin-4 in ovarian cancer is mediated by ADAM10 and ADAM17. J. Biol. Chem. 2017, 292, 6339–6351. [Google Scholar] [CrossRef]
- Heath, E.I.; Rosenberg, J.E. The biology and rationale of targeting nectin-4 in urothelial carcinoma. Nat. Rev. Urol. 2021, 18, 93–103. [Google Scholar] [CrossRef]
- Komlodi-Pasztor, E.; Sackett, D.; Wilkerson, J.; Fojo, T. Mitosis is not a key target of microtubule agents in patient tumors. Nat. Rev. Clin. Oncol. 2011, 8, 244–250. [Google Scholar] [CrossRef]
- Cimmino, G.; Cirillo, P. Tissue factor: Newer concepts in thrombosis and its role beyond thrombosis and hemostasis. Cardiovasc. Diagn. Ther. 2018, 8, 581–593. [Google Scholar] [CrossRef]
- Forster, Y.; Meye, A.; Albrecht, S.; Schwenzer, B. Tissue factor and tumor: Clinical and laboratory aspects. Clin. Chim. Acta 2006, 364, 12–21. [Google Scholar] [CrossRef]
- Drake, T.A.; Morrissey, J.H.; Edgington, T.S. Selective cellular expression of tissue factor in human tissues. Implications for disorders of hemostasis and thrombosis. Am. J. Pathol. 1989, 134, 1087–1097. [Google Scholar]
- Mandal, S.K.; Pendurthi, U.R.; Rao, L.V. Cellular localization and trafficking of tissue factor. Blood 2006, 107, 4746–4753. [Google Scholar] [CrossRef]
- Coleman, R.L.; Lorusso, D.; Gennigens, C.; González-Martín, A.; Randall, L.; Cibula, D.; Lund, B.; Woelber, L.; Pignata, S.; Forget, F.; et al. Efficacy and safety of tisotumab vedotin in previously treated recurrent or metastatic cervical cancer (innovaTV 204/GOG-3023/ENGOT-cx6): A multicentre, open-label, single-arm, phase 2 study. Lancet Oncol. 2021, 22, 609–619. [Google Scholar] [CrossRef]
- FDA, D.I.S.C.O. Burst Edition: FDA Approvals of Tivdak (Tisotumab Vedotin-Tftv) for Recurrent or Metastatic Cervical Cancer with Disease Progression on or after Chemotherapy, and Jakafi (Ruxolitinib) for Chronic Graft-versus-Host Disease after Failure of One or Two Lines of Systemic Therapy. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-disco-burst-edition-fda-approvals-tivdak-tisotumab-vedotin-tftv-recurrent-or-metastatic-cervical (accessed on 23 August 2022).
- Cheung, A.; Bax, H.J.; Josephs, D.H.; Ilieva, K.M.; Pellizzari, G.; Opzoomer, J.; Bloomfield, J.; Fittall, M.; Grigoriadis, A.; Figini, M.; et al. Targeting folate receptor alpha for cancer treatment. Oncotarget 2016, 7, 52553–52574. [Google Scholar] [CrossRef]
- Kamen, B.A.; Smith, A.K. A review of folate receptor alpha cycling and 5-methyltetrahydrofolate accumulation with an emphasis on cell models in vitro. Adv. Drug Deliv. Rev. 2004, 56, 1085–1097. [Google Scholar] [CrossRef]
- Bax, H.J.; Chauhan, J.; Stavraka, C.; Santaolalla, A.; Osborn, G.; Khiabany, A.; Grandits, M.; López-Abente, J.; Palhares, L.C.G.F.; Chan Wah Hak, C.; et al. Folate receptor alpha in ovarian cancer tissue and patient serum is associated with disease burden and treatment outcomes. Br. J. Cancer 2022, 128, 324–353. [Google Scholar] [CrossRef]
- Ab, O.; Whiteman, K.R.; Bartle, L.M.; Sun, X.; Singh, R.; Tavares, D.; LaBelle, A.; Payne, G.; Lutz, R.J.; Pinkas, J.; et al. IMGN853, a Folate Receptor-α (FRα)-Targeting Antibody-Drug Conjugate, Exhibits Potent Targeted Antitumor Activity against FRα-Expressing Tumors. Mol. Cancer Ther. 2015, 14, 1605–1613. [Google Scholar] [CrossRef]
- Moore, K.N.; Oza, A.M.; Colombo, N.; Oaknin, A.; Scambia, G.; Lorusso, D.; Konecny, G.E.; Banerjee, S.; Murphy, C.G.; Tanyi, J.L.; et al. Phase III, randomized trial of mirvetuximab soravtansine versus chemotherapy in patients with platinum-resistant ovarian cancer: Primary analysis of FORWARD I. Ann. Oncol. 2021, 32, 757–765. [Google Scholar] [CrossRef]
- Matulonis, U.A.; Oaknin, A.; Pignata, S.; Denys, H.; Colombo, N.; Gorp, T.V.; Konner, J.A.; Romeo, M.; Harter, P.; Murphy, C.G.; et al. Mirvetuximab soravtansine (MIRV) in patients with platinum-resistant ovarian cancer with high folate receptor alpha (FRα) expression: Characterization of antitumor activity in the SORAYA study. J. Clin. Oncol. 2022, 40 (Suppl. S16), 5512. [Google Scholar] [CrossRef]
- FDA Grants Accelerated Approval to Mirvetuximab Soravtansine-Gynx for FRα Positive, Platinum-Resistant Epithelial Ovarian, Fallopian Tube, or Peritoneal Cancer. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-mirvetuximab-soravtansine-gynx-fra-positive-platinum-resistant (accessed on 9 February 2023).
- Baselga, J.; Lewis Phillips, G.D.; Verma, S.; Ro, J.; Huober, J.; Guardino, A.E.; Samant, M.K.; Olsen, S.; de Haas, S.L.; Pegram, M.D. Relationship between Tumor Biomarkers and Efficacy in EMILIA, a Phase III Study of Trastuzumab Emtansine in HER2-Positive Metastatic Breast Cancer. Clin. Cancer Res. 2016, 22, 3755–3763. [Google Scholar] [CrossRef]
- Venetis, K.; Crimini, E.; Sajjadi, E.; Corti, C.; Guerini-Rocco, E.; Viale, G.; Curigliano, G.; Criscitiello, C.; Fusco, N. HER2 Low, Ultra-low, and Novel Complementary Biomarkers: Expanding the Spectrum of HER2 Positivity in Breast Cancer. Front. Mol. Biosci. 2022, 9, 834651. [Google Scholar] [CrossRef]
- MacNeil, I.A.; Burns, D.J.; Rich, B.E.; Soltani, S.M.; Kharbush, S.; Osterhaus, N.G.; Sullivan, B.F.; Hawkins, D.M.; Pietruska, J.R.; Laing, L.G. New HER2-negative breast cancer subtype responsive to anti-HER2 therapy identified. J. Cancer Res. Clin. Oncol. 2020, 146, 605–619. [Google Scholar] [CrossRef]
- McDonagh, C.F.; Kim, K.M.; Turcott, E.; Brown, L.L.; Westendorf, L.; Feist, T.; Sussman, D.; Stone, I.; Anderson, M.; Miyamoto, J.; et al. Engineered anti-CD70 antibody-drug conjugate with increased therapeutic index. Mol. Cancer Ther. 2008, 7, 2913–2923. [Google Scholar] [CrossRef]
- Polson, A.G.; Ho, W.Y.; Ramakrishnan, V. Investigational antibody-drug conjugates for hematological malignancies. Expert Opin. Investig. Drugs 2011, 20, 75–85. [Google Scholar] [CrossRef] [PubMed]
- FDA Approves Ado-Trastuzumab Emtansine for Early Breast Cancer. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-ado-trastuzumab-emtansine-early-breast-cancer (accessed on 9 February 2023).
- Jørgensen, J.T.; Winther, H.; Askaa, J.; Andresen, L.; Olsen, D.; Mollerup, J. A Companion Diagnostic With Significant Clinical Impact in Treatment of Breast and Gastric Cancer. Front. Oncol. 2021, 11, 676939. [Google Scholar] [CrossRef] [PubMed]
- Morris, R.T.; Joyrich, R.N.; Naumann, R.W.; Shah, N.P.; Maurer, A.H.; Strauss, H.W.; Uszler, J.M.; Symanowski, J.T.; Ellis, P.R.; Harb, W.A. Phase II study of treatment of advanced ovarian cancer with folate-receptor-targeted therapeutic (vintafolide) and companion SPECT-based imaging agent (99mTc-etarfolatide). Ann. Oncol. 2014, 25, 852–858. [Google Scholar] [CrossRef]
- Tijink, B.M.; Buter, J.; de Bree, R.; Giaccone, G.; Lang, M.S.; Staab, A.; Leemans, C.R.; van Dongen, G.A. A phase I dose escalation study with anti-CD44v6 bivatuzumab mertansine in patients with incurable squamous cell carcinoma of the head and neck or esophagus. Clin. Cancer Res. 2006, 12, 6064–6072. [Google Scholar] [CrossRef] [PubMed]
- Larson, R.A.; Sievers, E.L.; Stadtmauer, E.A.; Löwenberg, B.; Estey, E.H.; Dombret, H.; Theobald, M.; Voliotis, D.; Bennett, J.M.; Richie, M.; et al. Final report of the efficacy and safety of gemtuzumab ozogamicin (Mylotarg) in patients with CD33-positive acute myeloid leukemia in first recurrence. Cancer 2005, 104, 1442–1452. [Google Scholar] [CrossRef]
- Sievers, E.L.; Larson, R.A.; Stadtmauer, E.A.; Estey, E.; Löwenberg, B.; Dombret, H.; Karanes, C.; Theobald, M.; Bennett, J.M.; Sherman, M.L.; et al. Efficacy and safety of gemtuzumab ozogamicin in patients with CD33-positive acute myeloid leukemia in first relapse. J. Clin. Oncol. 2001, 19, 3244–3254. [Google Scholar] [CrossRef]
- Raponi, S.; Stefania De Propris, M.; Intoppa, S.; Laura Milani, M.; Vitale, A.; Elia, L.; Perbellini, O.; Pizzolo, G.; Foá, R.; Guarini, A. Flow cytometric study of potential target antigens (CD19, CD20, CD22, CD33) for antibody-based immunotherapy in acute lymphoblastic leukemia: Analysis of 552 cases. Leuk. Lymphoma 2011, 52, 1098–1107. [Google Scholar] [CrossRef]
- Shah, N.N.; Sokol, L. Targeting CD22 for the Treatment of B-Cell Malignancies. Immunotargets Ther. 2021, 10, 225–236. [Google Scholar] [CrossRef]
- Hamadani, M.; Radford, J.; Carlo-Stella, C.; Caimi, P.F.; Reid, E.; O’Connor, O.A.; Feingold, J.M.; Ardeshna, K.M.; Townsend, W.; Solh, M.; et al. Final results of a phase 1 study of loncastuximab tesirine in relapsed/refractory B-cell non-Hodgkin lymphoma. Blood 2021, 137, 2634–2645. [Google Scholar] [CrossRef]
- Kahl, B.S.; Hamadani, M.; Radford, J.; Carlo-Stella, C.; Caimi, P.; Reid, E.; Feingold, J.M.; Ardeshna, K.M.; Solh, M.; Heffner, L.T.; et al. A Phase I Study of ADCT-402 (Loncastuximab Tesirine), a Novel Pyrrolobenzodiazepine-Based Antibody-Drug Conjugate, in Relapsed/Refractory B-Cell Non-Hodgkin Lymphoma. Clin. Cancer Res. 2019, 25, 6986–6994. [Google Scholar] [CrossRef]
- Tilly, H.; Morschhauser, F.; Sehn, L.H.; Friedberg, J.W.; Trneny, M.; Sharman, J.P.; Herbaux, C.; Burke, J.M.; Matasar, M.; Rai, S.; et al. Polatuzumab Vedotin in Previously Untreated Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2022, 386, 351–363. [Google Scholar] [CrossRef]
- Dogan, A.; Siegel, D.; Tran, N.; Fu, A.; Fowler, J.; Belani, R.; Landgren, O. B-cell maturation antigen expression across hematologic cancers: A systematic literature review. Blood Cancer J. 2020, 10, 73. [Google Scholar] [CrossRef]
- Karube, K.; Kakimoto, Y.; Tonozuka, Y.; Ohshima, K. The expression of CD30 and its clinico-pathologic significance in peripheral T-cell lymphomas. Expert Rev. Hematol. 2021, 14, 777–787. [Google Scholar] [CrossRef]
- Connors, J.M.; Jurczak, W.; Straus, D.J.; Ansell, S.M.; Kim, W.S.; Gallamini, A.; Younes, A.; Alekseev, S.; Illes, A.; Picardi, M.; et al. Brentuximab Vedotin with Chemotherapy for Stage III or IV Hodgkin’s Lymphoma. N. Engl. J. Med. 2018, 378, 331–344. [Google Scholar] [CrossRef]
- Gravalos, C.; Jimeno, A. HER2 in gastric cancer: A new prognostic factor and a novel therapeutic target. Ann. Oncol. 2008, 19, 1523–1529. [Google Scholar] [CrossRef]
- Krop, I.E.; Kim, S.B.; Martin, A.G.; LoRusso, P.M.; Ferrero, J.M.; Badovinac-Crnjevic, T.; Hoersch, S.; Smitt, M.; Wildiers, H. Trastuzumab emtansine versus treatment of physician’s choice in patients with previously treated HER2-positive metastatic breast cancer (TH3RESA): Final overall survival results from a randomised open-label phase 3 trial. Lancet Oncol. 2017, 18, 743–754. [Google Scholar] [CrossRef]
- Li, B.T.; Smit, E.F.; Goto, Y.; Nakagawa, K.; Udagawa, H.; Mazieres, J.; Nagasaka, M.; Bazhenova, L.; Saltos, A.N.; Felip, E.; et al. Trastuzumab Deruxtecan in HER2-Mutant Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2022, 386, 241–251. [Google Scholar] [CrossRef]
- Bardia, A.; Messersmith, W.A.; Kio, E.A.; Berlin, J.D.; Vahdat, L.; Masters, G.A.; Moroose, R.; Santin, A.D.; Kalinsky, K.; Picozzi, V.; et al. Sacituzumab govitecan, a Trop-2-directed antibody-drug conjugate, for patients with epithelial cancer: Final safety and efficacy results from the phase I/II IMMU-132-01 basket trial. Ann. Oncol. 2021, 32, 746–756. [Google Scholar] [CrossRef]
- Powles, T.; Rosenberg, J.E.; Sonpavde, G.P.; Loriot, Y.; Durán, I.; Lee, J.L.; Matsubara, N.; Vulsteke, C.; Castellano, D.; Wu, C.; et al. Enfortumab Vedotin in Previously Treated Advanced Urothelial Carcinoma. N. Engl. J. Med. 2021, 384, 1125–1135. [Google Scholar] [CrossRef]
- Yu, E.Y.; Petrylak, D.P.; O’Donnell, P.H.; Lee, J.L.; van der Heijden, M.S.; Loriot, Y.; Stein, M.N.; Necchi, A.; Kojima, T.; Harrison, M.R.; et al. Enfortumab vedotin after PD-1 or PD-L1 inhibitors in cisplatin-ineligible patients with advanced urothelial carcinoma (EV-201): A multicentre, single-arm, phase 2 trial. Lancet Oncol. 2021, 22, 872–882. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, J.; Sridhar, S.S.; Zhang, J.; Smith, D.; Ruether, D.; Flaig, T.W.; Baranda, J.; Lang, J.; Plimack, E.R.; Sangha, R.; et al. EV-101: A Phase I Study of Single-Agent Enfortumab Vedotin in Patients With Nectin-4-Positive Solid Tumors, Including Metastatic Urothelial Carcinoma. J. Clin. Oncol. 2020, 38, 1041–1049. [Google Scholar] [CrossRef] [PubMed]
- Hisada, Y.; Mackman, N. Tissue Factor and Cancer: Regulation, Tumor Growth, and Metastasis. Semin. Thromb. Hemost. 2019, 45, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Koizume, S.; Miyagi, Y. Breast cancer phenotypes regulated by tissue factor-factor VII pathway: Possible therapeutic targets. World J. Clin. Oncol. 2014, 5, 908–920. [Google Scholar] [CrossRef]
- Cocco, E.; Varughese, J.; Buza, N.; Bellone, S.; Glasgow, M.; Bellone, M.; Todeschini, P.; Carrara, L.; Silasi, D.A.; Azodi, M.; et al. Expression of tissue factor in adenocarcinoma and squamous cell carcinoma of the uterine cervix: Implications for immunotherapy with hI-con1, a factor VII-IgGFc chimeric protein targeting tissue factor. BMC Cancer 2011, 11, 263. [Google Scholar] [CrossRef]
- Zhao, X.; Cheng, C.; Gou, J.; Yi, T.; Qian, Y.; Du, X.; Zhao, X. Expression of tissue factor in human cervical carcinoma tissue. Exp. Ther. Med. 2018, 16, 4075–4081. [Google Scholar] [CrossRef]
- Grover, S.P.; Mackman, N. Tissue Factor. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 709–725. [Google Scholar] [CrossRef]
- Osterud, B. Tissue factor expression in blood cells. Thromb. Res. 2010, 125 (Suppl. S1), S31–S34. [Google Scholar] [CrossRef]
- Hong, D.S.; Concin, N.; Vergote, I.; de Bono, J.S.; Slomovitz, B.M.; Drew, Y.; Arkenau, H.T.; Machiels, J.P.; Spicer, J.F.; Jones, R.; et al. Tisotumab Vedotin in Previously Treated Recurrent or Metastatic Cervical Cancer. Clin. Cancer Res. 2020, 26, 1220–1228. [Google Scholar] [CrossRef]
- O’Shannessy, D.J.; Somers, E.B.; Wang, L.C.; Wang, H.; Hsu, R. Expression of folate receptors alpha and beta in normal and cancerous gynecologic tissues: Correlation of expression of the beta isoform with macrophage markers. J. Ovarian Res. 2015, 8, 29. [Google Scholar] [CrossRef]
- Solanky, N.; Requena Jimenez, A.; D’Souza, S.W.; Sibley, C.P.; Glazier, J.D. Expression of folate transporters in human placenta and implications for homocysteine metabolism. Placenta 2010, 31, 134–143. [Google Scholar] [CrossRef]
- Parker, N.; Turk, M.J.; Westrick, E.; Lewis, J.D.; Low, P.S.; Leamon, C.P. Folate receptor expression in carcinomas and normal tissues determined by a quantitative radioligand binding assay. Anal. Biochem. 2005, 338, 284–293. [Google Scholar] [CrossRef]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Lin, K.; Rubinfeld, B.; Zhang, C.; Firestein, R.; Harstad, E.; Roth, L.; Tsai, S.P.; Schutten, M.; Xu, K.; Hristopoulos, M.; et al. Preclinical Development of an Anti-NaPi2b (SLC34A2) Antibody-Drug Conjugate as a Therapeutic for Non-Small Cell Lung and Ovarian Cancers. Clin. Cancer Res. 2015, 21, 5139–5150. [Google Scholar] [CrossRef]
- Syed, Y.Y. Sacituzumab Govitecan: First Approval. Drugs 2020, 80, 1019–1025. [Google Scholar] [CrossRef]
- Bardia, A.; Mayer, I.A.; Diamond, J.R.; Moroose, R.L.; Isakoff, S.J.; Starodub, A.N.; Shah, N.C.; O’Shaughnessy, J.; Kalinsky, K.; Guarino, M.; et al. Efficacy and Safety of Anti-Trop-2 Antibody Drug Conjugate Sacituzumab Govitecan (IMMU-132) in Heavily Pretreated Patients With Metastatic Triple-Negative Breast Cancer. J. Clin. Oncol. 2017, 35, 2141–2148. [Google Scholar] [CrossRef]
- Naing, A.; Thistlethwaite, F.; De Vries, E.G.E.; Eskens, F.; Uboha, N.; Ott, P.A.; LoRusso, P.; Garcia-Corbacho, J.; Boni, V.; Bendell, J.; et al. CX-072 (pacmilimab), a Probody ((R)) PD-L1 inhibitor, in advanced or recurrent solid tumors (PROCLAIM-CX-072): An open-label dose-finding and first-in-human study. J. Immunother. Cancer 2021, 9, e002447. [Google Scholar] [CrossRef]
- Johnson, M.; El-Khoueiry, A.; Hafez, N.; Lakhani, N.; Mamdani, H.; Rodon, J.; Sanborn, R.E.; Garcia-Corbacho, J.; Boni, V.; Stroh, M.; et al. Phase I, First-in-Human Study of the Probody Therapeutic CX-2029 in Adults with Advanced Solid Tumor Malignancies. Clin. Cancer Res. 2021, 27, 4521–4530. [Google Scholar] [CrossRef]
- Fujimori, K.; Covell, D.G.; Fletcher, J.E.; Weinstein, J.N. A modeling analysis of monoclonal antibody percolation through tumors: A binding-site barrier. J. Nucl. Med. 1990, 31, 1191–1198. [Google Scholar]
- Aithal, A.; Rauth, S.; Kshirsagar, P.; Shah, A.; Lakshmanan, I.; Junker, W.M.; Jain, M.; Ponnusamy, M.P.; Batra, S.K. MUC16 as a novel target for cancer therapy. Expert Opin. Ther. Targets 2018, 22, 675–686. [Google Scholar] [CrossRef]
- Gorbatenko, A.; Sokilde, R.; Sorensen, E.E.; Newie, I.; Persson, H.; Morancho, B.; Arribas, J.; Litman, T.; Rovira, C.; Pedersen, S.F. HER2 and p95HER2 differentially regulate miRNA expression in MCF-7 breast cancer cells and downregulate MYB proteins through miR-221/222 and miR-503. Sci. Rep. 2019, 9, 3352. [Google Scholar] [CrossRef] [PubMed]
- Chumsri, S.; Sperinde, J.; Liu, H.; Gligorov, J.; Spano, J.P.; Antoine, M.; Moreno Aspitia, A.; Tan, W.; Winslow, J.; Petropoulos, C.J.; et al. High p95HER2/HER2 Ratio Associated With Poor Outcome in Trastuzumab-Treated HER2-Positive Metastatic Breast Cancer NCCTG N0337 and NCCTG 98-32-52 (Alliance). Clin. Cancer Res. 2018, 24, 3053–3058. [Google Scholar] [CrossRef] [PubMed]
- Damoiseaux, J. The IL-2—IL-2 receptor pathway in health and disease: The role of the soluble IL-2 receptor. Clin. Immunol. 2020, 218, 108515. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Majhi, P.D.; Al-Mugotir, M.H.; Rachagani, S.; Sorgen, P.; Batra, S.K. Membrane proximal ectodomain cleavage of MUC16 occurs in the acidifying Golgi/post-Golgi compartments. Sci. Rep. 2015, 5, 9759. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ADC Name | Developer | Year of Approval | Indication | Antibody Isotype | Target | Target Description | Payload | Linker Type | Approximate DAR | Payload Mechanism |
|---|---|---|---|---|---|---|---|---|---|---|
| ADCs with Tubulin Inhibiting Payloads | ||||||||||
| Brentuximab vedotin (Adcetris®) | Seagen | 2011 (FDA) 2012 (EMA) | HL, Systemic ALCL | IgG1 | CD30 | Marker of activated lymphocytes | MMAE | Cleavable | 4 | Tubulin inhibition |
| Ado-trastuzumab emtansine (Kadcyla®) | Genentech | 2012 (FDA) 2013 (EMA) | HER2-positive breast cancer | IgG1 | HER2 | Growth Factor Receptor | DM-1 | Non-cleavable | 4 | Tubulin inhibition |
| Polatuzumab vedotin-piiq (Polivy®) | Genentech | 2019 (FDA) 2020 (EMA) | DLBCL | IgG1 | CD79b | B-cell Receptor component | MMAE | Cleavable | 3 | Tubulin inhibition |
| Enfortumab vedotin-ejfv (Padcev®) | Astellas/Seagen | 2019 (FDA*) 2022 (EMA) | Urothelial cancer | IgG1 | Nectin-4 | Adhesion Molecule | MMAE | Cleavable | 4 | Tubulin inhibition |
| Belantamab mafodotin-blmf (Blenrep®) | GlaxoSmithKline | 2020 (EMA), 2020 (FDA*, To be withdrawn) | MM | IgG1 | BCMA | Marker of mature B cells | MMAF | Non-cleavable | 4 | Tubulin inhibition |
| Tisotumab vedotin-tftv (Tivdak®) | Genmab/Seagen | 2021 (FDA) | Cervical cancer | IgG1 | Tissue factor | Blood Clotting Co-factor | MMAE | Cleavable | 4 | Tubulin inhibition |
| Mirvetuximab soravtansine-gynx (Elahere®) | ImmunoGen | 2022 (FDA*) | Platinum-resistant, FRα-positive epithelial ovarian, fallopian tube, or primary peritoneal cancer | IgG1 | FRα | Folic Acid Metabolic Receptor | DM-4 | Cleavable | 2 | Tubulin inhibition |
| ADCs with DNA-Interactive Payloads | ||||||||||
| Gemtuzumab ozogamicin (Mylotarg®) | Pfizer | 2000 (Withdrawn) 2017 (FDA) 2018 (EMA) | AML | IgG4 | CD33 | Myeloid-specific marker | Calicheamicin | Cleavable | 2 | DNA Cleaving |
| Inotuzumab ozogamicin (Besponsa®) | Pfizer | 2017 (EMA) 2017 (FDA) | ALL | IgG4 | CD22 | B-cell Receptor component and negative regulator of B-cell receptor signalling | Calicheamicin | Cleavable | 6 | DNA Cleaving |
| Trastuzumab deruxtecan-nxki (Enhertu®) | AstraZeneca/Daiichi Sankyo | 2019, 2021, 2022 (FDA*) 2021 (EMA) | HER2-positive metastatic breast cancer, HER2-mutated NSCLC, HER2-positive gastric or gastroesophogeal cancer | IgG1 | HER2 | Growth Factor Receptor | Dxd | Cleavable | 8 | Topoisomerase inhibition |
| Sacituzumab govitecan-hziy (Trodelvy®) | Immunomedics/Gilead Sciences | 2020 (FDA) 2021 (EMA) | TNBC, HR-Positive, HER2-negative breast cancer, Urothelial Carcinoma | IgG1 | TROP-2 | Transmembrane Glycoprotein | SN-38 | Cleavable | 8 | Topoisomerase inhibition |
| Loncastuximab tesirine-lpyl (Zynlonta®) | ADC Therapeutics | 2021 (FDA), 2022 (EMA) | DLBCL | IgG1 | CD19 | B-cell marker and positive regulator of B-cell receptor signalling | PBD Dimer | Cleavable | 2 | DNA Cross-Linking |
| ADC Name | Developer | Stage | Clinical Trial Number | Indication | Antibody Isotype | Target | Target Description | Payload | Payload Mechanism |
|---|---|---|---|---|---|---|---|---|---|
| ADCs with Tubulin Inhibiting Payloads | |||||||||
| ARX788 | Ambrx, NovoCodex Biopharmaceutical Co. | Phase II/III | CTR20200713 | Metastatic Breast Carcinoma | IgG1 | HER2 | Growth Factor Receptor | Auristatin | Tubulin inhibition |
| Tusamitamab ravtansine | ImmunoGen, Sanofi, Innovent Biologics (Suzhou) | Phase III | NCT04154956 | NSCLC | IgG1 | CEACAM5 | Cell Adhesion Molecule | DM-4 | Tubulin inhibition |
| Trastuzumab emtansine (Ujvira®) * | Zydus Cadila * | Phase III | N/A | HER2-Positive Metastatic Breast Cancer | IgG1 | HER2 | Growth Factor Receptor | DM-1 | Tubulin inhibition |
| Disitamab vedotin (Aidexi®) | RemeGen Co, Seagen | Phase IV | NCT05488353 | Urothelial Carcinoma | IgG1 | HER2 | Growth Factor Receptor | MMAE | Tubulin inhibition |
| Upifitamab rilsodotin | Mersana Therapeutics | Phase III | NCT05329545 | High-Grade Serous Ovarian Cancer | IgG1 | NaPi2b | Sodium/Phosphate Transporter | Auristatin | Tubulin inhibition |
| Telisotuzumab vedotin | Abbvie | Phase III | NCT04928846 | NSCLC | IgG1 | c-Met | Growth Factor Receptor | MMAE | Tubulin inhibition |
| Zilovertamab vedotin | Velos Bio, Merck | Phase II/III | NCT05139017 | Relapsed or refractory DLBCL | IgG1 | ROR-1 | Receptor Tyrosine Kinase-Like Receptor | MMAE | Tubulin inhibition |
| ADCs with DNA-Interactive Payloads | |||||||||
| SKB264 | Klus Pharma, Merck | Phase III | NCT05347134 | Locally Advanced, Recurrent or Metastatic TNBC | Undisclosed | TROP2 | Transmembrane Glycoprotein | Belotecan Derivative | Topoisomerase inhibition |
| Datopotamab deruxtecan | Daiichi Sankyo, AstraZeneca | Phase III | NCT05104866 | HR-positive, HER2-Negative Breast Cancer | IgG1 | TROP-2 | Transmembrane Glycoprotein | Dxd | Topoisomerase inhibition |
| Patritumab deruxtecan | Daiichi Sankyo | Phase III | NCT05338970 | NSCLC | IgG1 | HER2 | Growth Factor Receptor | Dxd | Topoisomerase inhibition |
| Pivekimab sunirine (IMGN632) | ImmunoGen | Phase I/II | NCT03386513 | BPDCN, AML | IgG1 | CD123 | IL-3 Receptor | DGN549 | DNA Guanine Mono-Alkylation |
| Trastuzumab duocarmazine | Byondis | Phase III | NCT03262935 | Metastatic Breast Cancer | IgG1 | HER2 | Growth Factor Receptor | Duocarmycin | DNA Adenine Mono-Alkylation |
| Trastuzumab rezetecan | Jiangsu HengRui Medicine, Luzsana Biotechnology | Phase III | NCT05424835 | HER2-Positive Metastatic Breast Cancer | IgG1 | HER2 | Growth Factor Receptor | Camptothecin | Topoisomerase inhibition |
| Vobramitamab duocarmazine | Byondis, MacroGenics | Phase II/III | NCT05551117 | Prostate Cancer | IgG1 | B7-H3 | Immunoregulatory Glycoprotein | Duocarmycin | DNA Adenine Mono-Alkylation |
| Feature | Ado-Trastuzumab Emtansine (Kadcyla®) | Trastuzumab Deruxtecan (Enhertu®) |
|---|---|---|
| Antibody | Trastuzumab | Trastuzumab |
| Target Antigen | HER2 | HER2 |
| Linker Type | Non-cleavable | Cleavable |
| Payload | DM-1 | Dxd |
| Payload Mechanism | Tubulin inhibition | Topoisomerase inhibition |
| Drug Antibody Ratio (DAR) | 3.5 | 7.7 |
| Approved Indications | HER2-positive breast cancer | HER2-positive metastatic breast cancer, HER2-mutated NSCLC, HER2-positive gastric or gastroesophageal cancer |
| Antigen | Cancer Types with High Expression | Normal Tissue Distribution | Commonly Observed Toxicities for ADC Treatment |
|---|---|---|---|
| CD33 | AML [60] | Salivary Gland Kidney Epididymis Spleen Lymph Node Tonsil Testis Duodenum Bone Marrow | Gemtuzumab ozogamicin (Mylotarg®) Hepatic Veno-occlusive Disease [62] Elevated Aspartate/ Alanine Aminotransferase [182] Hyperbilirubinemia [182] Pneumonia [182] Dyspnoea [182] Neutropenia [182,183] Thrombocytopenia [183] Fever [182,183] Chills [182,183] Nausea [182] Hypertension [182] Hypotension [182] Sepsis [182] |
| CD22 | B-ALL [184] B-NHL [185] | Appendix Lymph Node Testis Spleen Tonsil | Inotuzumab ozogamicin (Besponsa®) Hepatic Veno-occlusive disease [75] Thrombocytopenia [75] Febrile Neutropenia [75] Pneumonia [75] |
| CD19 | B-NHL [185] | Bone Marrow Appendix Spleen Lymph Node Tonsil | Loncastuximab tesirine (Zynlonta®) Neutropenia [82,186,187] Anaemia [82,186,187] Thrombocytopenia [82,186,187] Lymphopenia [82,186] Leukopenia [82] Elevated gamma-glutamyl transferase [82,186,187] Elevated Blood Alkaline Phosphatase [82,186,187] Hypokalaemia [82,186] Hypophosphatemia [82] Fatigue [187] Dyspnoea [82] |
| CD79b | B-NHL [83] | Appendix Spleen Lymph Node Tonsil | Polatuzumab vedotin (Polivy®) Anaemia [89,188] Neutropenia [89,188] Lymphopenia [89] Thrombocytopenia [89] |
| BCMA * | MM [189] CLL [189] B-ALL [189] B-NHL [189] HL [189] | Plasma Cells [94] Mature B Cells [94] | Belantamab mafodotin (Blenrep®) Keratopathy [34] Thrombocytopenia [34] Anaemia [34] Nausea [34] Pyrexia [34] Blurred Vision [34] Increased Aspartate Aminotransferase [34] Fatigue [34] Dry Eyes [34] Neutropenia [34] Hyperkalaemia [34] Lymphopenia [34] Increased Gamma Gliutamyltransferase [34] Hypophosphataemia [34] Pneumonia [34] |
| CD30 | HL [100] CTCL [190] PTCL-NOS [190] | Testis Fallopian Tube Appendix Lymph Node Tonsil | Brentuximab vedotin (Adcetris®) Neutropenia [191] Peripheral Neuropathy [191] |
| HER2 | Biliary Tract Cancer [107] Colorectal Cancer [107] NSCLC [107] Breast Cancer [108] Gastric Cancer [192] | Nasopharynx Lung Urinary bladder Testis Fallopian Tube Endometrium Cervix Placenta Breast Heart muscle Skeletal muscle Skin Appendix Thyroid gland Parathyroid Gland Bronchus Salivary gland Oesophagus Small intestine Rectum Liver Kidney Seminal vesicle Prostate Vagina Tonsil Bone marrow Cerebral cortex | Trastuzumab emtansine (Kadcyla®) Thrombocytopenia [125,126,127,193] Elevated Aspartate Aminotransferase [127] Trastuzumab deruxtecan (Enhertu®) Nausea [128,130,194] Fatigue [128,130,194] Neutropenia [128,130,194] Anorexia [128] Anaemia [128,130,194] Lymphopenia [128] Leukopenia [128] Thrombocytopenia [130] |
| TROP-2 | Breast, Pancreatic, Urothelial and Ovarian Squamous Cell Carcinomas [138] | Nasopharynx Bronchus Oral mucosa Oesophagus Kidney Urinary bladder Seminal vesicle Cervix Skin Salivary gland Rectum Gallbladder Pancreas Epididymis Vagina Fallopian tube Endometrium Breast Tonsil | Sacituzumab govitecan (Trodelvy®) Nausea [145,195] Neutropenia [147,152,195] Leukopenia [147,152] Lymphopenia [152] Anaemia [152,195] Fatigue [145,195] Diarrhoea [145,152,195] Hypophosphatemia [145] |
| Nectin-4 | Bladder Cancer [155] Breast Cancer [155] Pancreatic Cancer [155] Ovarian Cancer [155] Head and Neck Cancer [155] Oesophageal Cancer [155] | Oral mucosa Oesophagus Urinary bladder Placenta Breast Skin Tonsil Pancreas Kidney Testis Epididymis Seminal vesicle Prostate Vagina Endometrium Cervix | Enfortumab vedotin (Padcev®) Fatigue [196] Maculopapular Rash [196,197] Neutropenia [196,197] Anaemia [197] Diarrhoea [197] Hyperglycaemia [197,198] Increased Lipase [197] Anorexia [197] |
| TF * | Glioblastoma [166,199] Breast Cancer [161,199,200] Colorectal Cancer [166,199] Pancreatic Cancer [166,199] Lung Cancer [161] Cervical Cancer [201,202] | Monocytes [203] Platelets [203] Endothelial Cells [204] | Tisotumab vedotin (Tivdak®) Fatigue [205] Nausea [205] Vomiting [205] Abdominal Pain [205] Anaemia [205] Hypokalaemia [205] |
| FRα * | Ovarian Cancer [166,168] Lung Cancer [166] Breast Cancer [166] | Fallopian Tube Epithelium [206] Placental Epithelium [207] Kidney [208] Lung [208] | Mirvetuximab soravtansine (Elahere®) Diarrhoea [170,171] Blurred Vision [170,171] Keratopathy [170,171] |
| Antigen-Related Feature | Factors Increasing ADC Effectiveness | Factors Reducing ADC Effectiveness | Examples of Relevant Antigens |
|---|---|---|---|
| Physiological Functions of the Target Antigen | If the target antigen promotes pro-tumour signalling pathways, targeting with an ADC may also exert antitumour effects by inhibiting oncogenic signalling pathways. | If signalling mediated by the target antigen is required for essential normal functions, inhibition of this signalling and killing of non-malignant cells expressing the antigen may lead to toxicity. | Antigens Promoting Pro-Tumour Signalling Pathways BCMA [94] CD30 [100,102] HER2 [108] TROP-2 [139,140] FRα [166] Antigens that when targeted by an ADC lead to target-mediated toxicity by inhibiting essential normal functions HER2 [120,121,122] |
| Antigen Shedding | Reduction in ‘Binding-Site Barrier’ for greater penetration of ADC. | ‘Sink effect’ potentiated by ADC binding to shed antigen. | Highly Shed: HER2 [113,118,217] Nectin-4 [156] CD25 [219] MUC16 [220] TROP-2 [140] FRα [168] Limited Shedding: CD19 [41] CD22 [73] |
| Recycling of Antigen to the Plasma Membrane | A high degree of ADC recycling could counteract the sink effect caused by high rates of antigen shedding. | Direction of internalised ADC to the plasma membrane without potentiation of cytotoxicity. | Antigens which may be recycled to the plasma membrane: CD22 [73] HER2 [116] TROP-2 [142] |
| High Expression on Malignant Cells, Low or Absent Expression on Normal Cells | Therapeutic window for targeting malignant cells while sparing healthy cells. | - | Antigens for which this paradigm does not appear to directly apply: TROP-2 [137,139] SLC34A2 [210] HER2 [174] CD70 [176] |
| Rapid de novo synthesis of antigen and restoration of cell surface expression | More rapid repeat targeting of the same cell by an ADC | - | Antigens rapidly synthesised de novo: CD33 [58] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esapa, B.; Jiang, J.; Cheung, A.; Chenoweth, A.; Thurston, D.E.; Karagiannis, S.N. Target Antigen Attributes and Their Contributions to Clinically Approved Antibody-Drug Conjugates (ADCs) in Haematopoietic and Solid Cancers. Cancers 2023, 15, 1845. https://doi.org/10.3390/cancers15061845
Esapa B, Jiang J, Cheung A, Chenoweth A, Thurston DE, Karagiannis SN. Target Antigen Attributes and Their Contributions to Clinically Approved Antibody-Drug Conjugates (ADCs) in Haematopoietic and Solid Cancers. Cancers. 2023; 15(6):1845. https://doi.org/10.3390/cancers15061845
Chicago/Turabian StyleEsapa, Benjamina, Jiexuan Jiang, Anthony Cheung, Alicia Chenoweth, David E. Thurston, and Sophia N. Karagiannis. 2023. "Target Antigen Attributes and Their Contributions to Clinically Approved Antibody-Drug Conjugates (ADCs) in Haematopoietic and Solid Cancers" Cancers 15, no. 6: 1845. https://doi.org/10.3390/cancers15061845
APA StyleEsapa, B., Jiang, J., Cheung, A., Chenoweth, A., Thurston, D. E., & Karagiannis, S. N. (2023). Target Antigen Attributes and Their Contributions to Clinically Approved Antibody-Drug Conjugates (ADCs) in Haematopoietic and Solid Cancers. Cancers, 15(6), 1845. https://doi.org/10.3390/cancers15061845