
The Melanoma-Associated Antigen Family A (MAGE-A): A Promising Target for Cancer Immunotherapy?


Simple Summary
Abstract
1. Introduction
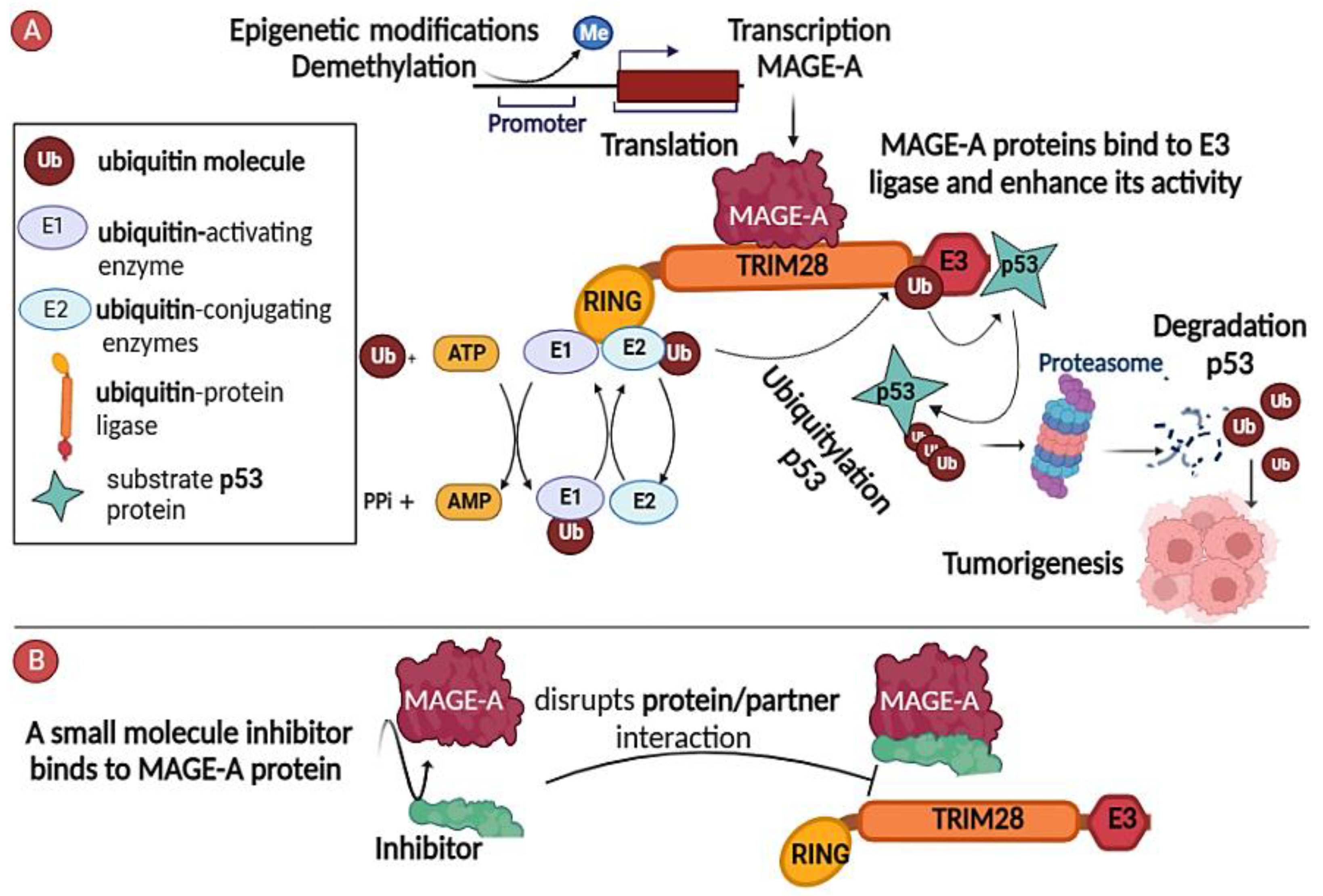
MAGE-A Antigens and Cancer
2. Cancer-Fighting MAGE-A-Targeting Strategies
2.1. Inhibitors of MAGE-A/Partner Interaction
2.2. Targeting the Regulatory Pathways Responsible for Functional Expression
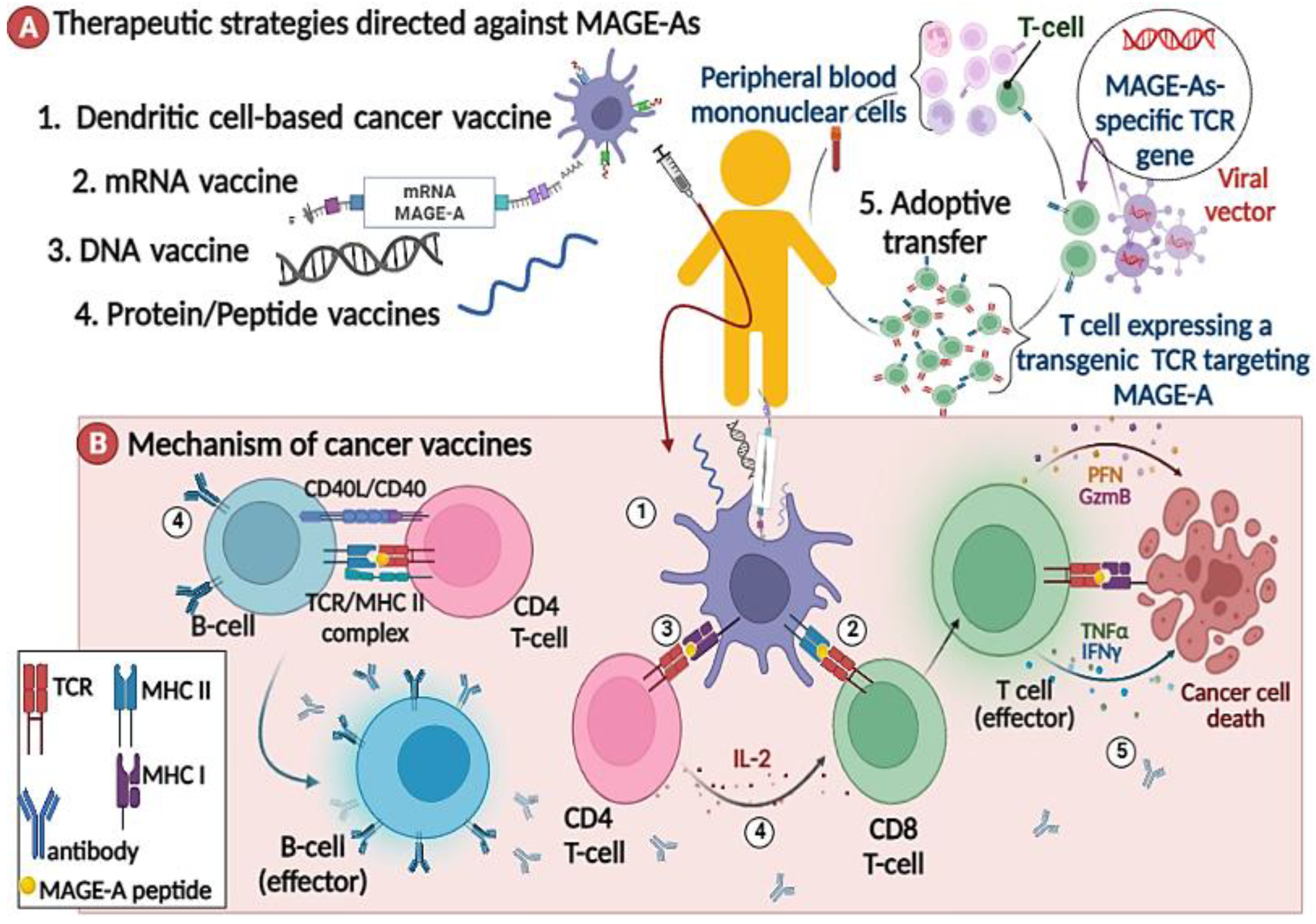
2.3. Immunotherapy against MAGE-As
2.3.1. MAGE-As Antigen Vaccination


2.3.2. MAGE-A-Directed Adoptive T Cell Therapy
3. Future Perspective and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wagner, S.; Mullins, C.S.; Linnebacher, M. Colorectal Cancer Vaccines: Tumor-Associated Antigens vs Neoantigens. World J. Gastroenterol. 2018, 24, 5418–5432. [Google Scholar] [CrossRef]
- Gjerstorff, M.F.; Andersen, M.H.; Ditzel, H.J. Oncogenic Cancer/Testis Antigens: Prime Candidates for Immunotherapy. Oncotarget 2015, 6, 15772–15787. [Google Scholar] [CrossRef]
- Suri, A.; Jagadish, N.; Saini, S.; Gupta, N. Targeting Cancer Testis Antigens for Biomarkers and Immunotherapy in Colorectal Cancer: Current Status and Challenges. World J. Gastrointest. Oncol. 2015, 7, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Zendman, A.J.W.; Ruiter, D.J.; Van Muijen, G.N.P. Cancer/Testis-Associated Genes: Identification, Expression Profile, and Putative Function. J. Cell. Physiol. 2003, 194, 272–288. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, O.; Caballero, O.L.; Stevenson, B.J.; Chen, Y.-T.; Cohen, T.; Chua, R.; Maher, C.A.; Panji, S.; Schaefer, U.; Kruger, A.; et al. Genome-Wide Analysis of Cancer/Testis Gene Expression. Proc. Natl. Acad. Sci. USA 2008, 105, 20422–20427. [Google Scholar] [CrossRef]
- Peng, J.; Chen, H.; Mou, D.; Cao, J.; Cong, X.; Qin, L.; Wei, L.; Leng, X.; Wang, Y.; Chen, W. Expression of Cancer/Testis (CT) Antigens in Chinese Hepatocellular Carcinoma and Its Correlation with Clinical Parameters. Cancer Lett. 2005, 219, 223–232. [Google Scholar] [CrossRef]
- Chen, Y.-T.; Chadburn, A.; Lee, P.; Hsu, M.; Ritter, E.; Chiu, A.; Gnjatic, S.; Pfreundschuh, M.; Knowles, D.M.; Old, L.J. Expression of Cancer Testis Antigen CT45 in Classical Hodgkin Lymphoma and Other B-Cell Lymphomas. Proc. Natl. Acad. Sci. USA 2010, 107, 3093–3098. [Google Scholar] [CrossRef] [PubMed]
- Kalejs, M.; Erenpreisa, J. Cancer/Testis Antigens and Gametogenesis: A Review and “Brain-Storming” Session. Cancer Cell Int. 2005, 5, 4. [Google Scholar] [CrossRef]
- van der Bruggen, P.; Traversari, C.; Chomez, P.; Lurquin, C.; De Plaen, E.; Van den Eynde, B.; Knuth, A.; Boon, T. A Gene Encoding an Antigen Recognized by Cytolytic T Lymphocytes on a Human Melanoma. Science 1991, 254, 1643–1647. [Google Scholar] [CrossRef] [PubMed]
- Chomez, P.; De Backer, O.; Bertrand, M.; De Plaen, E.; Boon, T.; Lucas, S. An Overview of the MAGE Gene Family with the Identification of All Human Members of the Family. Cancer Res. 2001, 61, 5544–5551. [Google Scholar]
- Weon, J.L.; Potts, P.R. The MAGE Protein Family and Cancer. Curr. Opin. Cell Biol. 2015, 37, 1–8. [Google Scholar] [CrossRef]
- Barker, P.A.; Salehi, A. The MAGE Proteins: Emerging Roles in Cell Cycle Progression, Apoptosis, and Neurogenetic Disease. J. Neurosci. Res. 2002, 67, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Doyle, J.M.; Gao, J.; Wang, J.; Yang, M.; Potts, P.R. MAGE-RING Protein Complexes Comprise a Family of E3 Ubiquitin Ligases. Mol. Cell 2010, 39, 963–974. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.W.; Huang, X.; Lin, W.; Min, J.; Miller, D.J.; Mayasundari, A.; Rodrigues, P.; Griffith, E.C.; Gee, C.T.; Li, L.; et al. Structural Basis for Substrate Recognition and Chemical Inhibition of Oncogenic MAGE Ubiquitin Ligases. Nat. Commun. 2020, 11, 4931. [Google Scholar] [CrossRef] [PubMed]
- Pineda, C.T.; Ramanathan, S.; Fon Tacer, K.; Weon, J.L.; Potts, M.B.; Ou, Y.-H.; White, M.A.; Potts, P.R. Degradation of AMPK by a Cancer-Specific Ubiquitin Ligase. Cell 2015, 160, 715–728. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, Y.; Sieverling, L.; Hanif, F.; Anton, J.; Dickinson, E.R.; Bui, T.T.T.; Andreeva, A.; Barran, P.E.; Cota, E.; Nikolova, P.V. Consequences of Point Mutations in Melanoma-Associated Antigen 4 (MAGE-A4) Protein: Insights from Structural and Biophysical Studies. Sci. Rep. 2016, 6, 25182. [Google Scholar] [CrossRef] [PubMed]
- Brasseur, F.; Rimoldi, D.; Liénard, D.; Lethé, B.; Carrel, S.; Arienti, F.; Suter, L.; Vanwijck, R.; Bourlond, A.; Humblet, Y.; et al. Expression of MAGE Genes in Primary and Metastatic Cutaneous Melanoma. Int. J. Cancer 1995, 63, 375–380. [Google Scholar] [CrossRef]
- Mori, M.; Inoue, H.; Mimori, K.; Shibuta, K.; Baba, K.; Nakashima, H.; Haraguchi, M.; Tsuji, K.; Ueo, H.; Barnard, G.F.; et al. Expression of MAGE Genes in Human Colorectal Carcinoma. Ann. Surg. 1996, 224, 183–188. [Google Scholar] [CrossRef]
- Otte, M.; Zafrakas, M.; Riethdorf, L.; Pichlmeier, U.; Löning, T.; Jänicke, F.; Pantel, K. MAGE-A Gene Expression Pattern in Primary Breast Cancer. Cancer Res. 2001, 61, 6682–6687. [Google Scholar]
- Daudi, S.; Eng, K.H.; Mhawech-Fauceglia, P.; Morrison, C.; Miliotto, A.; Beck, A.; Matsuzaki, J.; Tsuji, T.; Groman, A.; Gnjatic, S.; et al. Expression and Immune Responses to MAGE Antigens Predict Survival in Epithelial Ovarian Cancer. PLoS ONE 2014, 9, e104099. [Google Scholar] [CrossRef]
- Kim, Y.-D.; Park, H.-R.; Song, M.-H.; Shin, D.-H.; Lee, C.-H.; Lee, M.-K.; Lee, S.-Y. Pattern of Cancer/Testis Antigen Expression in Lung Cancer Patients. Int. J. Mol. Med. 2012, 29, 656–662. [Google Scholar] [CrossRef] [PubMed]
- Gure, A.O.; Chua, R.; Williamson, B.; Gonen, M.; Ferrera, C.A.; Gnjatic, S.; Ritter, G.; Simpson, A.J.G.; Chen, Y.-T.; Old, L.J.; et al. Cancer-Testis Genes Are Coordinately Expressed and Are Markers of Poor Outcome in Non–Small Cell Lung Cancer. Clin. Cancer Res. 2005, 11, 8055–8062. [Google Scholar] [CrossRef]
- Zhang, S.; Zhai, X.; Wang, G.; Feng, J.; Zhu, H.; Xu, L.; Mao, G.; Huang, J. High Expression of MAGE-A9 in Tumor and Stromal Cells of Non-Small Cell Lung Cancer Was Correlated with Patient Poor Survival. Int. J. Clin. Exp. Pathol. 2015, 8, 541–550. [Google Scholar] [PubMed]
- Gu, X.; Fu, M.; Ge, Z.; Zhan, F.; Ding, Y.; Ni, H.; Zhang, W.; Zhu, Y.; Tang, X.; Xiong, L.; et al. High Expression of MAGE-A9 Correlates with Unfavorable Survival in Hepatocellular Carcinoma. Sci. Rep. 2014, 4, 6625. [Google Scholar] [CrossRef]
- Monte, M.; Simonatto, M.; Peche, L.Y.; Bublik, D.R.; Gobessi, S.; Pierotti, M.A.; Rodolfo, M.; Schneider, C. MAGE-A Tumor Antigens Target P53 Transactivation Function through Histone Deacetylase Recruitment and Confer Resistance to Chemotherapeutic Agents. Proc. Natl. Acad. Sci. USA 2006, 103, 11160–11165. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; O’Herrin, S.M.; Wu, J.; Reagan-Shaw, S.; Ma, Y.; Bhat, K.M.R.; Gravekamp, C.; Setaluri, V.; Peters, N.; Hoffmann, F.M.; et al. MAGE-A, MMage-b, and MAGE-C Proteins Form Complexes with KAP1 and Suppress P53-Dependent Apoptosis in MAGE-Positive Cell Lines. Cancer Res. 2007, 67, 9954–9962. [Google Scholar] [CrossRef]
- Schwarzenbach, H.; Eichelser, C.; Steinbach, B.; Tadewaldt, J.; Pantel, K.; Lobanenkov, V.; Loukinov, D. Differential Regulation of MAGE-A1 Promoter Activity by BORIS and Sp 1, Both Interacting with the TATA Binding Protein. BMC Cancer 2014, 14, 796. [Google Scholar] [CrossRef]
- Wischnewski, F.; Pantel, K.; Schwarzenbach, H. Promoter Demethylation and Histone Acetylation Mediate Gene Expression of MAGE-A1, -A2, -A3, and -A12 in Human Cancer Cells. Mol. Cancer Res. 2006, 4, 339–349. [Google Scholar] [CrossRef]
- De Smet, C.; Lurquin, C.; Lethé, B.; Martelange, V.; Boon, T. DNA Methylation Is the Primary Silencing Mechanism for a Set of Germ Line- and Tumor-Specific Genes with a CpG-Rich Promoter. Mol. Cell. Biol. 1999, 19, 7327–7335. [Google Scholar] [CrossRef]
- De Smet, C.; Courtois, S.J.; Faraoni, I.; Lurquin, C.; Szikora, J.P.; De Backer, O.; Boon, T. Involvement of Two Ets Binding Sites in the Transcriptional Activation of the MAGE1 Gene. Immunogenetics 1995, 42, 282–290. [Google Scholar] [CrossRef]
- Bhatia, N.; Yang, B.; Xiao, T.Z.; Peters, N.; Hoffmann, M.F.; Longley, B.J. Identification of Novel Small Molecules That Inhibit Protein-Protein Interactions between MAGE and KAP-1. Arch. Biochem. Biophys. 2011, 508, 217–221. [Google Scholar] [CrossRef]
- De Smet, C.; De Backer, O.; Faraoni, I.; Lurquin, C.; Brasseur, F.; Boon, T. The Activation of Human Gene MAGE-1 in Tumor Cells Is Correlated with Genome-Wide Demethylation. Proc. Natl. Acad. Sci. USA 1996, 93, 7149–7153. [Google Scholar] [CrossRef]
- Weber, J.; Salgaller, M.; Samid, D.; Johnson, B.; Herlyn, M.; Lassam, N.; Treisman, J.; Rosenberg, S.A. Expression of the MAGE-1 Tumor Antigen Is up-Regulated by the Demethylating Agent 5-Aza-2′-Deoxycytidine. Cancer Res. 1994, 54, 1766–1771. [Google Scholar]
- Chaux, P.; Vantomme, V.; Coulie, P.; Boon, T.; van der Bruggen, P. Estimation of the Frequencies of Anti-MAGE-3 Cytolytic T-Lymphocyte Precursors in Blood from Individuals without Cancer. Int. J. Cancer 1998, 77, 538–542. [Google Scholar] [CrossRef]
- Tahara, K.; Mori, M.; Sadanaga, N.; Sakamoto, Y.; Kitano, S.; Makuuchi, M. Expression of the MAGE Gene Family in Human Hepatocellular Carcinoma. Cancer 1999, 85, 1234–1240. [Google Scholar] [CrossRef]
- Schooten, E.; Di Maggio, A.; van Bergen En Henegouwen, P.M.P.; Kijanka, M.M. MAGE-A Antigens as Targets for Cancer Immunotherapy. Cancer Treat. Rev. 2018, 67, 54–62. [Google Scholar] [CrossRef]
- Kruit, W.H.; Suciu, S.; Dreno, B.; Mortier, L.; Robert, C.; Chiarion-Sileni, V.; Maio, M.; Testori, A.; Dorval, T.; Grob, J.-J. Selection of Immunostimulant AS15 for Active Immunization with MAGE-A3 Protein: Results of a Randomized Phase II Study of the European Organisation for Research and Treatment of Cancer Melanoma Group in Metastatic Melanoma. J. Clin. Oncol. 2013, 31, 2413–2420. [Google Scholar] [CrossRef] [PubMed]
- Dreno, B.; Thompson, J.F.; Smithers, B.M.; Santinami, M.; Jouary, T.; Gutzmer, R.; Levchenko, E.; Rutkowski, P.; Grob, J.-J.; Korovin, S.; et al. MAGE-A3 Immunotherapeutic as Adjuvant Therapy for Patients with Resected, MAGE-A3-Positive, Stage III Melanoma (DERMA): A Double-Blind, Randomised, Placebo-Controlled, Phase 3 Trial. Lancet. Oncol. 2018, 19, 916–929. [Google Scholar] [CrossRef]
- Grob, J.-J.; Mortier, L.; D’Hondt, L.; Grange, F.; Baurain, J.; Dréno, B.; Lebbe, C.; Robert, C.; Dompmartin, A.; Neyns, B.; et al. Safety and Immunogenicity of MAGE-A3 Cancer Immunotherapeutic with Dacarbazine in Patients with MAGE-A3-Positive Metastatic Cutaneous Melanoma: An Open Phase I/II Study with a First Assessment of a Predictive Gene Signature. ESMO Open 2017, 2, e000203. [Google Scholar] [CrossRef] [PubMed]
- Vansteenkiste, J.F.; Cho, B.C.; Vanakesa, T.; De Pas, T.; Zielinski, M.; Kim, M.S.; Jassem, J.; Yoshimura, M.; Dahabreh, J.; Nakayama, H.; et al. Efficacy of the MAGE-A3 Cancer Immunotherapeutic as Adjuvant Therapy in Patients with Resected MAGE-A3-Positive Non-Small-Cell Lung Cancer (MAGRIT): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2016, 17, 822–835. [Google Scholar] [CrossRef]
- Duperret, E.K.; Liu, S.; Paik, M.; Trautz, A.; Stoltz, R.; Liu, X.; Ze, K.; Perales-Puchalt, A.; Reed, C.; Yan, J.; et al. A Designer Cross-Reactive DNA Immunotherapeutic Vaccine That Targets Multiple MAGE-A Family Members Simultaneously for Cancer Therapy. Clin. Cancer Res. 2018, 24, 6015–6027. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Wei, J.; Chen, Y.; Huang, A.; Li, K.K.-W.; Zhang, W. Induction of Antigen-Specific Immune Responses by Dendritic Cells Transduced with a Recombinant Lentiviral Vector Encoding MAGE-A3 Gene. J. Cancer Res. Clin. Oncol. 2014, 140, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Batchu, R.B.; Gruzdyn, O.V.; Moreno-Bost, A.M.; Szmania, S.; Jayandharan, G.; Srivastava, A.; Kolli, B.K.; Weaver, D.W.; van Rhee, F.; Gruber, S.A. Efficient Lysis of Epithelial Ovarian Cancer Cells by MAGE-A3-Induced Cytotoxic T Lymphocytes Using RAAV-6 Capsid Mutant Vector. Vaccine 2014, 32, 938–943. [Google Scholar] [CrossRef]
- Wilgenhof, S.; Van Nuffel, A.M.T.; Corthals, J.; Heirman, C.; Tuyaerts, S.; Benteyn, D.; De Coninck, A.; Van Riet, I.; Verfaillie, G.; Vandeloo, J.; et al. Therapeutic Vaccination With an Autologous MRNA Electroporated Dendritic Cell Vaccine in Patients With Advanced Melanoma. J. Immunother. 2011, 34, 448–456. [Google Scholar] [CrossRef]
- Bonehill, A.; Van Nuffel, A.M.T.; Corthals, J.; Tuyaerts, S.; Heirman, C.; François, V.; Colau, D.; van der Bruggen, P.; Neyns, B.; Thielemans, K. Single-Step Antigen Loading and Activation of Dendritic Cells by MRNA Electroporation for the Purpose of Therapeutic Vaccination in Melanoma Patients. Clin. Cancer Res. 2009, 15, 3366–3375. [Google Scholar] [CrossRef] [PubMed]
- Kranz, L.M.; Diken, M.; Haas, H.; Kreiter, S.; Loquai, C.; Reuter, K.C.; Meng, M.; Fritz, D.; Vascotto, F.; Hefesha, H.; et al. Systemic RNA Delivery to Dendritic Cells Exploits Antiviral Defence for Cancer Immunotherapy. Nature 2016, 534, 396–401. [Google Scholar] [CrossRef]
- Takahashi, N.; Ohkuri, T.; Homma, S.; Ohtake, J.; Wakita, D.; Togashi, Y.; Kitamura, H.; Todo, S.; Nishimura, T. First Clinical Trial of Cancer Vaccine Therapy with Artificially Synthesized Helper/Killer-Hybrid Epitope Long Peptide of MAGE-A4 Cancer Antigen. Cancer Sci. 2012, 103, 150–153. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- De Keersmaecker, B.; Claerhout, S.; Carrasco, J.; Bar, I.; Corthals, J.; Wilgenhof, S.; Neyns, B.; Thielemans, K. TriMix and Tumor Antigen MRNA Electroporated Dendritic Cell Vaccination plus Ipilimumab: Link between T-Cell Activation and Clinical Responses in Advanced Melanoma. J. Immunother. Cancer 2020, 8, e000329. [Google Scholar] [CrossRef]
- Liu, J.; Fu, M.; Wang, M.; Wan, D.; Wei, Y.; Wei, X. Cancer Vaccines as Promising Immuno-Therapeutics: Platforms and Current Progress. J. Hematol. Oncol. 2022, 15, 28. [Google Scholar] [CrossRef]
- Krishnadas, D.K.; Shusterman, S.; Bai, F.; Diller, L.; Sullivan, J.E.; Cheerva, A.C.; George, R.E.; Lucas, K.G. A Phase I Trial Combining Decitabine/Dendritic Cell Vaccine Targeting MAGE-A1, MAGE-A3 and NY-ESO-1 for Children with Relapsed or Therapy-Refractory Neuroblastoma and Sarcoma. Cancer Immunol. Immunother. 2015, 64, 1251–1260. [Google Scholar] [CrossRef] [PubMed]
- Dudley, M.E.; Yang, J.C.; Sherry, R.; Hughes, M.S.; Royal, R.; Kammula, U.; Robbins, P.F.; Huang, J.; Citrin, D.E.; Leitman, S.F.; et al. Adoptive Cell Therapy for Patients with Metastatic Melanoma: Evaluation of Intensive Myeloablative Chemoradiation Preparative Regimens. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 5233–5239. [Google Scholar] [CrossRef] [PubMed]
- Cruz, C.R.; Gerdemann, U.; Leen, A.M.; Shafer, J.A.; Ku, S.; Tzou, B.; Horton, T.M.; Sheehan, A.; Copeland, A.; Younes, A.; et al. Improving T-Cell Therapy for Relapsed EBV-Negative Hodgkin Lymphoma by Targeting Upregulated MAGE-A4. Clin. Cancer Res. 2011, 17, 7058–7066. [Google Scholar] [CrossRef] [PubMed]
- Westwood, J.A.; Kershaw, M.H. Genetic Redirection of T Cells for Cancer Therapy. J. Leukoc. Biol. 2010, 87, 791–803. [Google Scholar] [CrossRef] [PubMed]
- Shirakura, Y.; Mizuno, Y.; Wang, L.; Imai, N.; Amaike, C.; Sato, E.; Ito, M.; Nukaya, I.; Mineno, J.; Takesako, K.; et al. T-Cell Receptor Gene Therapy Targeting Melanoma-Associated Antigen-A4 Inhibits Human Tumor Growth in Non-Obese Diabetic/SCID/Γcnull Mice. Cancer Sci. 2012, 103, 17–25. [Google Scholar] [CrossRef]
- Kageyama, S.; Ikeda, H.; Miyahara, Y.; Imai, N.; Ishihara, M.; Saito, K.; Sugino, S.; Ueda, S.; Ishikawa, T.; Kokura, S.; et al. Adoptive Transfer of MAGE-A4 T-Cell Receptor Gene-Transduced Lymphocytes in Patients with Recurrent Esophageal Cancer. Clin. cancer Res. 2015, 21, 2268–2277. [Google Scholar] [CrossRef]
- Shah, N.N.; Maatman, T.; Hari, P.; Johnson, B. Multi Targeted CAR-T Cell Therapies for B-Cell Malignancies. Front. Oncol. 2019, 9, 146. [Google Scholar] [CrossRef]
- Morgan, R.A.; Chinnasamy, N.; Abate-Daga, D.; Gros, A.; Robbins, P.F.; Zheng, Z.; Dudley, M.E.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; et al. Cancer Regression and Neurological Toxicity Following Anti-MAGE-A3 TCR Gene Therapy. J. Immunother. 2013, 36, 133–151. [Google Scholar] [CrossRef]
- Cameron, B.J.; Gerry, A.B.; Dukes, J.; Harper, J.V.; Kannan, V.; Bianchi, F.C.; Grand, F.; Brewer, J.E.; Gupta, M.; Plesa, G.; et al. Identification of a Titin-Derived HLA-A1-Presented Peptide as a Cross-Reactive Target for Engineered MAGE A3-Directed T Cells. Sci. Transl. Med. 2013, 5, 197ra103. [Google Scholar] [CrossRef]
- Linette, G.P.; Stadtmauer, E.A.; Maus, M.V.; Rapoport, A.P.; Levine, B.L.; Emery, L.; Litzky, L.; Bagg, A.; Carreno, B.M.; Cimino, P.J.; et al. Cardiovascular Toxicity and Titin Cross-Reactivity of Affinity-Enhanced T Cells in Myeloma and Melanoma. Blood 2013, 122, 863–871. [Google Scholar] [CrossRef]
- Yao, X.; Lu, Y.-C.; Parker, L.L.; Li, Y.F.; El-Gamil, M.; Black, M.A.; Xu, H.; Feldman, S.A.; van der Bruggen, P.; Rosenberg, S.A.; et al. Isolation and Characterization of an HLA-DPB1*04: 01-Restricted MAGE-A3 T-Cell Receptor for Cancer Immunotherapy. J. Immunother. 2016, 39, 191–201. [Google Scholar] [CrossRef]
- Lu, Y.-C.; Parker, L.L.; Lu, T.; Zheng, Z.; Toomey, M.A.; White, D.E.; Yao, X.; Li, Y.F.; Robbins, P.F.; Feldman, S.A.; et al. Treatment of Patients With Metastatic Cancer Using a Major Histocompatibility Complex Class II-Restricted T-Cell Receptor Targeting the Cancer Germline Antigen MAGE-A3. J. Clin. Oncol. 2017, 35, 3322–3329. [Google Scholar] [CrossRef]
- Blumenschein, G.R.; Devarakonda, S.; Johnson, M.; Moreno, V.; Gainor, J.; Edelman, M.J.; Heymach, J.V.; Govindan, R.; Bachier, C.; Doger de Spéville, B.; et al. Phase I Clinical Trial Evaluating the Safety and Efficacy of ADP-A2M10 SPEAR T Cells in Patients with MAGE-A10(+) Advanced Non-Small Cell Lung Cancer. J. Immunother. cancer 2022, 10, e003581. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Butler, M.O.; Pachynski, R.K.; Sullivan, R.; Kebriaei, P.; Boross-Harmer, S.; Ghobadi, A.; Frigault, M.J.; Dumbrava, E.E.; Sauer, A.; et al. Phase 1 Clinical Trial Evaluating the Safety and Anti-Tumor Activity of ADP-A2M10 SPEAR T-Cells in Patients With MAGE-A10+ Head and Neck, Melanoma, or Urothelial Tumors. Front. Oncol. 2022, 12, 818679. [Google Scholar] [CrossRef]
- Sanderson, J.P.; Crowley, D.J.; Wiedermann, G.E.; Quinn, L.L.; Crossland, K.L.; Tunbridge, H.M.; Cornforth, T.V.; Barnes, C.S.; Ahmed, T.; Howe, K.; et al. Preclinical Evaluation of an Affinity-Enhanced MAGE-A4-Specific T-Cell Receptor for Adoptive T-Cell Therapy. Oncoimmunology 2020, 9, 1682381. [Google Scholar] [CrossRef]
- Hong, D.S.; Tine, B.A.; Van Olszanski, A.J.; Johnson, M.L.; Liebner, D.A.; Trivedi, T.I.; Lin, Q.D.; Elefant, E.; Dryer-Minnerly, R.; Navenot, J.-M.; et al. Phase I Dose Escalation and Expansion Trial to Assess the Safety and Efficacy of ADP-A2M4 SPEAR T Cells in Advanced Solid Tumors. J. Clin. Oncol. 2020, 38, 102. [Google Scholar] [CrossRef]
- Willemsen, R.A.; Debets, R.; Hart, E.; Hoogenboom, H.R.; Bolhuis, R.L.H.; Chames, P. A Phage Display Selected Fab Fragment with MHC Class I-Restricted Specificity for MAGE-A1 Allows for Retargeting of Primary Human T Lymphocytes. Gene Ther. 2001, 8, 1601–1608. [Google Scholar] [CrossRef]
- Akatsuka, Y. TCR-Like CAR-T Cells Targeting MHC-Bound Minor Histocompatibility Antigens. Front. Immunol. 2020, 11, 257. [Google Scholar] [CrossRef]
- Chiavenna, S.M.; Jaworski, J.P.; Vendrell, A. State of the Art in Anti-Cancer MAbs. J. Biomed. Sci. 2017, 24, 15. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Identifier | Phase | Type of Tumor | MAGE-A | HLA | Formulation of Therapy | Results |
|---|---|---|---|---|---|---|
| NCT00086866 [37] | II | Melanoma | MAGE-A3 | Recombinant protein |
| |
| NCT00849875 [39] | I/II | Peptide vaccine + Dacarbazine |
| |||
| PMC6319943 [41] | In vivo | Melanoma model | Consensus sequence shared of (MAGE-A1, A2, A3, A5, A6, and A8) | DNA vaccine |
| |
| PMID: 24322180 [42] | In vitro | Melanoma | MAGE-A3-transduced dendritic cells by lentiviral vectors (rLV/MAGE-A3) | Dendritic cell (DC) |
| |
| PMID: 24406390 [43] | MAGE-A3-transduced dendritic cells by rAAV-6 capsid mutant vector Y445F (rAAV-6-MAGE-A3) |
| ||||
| PMID: 19417017 [45] | I | Co-electroporate-Trimix Dcs with mRNA encoding MAGE-A3 | TriMix-DC vaccine |
| ||
| NCT02410733 [46] | I | MAGE-A3 plus NY-ESO1 | RNA-LPX vaccine |
| ||
| PMID: 22221328 [47] | I | Helper/killer-hybrid epitope long peptide MAGE-A4 | Peptide vaccine |
| ||
| NCT01302496 [49] | II | Co-electroporated-TriMix DCs with mRNA encoding MAGE-A3 and other TAA | DCs vaccine + Ipilimumab (anti-CTLA-4) |
| ||
| NCT01241162 [51] | I | Sarcoma Neuroblastoma | MAGE-A1, MAGE-A3, and NY-ESO-1-pulsed DCs | DCs vaccine + Decitabine (DAC) |
| |
| PMC3218253 [51] | In vitro | MAGEA-4-pulsed DCs co-cultured with autologous T -cells | Preparing of autologous MAGE-A4 specific T- cells (in vitro) + Decitabine vaccine in patients |
| ||
| PMID: 21951605 [56] | In vivo | Model tumor NOG mice injected with the cell line QG56 of human lung cancer (MAGE-A4 + HLA-A*2402−) or KE4 of human esophageal cancer (MAGE-A4 + HLA-A*2402+) | MAGE-A4 143–151 peptide (NYKRCFPVI) | HLA-A*2402 | Adoptive transfer (TCR- modified T cells) + Peptide vaccine |
|
| PMID: 25855804 [57] | I | Esophageal cancer | Adoptive transfer (TCR-modified T cells) |
| ||
| NCT01273181 [58] | I//II | Melanoma Synovial sarcoma Esophageal cancer | MAGE-A3 112–120 (KVAELVHFL) | HLA-A*0201 | Adoptive transfer (TCR-modified T cells) + chemotherapy |
|
| NCT01352286 [60] | Melanoma | MAGE-A3 peptide (EVDPIGHLY) | HLA-A*01 |
| ||
| NCT02111850 [62] | Cervical cancer Esophageal cancer Urothelial cancer Osteosarcoma | MAGE-A3 243–258 peptide (KKLLTQHFVQENYLEY) | HLA-DPB1*04:01 |
| ||
| NCT02592577 NCT02989064 [64,66] | NSCLC Head and neck squamous cell carcinoma | MAGE-A10 | HLA-A*02 |
| ||
| NCT03132922 [66] | I | Synovial sarcoma Non-small cell lung cancer Head and neck cancer | MAGE-A4 | HLA-A2 | Adoptive transfer (TCR-modified T cells) + Low-dose radiation |
|
| PMID: 11894998 [67] | In vitro | Melanoma cells | MAGE-A1160–169 | HLA-A1 | Fab-based chimeric receptor, specific for MAGE-A1/HLA-A1 |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alsalloum, A.; Shevchenko, J.A.; Sennikov, S. The Melanoma-Associated Antigen Family A (MAGE-A): A Promising Target for Cancer Immunotherapy? Cancers 2023, 15, 1779. https://doi.org/10.3390/cancers15061779
Alsalloum A, Shevchenko JA, Sennikov S. The Melanoma-Associated Antigen Family A (MAGE-A): A Promising Target for Cancer Immunotherapy? Cancers. 2023; 15(6):1779. https://doi.org/10.3390/cancers15061779
Chicago/Turabian StyleAlsalloum, Alaa, Julia A. Shevchenko, and Sergey Sennikov. 2023. "The Melanoma-Associated Antigen Family A (MAGE-A): A Promising Target for Cancer Immunotherapy?" Cancers 15, no. 6: 1779. https://doi.org/10.3390/cancers15061779
APA StyleAlsalloum, A., Shevchenko, J. A., & Sennikov, S. (2023). The Melanoma-Associated Antigen Family A (MAGE-A): A Promising Target for Cancer Immunotherapy? Cancers, 15(6), 1779. https://doi.org/10.3390/cancers15061779