

Non-Interventional Management of Advanced Pancreatic Neuroendocrine Neoplasms in Patients with von Hippel-Lindau Disease

Abstract
Simple Summary
Abstract
1. Introduction
2. VHL-Mechanism-Based Interventions for vPNEN
2.1. Tyrosine Kinase Inhibitors (TKIs) with Vascular Endothelial Growth Factor (VEGF) Receptor Inhibition Ability
2.1.1. Sunitinib
2.1.2. Vandetanib
2.1.3. Pazopanib
2.2. Hypoxia-Inducible Factors (HIF) Inhibitors
Belzutifan
3. Non-VHL-Mechanism-Related Treatments for vPNEN
3.1. Somatostatin Analogues (SSA)
3.2. SSA-Base-Based Peptide Receptor Radionuclide Therapy (PRRT)
3.3. Mechanistic Target of Rapamycin (mTOR) Inhibitors
Everolimus
3.4. Chemotherapy
Capecitabine and Temozolomide (CAPTEM)
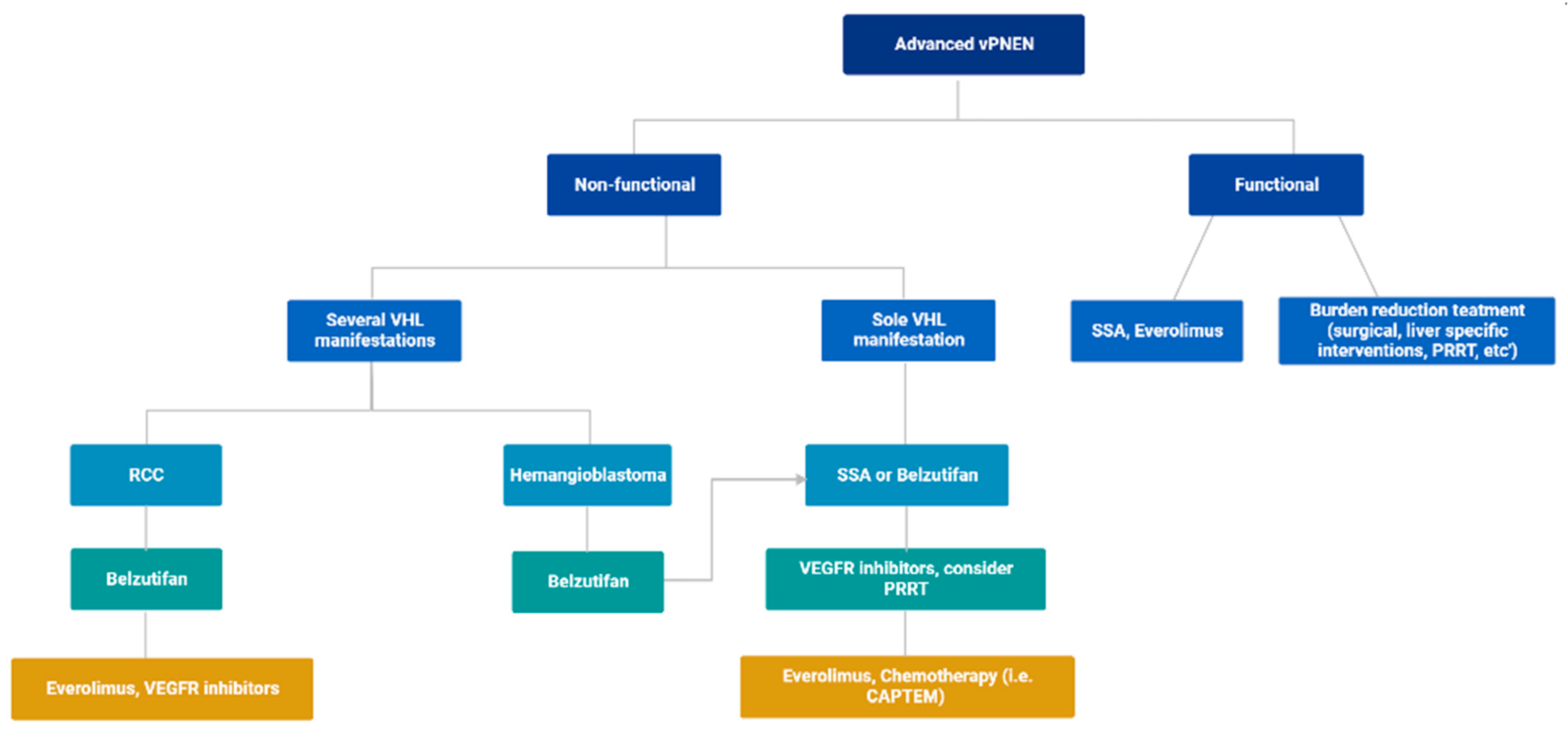
4. Proposed Treatment Algorithm
5. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Dasari, A.; Shen, C.; Halperin, D.; Zhao, B.; Zhou, S.; Xu, Y.; Shih, T.; Yao, J.C. Trends in the Incidence, Prevalence, and Survival Outcomes in Patients with Neuroendocrine Tumors in the United States. JAMA Oncol. 2017, 3, 1335–1342. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.B.; Edge, S.B.; Greene, F.L.; Page, D.L.; Fleming, I.D.; Frizt, A.G.; Balch, C.M.; Haller, D.G.; Morrow, M. AJCC Cancer Staging Manual; Springer International Publishing: New York, NY, USA, 2018. [Google Scholar]
- Falconi, M.; Eriksson, B.; Kaltsas, G.; Bartsch, D.K.; Capdevila, J.; Caplin, M.; Kos-Kudla, B.; Kwekeboom, D.; Rindi, G.; Kloppel, G.; et al. ENETS Consensus Guidelines Update for the Management of Patients with Functional Pancreatic Neuroendocrine Tumors and Non-Functional Pancreatic Neuroendocrine Tumors. Neuroendocrinology 2016, 103, 153–171. [Google Scholar] [CrossRef] [PubMed]
- Lonser, R.R.; Glenn, G.M.; Walther, M.; Chew, E.Y.; Libutti, S.K.; Linehan, W.M.; Oldfield, E.H. Von Hippel-Lindau disease. Lancet 2003, 361, 2059–2067. [Google Scholar] [CrossRef] [PubMed]
- Maher, E.R.; Webster, A.R.; Richards, F.M.; Green, J.S.; Crossey, P.A.; Payne, S.J.; Moore, A.T. Phenotypic expression in von Hippel-Lindau disease: Correlations with germline VHL gene mutations. J. Med. Genet. 1996, 33, 328–332. [Google Scholar] [CrossRef]
- Maher, E.R.; Neumann, H.P.; Richard, S. von Hippel–Lindau disease: A clinical and scientific review. Eur. J. Hum. Genet. 2011, 19, 617–623. [Google Scholar] [CrossRef]
- Binderup, M.L.M.; Smerdel, M.; Borgwadt, L.; Beck Nielsen, S.S.; Madsen, M.G.; Moller, H.U.; Kiilgaard, J.F.; Friis-Hansen, L.; Harbud, V.; Cortnum, S.; et al. von Hippel-Lindau disease: Updated guideline for diagnosis and surveillance. Eur. J. Med. Genet. 2022, 65, 104538. [Google Scholar] [CrossRef]
- Bender, B.U.; Eng, C.; Olschewski, M.; Berger, D.P.; Laubenberger, J.; Alterhofer, C.; Kirste, G.; Orszagh, M.; van Velthoven, V.; Milosczka, H.; et al. VHL c.505 T>C mutation confers a high age related penetrance but no increased overall mortality. J. Med. Genet. 2001, 38, 508–514. [Google Scholar] [CrossRef]
- Salama, Y.; Albanyan, S.; Szybowska, M.; Bullivant, G.; Gallinger, B.; Giles, R.H.; Asa, S.; Badduke, C.; Chiorean, A.; Druker, H.; et al. Comprehensive characterization of a Canadian cohort of von Hippel-Lindau disease patients. Clin. Genet. 2019, 96, 461–467. [Google Scholar] [CrossRef]
- Blansfield, J.A.; Choyke, L.; Morita, S.Y.; Choyke, P.L.; Pingpank, J.F.; Alexander, H.R.; Seidel, G.; Shutack, Y.; Yuldasheva, N.; Eugeni, M.; et al. Clinical, genetic and radiographic analysis of 108 patients with von Hippel-Lindau disease (VHL) manifested by pancreatic neuroendocrine neoplasms (PNETs). Surgery 2007, 142, 814–818, discussion 818.e1–2. [Google Scholar] [CrossRef]
- Libutti, S.K.; Choyke, P.L.; Bartlett, D.L.; Vargas, H.; Walther, M.; Lubensky, I.; Glenn, G.; Linehan, W.M.; Alexander, H.R. Pancreatic neuroendocrine tumors associated with von Hippel Lindau disease: Diagnostic and management recommendations. Surgery 1998, 124, 1153–1159. [Google Scholar] [CrossRef]
- De Mestier, L.; Gaujoux, S.; Cros, J.; Hentic, O.; Vullierma, M.P.; Couvelard, A.; Cadiot, G.; Sauvanet, A.; Ruszniewski, P.; Richard, S.; et al. Long-term Prognosis of Resected Pancreatic Neuroendocrine Tumors in von Hippel-Lindau Disease Is Favorable and Not Influenced by Small Tumors Left in Place. Ann. Surg. 2015, 262, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Erlic, Z.; Ploeckinger, U.; Cascon, A.; Hoffmann, M.M.; Von Duecker, L.; Winter, A.; Kammel, G.; Bacher, J.; Sullivan, M.; Isermann, B.; et al. Systematic comparison of sporadic and syndromic pancreatic islet cell tumors. Endocr. Relat. Cancer 2010, 17, 875–883. [Google Scholar] [CrossRef] [PubMed]
- Hammel, P.R.; Vilgrain, V.; Terris, B.; Penfornis, A.; Sauvanet, A.; Correas, J.M.; Chauveau, D.; Balian, A.; Beigelman, C.; O’Toole, D.; et al. Pancreatic involvement in von Hippel-Lindau disease. The Groupe Francophone d’Etude de la Maladie de von Hippel-Lindau. Gastroenterology 2000, 119, 1087–1095. [Google Scholar] [CrossRef]
- Sala Hernández, Á.; Montalvá Orón, E.M.; Pareja Ibars, E.; Ballester Pla, N.; López Andújar, R. Management of pancreatic gastrinoma associated with Von Hippel-Lindau disease: A case report. Rev. Española Enferm. Dig. 2017, 109, 154–157. [Google Scholar] [CrossRef]
- Tirosh, A.; Sadowski, S.M.; Linehan, W.M.; Libutti, S.K.; Patel, D.; Nilubol, N.; Kebebew, E. Association of VHL genotype with pancreatic neuroendocrine tumor phenotype in patients with von hippel-lindau disease. JAMA Oncol. 2018, 4, 124–126. [Google Scholar] [CrossRef] [PubMed]
- Arnon, L.; Halperin, R.; Tirosh, A. Impact of Pancreatic Neuroendocrine Tumor on Mortality in Patients With von Hippel-Lindau Disease. Endocr. Pract. 2021, 27, 1040–1045. [Google Scholar] [CrossRef]
- Krauss, T.; Ferrara, A.M.; Links, T.P.; Wellner, U.; Bancos, I.; Kvachenyuk, A.; Villar Gómez de Las Heras, K.; Yukina, M.Y.; Petrov, R.; Bullivant, G.; et al. Preventive medicine of von Hippel-Lindau disease-associated pancreatic neuroendocrine tumors. Endocr. Relat. Cancer 2018, 25, 783–793. [Google Scholar] [CrossRef]
- Libutti, S.K.; Choyke, P.L.; Alexander, H.R.; Glenn, G.; Bartlett, D.L.; Zbar, B.; Lubensky, I.; McKee, S.A.; Maher, E.R.; Linehan, W.M.; et al. Clinical and genetic analysis of patients with pancreatic neuroendocrine tumors associated with von Hippel-Lindau disease. Surgery 2000, 128, 1022–1028. [Google Scholar] [CrossRef]
- Laks, S.; van Leeuwaarde, R.; Patel, D.; Keutgen, X.M.; Hammel, P.; Nilubol, N.; Links, T.P.; Halfdanarson, T.; Daniels, A.B.; Tirosh, A.; et al. Management recommendations for pancreatic manifestations of von Hippel-Lindau disease. Cancer 2022, 128, 435–446. [Google Scholar] [CrossRef]
- Maharjan, C.K.; Ear, P.H.; Tran, C.G.; Howe, J.R.; Chandrasekharan, C.; Quelle, D.E. Pancreatic Neuroendocrine Tumors: Molecular Mechanisms and Therapeutic Targets. Cancers 2021, 13, 5117. [Google Scholar] [CrossRef]
- Jonasch, E.; McCutcheon, I.E.; Waguespack, S.G.; Wen, S.; Davis, D.W.; Smith, L.A.; Tannir, N.M.; Gombos, D.S.; Fuller, G.N.; Matin, S.F. Pilot trial of sunitinib therapy in patients with von Hippel-Lindau disease. Ann. Oncol. 2011, 22, 2661–2666. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrials.gov. Phase II Study of Vandetanib in Individuals with Kidney Cancer. Study Results. Available online: https://clinicaltrials.gov/ct2/show/NCT00566995 (accessed on 3 December 2022).
- Jonasch, E.; McCutcheon, I.E.; Gombos, D.S.; Ahrar, K.; Perrier, N.D.; Liu, D.; Robichaux, C.C.; Villarreal, M.F.; Weldon, J.A.; Woodson, A.H.; et al. Pazopanib in patients with von Hippel-Lindau disease: A single-arm, single-centre, phase 2 trial. Lancet Oncol. 2018, 19, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Jonasch, E.; Donskov, F.; Iliopoulos, O.; Rathmell, W.K.; Narayan, V.K.; Maughan, B.L.; Oudard, S.; Else, T.; Maranchie, J.K.; Welsh, S.J.; et al. Belzutifan for Renal Cell Carcinoma in von Hippel-Lindau Disease. N. Engl. J. Med. 2021, 385, 2036–2046. [Google Scholar] [CrossRef] [PubMed]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Hong, B.; Zhou, J.; Gong, Y.; Wang, J.; Liu, S.; Peng, X.; Zhou, B.; Zhang, J.; Xie, H.; et al. The Efficacy and Safety of Tyrosine Kinase Inhibitors for Von Hippel-Lindau Disease: A Retrospective Study of 32 Patients. Front. Oncol. 2019, 9, 1122. [Google Scholar] [CrossRef]
- Raymond, E.; Dahan, L.; Raoul, J.-L.; Bang, Y.J.; Borbath, I.; Lombard-Bohas, C.; Valle, J.; Metrakos, P.; Smith, D.; Vinik, A.; et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N. Engl. J. Med. 2011, 364, 501–513. [Google Scholar] [CrossRef]
- Jimenez, C.; Cabanillas, M.E.; Santarpia, L.; Jonasch, E.; Kyle, K.l.; Lano, E.A.; Matin, S.F.; Nunez, R.F.; Perrier, N.D.; Phan, A.; et al. Use of the tyrosine kinase inhibitor sunitinib in a patient with von Hippel-Lindau disease: Targeting angiogenic factors in pheochromocytoma and other von Hippel-Lindau disease-related tumors. J. Clin. Endocrinol. Metab. 2009, 94, 386–391. [Google Scholar] [CrossRef]
- Wang, W.; Jiang, C.Y.; Wang, H.W. Use of sunitinib in a 30-year-old woman with pancreatic neuroendocrine tumors associated with Von Hippel-Lindau syndrome. J. Clin. Gastroenterol. 2015, 49, 89–90. [Google Scholar] [CrossRef]
- Yuan, G.; Liu, Q.; Tong, D.; Liu, G.; Yi, Y.; Zhang, J.; Zhang, Y.; Wang, L.A.; Wang, L.; Chen, R.; et al. A retrospective case study of sunitinib treatment in three patients with Von Hippel-Lindau disease. Cancer Biol. Ther. 2018, 19, 766–772. [Google Scholar] [CrossRef]
- Ali, T.; Kandil, D.; Piperdi, B. Long-term disease control with sunitinib in a patient with metastatic pancreatic neuroendocrine tumor (NET) associated with Von Hippel-Lindau syndrome (VHL). Pancreas 2012, 41, 492–493. [Google Scholar] [CrossRef]
- Carlomagno, F.; Vitagliano, D.; Guida, T.; Ciardiello, F.; Tortora, G.; Vecchio, G.; Ryan, A.J.; Fontanini, G.; Fusco, A.; Santoro, M. ZD6474, an orally available inhibitor of KDR tyrosine kinase activity, efficiently blocks oncogenic RET kinases. Cancer Res. 2002, 62, 7284–7290. [Google Scholar] [PubMed]
- Wells, S.A.J.; Robinson, B.G.; Gagel, R.F.; Dralle, H.; Fagin, J.A.; Santoro, M.; Baudin, E.; Elisei, R.; Jarzab, B.; Vasselli, J.R.; et al. Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: A randomized, double-blind phase III trial. J. Clin. Oncol. 2012, 30, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Knick, V.B.; Rudolph, S.K.; Johnson, J.H.; Crosby, R.M.; Crouthamel, M.C.; Hopper, T.M.; Miller, C.G.; Harrington, L.E.; Onori, J.A.; et al. Pharmacokinetic-pharmacodynamic correlation from mouse to human with pazopanib, a multikinase angiogenesis inhibitor with potent antitumor and antiangiogenic activity. Mol. Cancer Ther. 2007, 6, 2012–2021. [Google Scholar] [CrossRef] [PubMed]
- Phan, A.T.; Halperin, D.M.; Chan, J.A.; Fogelman, D.R.; Hess, K.R.; Malinowski, P.; Regan, E.; Ng, C.S.; Yao, J.C.; Kulke, M.H. Pazopanib and depot octreotide in advanced, well-differentiated neuroendocrine tumours: A multicentre, single-group, phase 2 study. Lancet Oncol. 2015, 16, 695–703. [Google Scholar] [CrossRef]
- NCT04924075. Belzutifan/MK-6482 for the Treatment of Advanced Pheochromocytoma/Paraganglioma (PPGL), Pancreatic Neuroendocrine Tumor (pNET), or Von Hippel-Lindau (VHL) Disease-Associated Tumors (MK-6482-015). Available online: https://clinicaltrials.gov/ct2/show/NCT04924075 (accessed on 3 December 2022).
- Rinke, A.; Müller, H.-H.; Schade-Brittinger, C.; Klose, K.J.; Barth, P.; Wied, M.; Mayer, C.; Aminossadati, B.; Pape, U.F.; Bläker, M.; et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: A report from the PROMID Study Group. J. Clin. Oncol. 2009, 27, 4656–4663. [Google Scholar] [CrossRef]
- Caplin, M.E.; Pavel, M.; Ćwikła, J.B.; Phan, A.T.; Raderer, M.; Sedláčková, E.; Cadiot, G.; Wolin, E.M.; Capdevila, J.; Wall, L.; et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N. Engl. J. Med. 2014, 371, 224–233. [Google Scholar] [CrossRef]
- Yao, J.C.; Shah, M.H.; Ito, T.; Bohas, C.L.; Wolin, E.M.; Van Cutsem, E.; Hobday, J.M.; Okusaka, T.; Capdevila, J.; de Vries, E.G.E.; et al. Everolimus for advanced pancreatic neuroendocrine tumors. N. Engl. J. Med. 2011, 364, 514–523. [Google Scholar] [CrossRef]
- Strosberg, J.; El-Haddad, G.; Wolin, E.; Hendifar, A.; Yao, J.; Chasen, B.; Mittra, E.; Mittra, P.L.; Kulke, M.H.; Jacene, H.; et al. Phase 3 Trial of (177)Lu-Dotatate for Midgut Neuroendocrine Tumors. N. Engl. J. Med. 2017, 376, 125–135. [Google Scholar] [CrossRef]
- Kunz, P.L.; Graham, N.T.; Catalano, P.J.; Nimeiri, H.S.; Fisher, G.A.; Longacre, T.A.; Suarez, C.J.; Martin, B.A.; Yao, J.C.; Kulke, M.H.; et al. A Randomized Study of Temozolomide or Temozolomide and Capecitabine in Patients with Advanced Pancreatic Neuroendocrine Tumors (ECOG-ACRIN E2211). J. Clin. Oncol. 2023, 41, 1359–1369. [Google Scholar] [CrossRef]
- Hofland, J.; Brabander, T.; Verburg, F.A.; Feelders, R.A.; de Herder, W.W. Peptide Receptor Radionuclide Therapy. J. Clin. Endocrinol. Metab. 2022, 107, 3199–3208. [Google Scholar] [CrossRef]
- Strosberg, J.R.; Caplin, M.E.; Kunz, P.L.; Ruszniewski, P.B.; Bodei, L.; Hendifar, A.; Mittra, E.; Wolin, E.M.; Yao, J.C.; Pavel, M.E.; et al. (177)Lu-Dotatate plus long-acting octreotide versus high-dose long-acting octreotide in patients with midgut neuroendocrine tumours (NETTER-1): Final overall survival and long-term safety results from an open-label, randomised, controlled, phase 3 trial. Lancet Oncol. 2021, 22, 1752–1763. [Google Scholar] [CrossRef] [PubMed]
- Alsadik, S.; Yusuf, S.; Al-Nahhas, A. Peptide Receptor Radionuclide Therapy for Pancreatic Neuroendocrine Tumours. Curr. Radiopharm. 2019, 12, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Brabander, T.; van der Zwan, W.A.; Teunissen, J.J.M.; Kam, B.L.R.; Feelders, R.A.; de Herder, W.W.; van Eijck, C.H.J.; Franssen, G.J.H.; Krenning, E.P.; Kwekkeboom, D.J. Long-Term Efficacy, Survival, and Safety of [(177)Lu-DOTA(0),Tyr(3)]octreotate in Patients with Gastroenteropancreatic and Bronchial Neuroendocrine Tumors. Clin. Cancer Res. 2017, 23, 4617–4624. [Google Scholar] [CrossRef] [PubMed]
- Pusceddu, S.; Prinzi, N.; Tafuto, S.; Ibrahim, T.; Filice, A.; Brizzi, M.P.; Panzuto, F.; Baldari, S.; Grana, C.M.; Campana, D.; et al. Association of Upfront Peptide Receptor Radionuclide Therapy With Progression-Free Survival Among Patients With Enteropancreatic Neuroendocrine Tumors. JAMA Netw. Open 2022, 5, e220290. [Google Scholar] [CrossRef] [PubMed]
- Satapathy, S.; Mittal, B.R. 177Lu-DOTATATE peptide receptor radionuclide therapy versus Everolimus in advanced pancreatic neuroendocrine tumors: A systematic review and meta-analysis. Nucl. Med. Commun. 2019, 40, 1195–1203. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Shi, C.; Edil, B.H.; de Wilde, R.F.; Klimstra, D.S.; Maitra, A.; Schulick, R.D.; Tang, L.H.; Wolfgang, C.L.; Choti, M.A.; et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 2011, 331, 1199–1203. [Google Scholar] [CrossRef] [PubMed]
- Scarpa, A.; Chang, D.K.; Nones, K.; Corbo, V.; Patch, A.M.; Bailey, P.; Lawlor, R.T.; Johns, A.L.; Miller, D.K.; Mafficini, A.; et al. Whole-genome landscape of pancreatic neuroendocrine tumours. Nature 2017, 543, 65–71. [Google Scholar] [CrossRef]
- Calvo, E.; Escudier, B.; Motzer, R.J.; Oudard, S.; Hutson, T.E.; Porta, C.; Bracarda, S.; Grünwald, V.; Thompson, J.A.; Ravaud, A.; et al. Everolimus in metastatic renal cell carcinoma: Subgroup analysis of patients with 1 or 2 previous vascular endothelial growth factor receptor-tyrosine kinase inhibitor therapies enrolled in the phase III RECORD-1 study. Eur. J. Cancer 2012, 48, 333–339. [Google Scholar] [CrossRef]
- Motzer, R.; Alekseev, B.; Rha, S.-Y.; Porta, C.; Eto, M.; Powles, T.; Grünwald, V.; Hutson, T.E.; Kopyltsov, E.; Méndez-Vidal, M.J.; et al. Lenvatinib plus Pembrolizumab or Everolimus for Advanced Renal Cell Carcinoma. N. Engl. J. Med. 2021, 384, 1289–1300. [Google Scholar] [CrossRef]
- Nuñez, J.E.; Donadio, M.; Filho, D.R.; Rego, J.F.; Barros, M.; Formiga, M.N.; Lopez, R.; Riechelmann, R. The efficacy of everolimus and sunitinib in patients with sporadic or germline mutated metastatic pancreatic neuroendocrine tumors. J. Gastrointest Oncol. 2019, 10, 645–651. [Google Scholar] [CrossRef]
- Pavel, M.; O’Toole, D.; Costa, F.; Capdevila, J.; Gross, D.; Kianmanesh, R.; Krenning, E.; Knigge, U.; Salazar, R.; Pape, U.F.; et al. ENETS Consensus Guidelines Update for the Management of Distant Metastatic Disease of Intestinal, Pancreatic, Bronchial Neuroendocrine Neoplasms (NEN) and NEN of Unknown Primary Site. Neuroendocrinology 2016, 103, 172–185. [Google Scholar] [CrossRef] [PubMed]
- Bergsma, H.; van Lom, K.; Raaijmakers, M.H.G.P.; Konijnenberg, M.; Kam, B.L.R.; Teunissen, J.J.M.; de Herder, W.W.; Krenning, E.P.; Kwekkeboom, D.J. Persistent Hematologic Dysfunction after Peptide Receptor Radionuclide Therapy with (177)Lu-DOTATATE: Incidence, Course, and Predicting Factors in Patients with Gastroenteropancreatic Neuroendocrine Tumors. J. Nucl. Med. 2018, 59, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, A.; Del Rivero, J.; Ramirez, R.A.; Soares, H.P.; Li, D. Treatment Sequencing Strategies in Advanced Neuroendocrine Tumors: A Review. Cancers 2022, 14, 5248. [Google Scholar] [CrossRef] [PubMed]
- Binderup, M.L.M.; Jensen, A.M.; Budtz-Jørgensen, E.; Bisgaard, M.L. Survival and causes of death in patients with von Hippel-Lindau disease. J. Med. Genet. 2017, 54, 11–18. [Google Scholar] [CrossRef]
- Keutgen, X.M.; Hammel, P.; Choyke, P.L.; Libutti, S.K.; Jonasch, E.; Kebebew, E. Evaluation and management of pancreatic lesions in patients with von Hippel-Lindau disease. Nat. Rev. Clin. Oncol. 2016, 13, 537–549. [Google Scholar] [CrossRef]
- Chan, J.A.; Faris, J.E.; Murphy, J.E.; Blaszkowsky, L.; Kwak, E.L.; McCleary, N.J.; Fuchs, C.S.; Meyerhardt, J.A.; Ng, K.; Zhu, A.X.; et al. Phase II trial of cabozantinib in patients with carcinoid and pancreatic neuroendocrine tumors (pNET). J. Clin Oncol. 2017, 35 (Suppl. 4), 228. [Google Scholar] [CrossRef]
- Kumar, S.K.; Callander, N.S.; Adekola, K.; Anderson, L.D.; Baljevic, M.; Campagnaro, E.; Castillo, J.J.; Costello, C.; D’Angelo, C.; Devarakonda, S.; et al. Kidney Cancer, Version 3.2022, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2022, 20, 67–81. [Google Scholar] [CrossRef]
- Bergsma, H.; Konijnenberg, M.W.; Kam, B.L.R.; Teunissen, J.J.M.; Kooij, P.P.; de Herder, W.W.; Franssen, G.J.H.; van Eijck, C.H.J.; Krenning, E.P.; Kwekkeboom, D.J. Subacute haematotoxicity after PRRT with (177)Lu-DOTA-octreotate: Prognostic factors, incidence and course. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 453–463. [Google Scholar] [CrossRef]
- Grozinsky-Glasberg, S.; Davar, J.; Hofland, J.; Dobson, R.; Prasad, V.; Pascher, A.; Denecke, T.; Tesselaar, M.E.T.; Panzuto, F.; Albåge, A.; et al. European Neuroendocrine Tumor Society (ENETS) 2022 Guidance Paper for Carcinoid Syndrome and Carcinoid Heart Disease. J. Neuroendocrinol. 2022, 34, e13146. [Google Scholar] [CrossRef]
- Kulke, M.H.; Bergsland, E.K.; Yao, J.C. Glycemic control in patients with insulinoma treated with everolimus. N. Engl. J. Med. 2009, 360, 195–197. [Google Scholar] [CrossRef]
- Bernard, V.; Lombard-Bohas, C.; Taquet, M.-C.; Caroli-Bosc, F.X.; Ruszniewski, P.; Niccoli, P.; Guimbaud, R.; Chougnet, C.N.; Goichot, B.; Rohmer, V.; et al. Efficacy of everolimus in patients with metastatic insulinoma and refractory hypoglycemia. Eur. J. Endocrinol. 2013, 168, 665–674. [Google Scholar] [CrossRef] [PubMed]
- King, J.; Quinn, R.; Glenn, D.M.; Janssen, J.; Tong, D.; Liaw, W.; Morris, D.L. Radioembolization with selective internal radiation microspheres for neuroendocrine liver metastases. Cancer 2008, 113, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Fiore, F.; Del Prete, M.; Franco, R.; Marotta, V.; Ramundo, V.; Marciello, F.; Di Sarno, A.; Carratù, A.C.; de Luca di Roseto, C.; Colao, A.; et al. Transarterial embolization (TAE) is equally effective and slightly safer than transarterial chemoembolization (TACE) to manage liver metastases in neuroendocrine tumors. Endocrine 2014, 47, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Mavligit, G.M.; Pollock, R.E.; Evans, H.L.; Wallace, S. Durable hepatic tumor regression after arterial chemoembolization-infusion in patients with islet cell carcinoma of the pancreas metastatic to the liver. Cancer 1993, 72, 375–380. [Google Scholar] [CrossRef]
- De Herder, W.W.; Zandee, W.T.; Hofland, J. Insulinoma. In Endotext; Feingold, K.R., Anawalt, B., Blackman, M.R., Boyce, E., Chrousos, G., Corpas, E., de Herder, W.W., Dhatariya, K., Hofland, J., Dungan, K., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]


{kind=link}
| Treatment | Mechanism of Action | n (n vPNEN) | Efficacy n Response/Total Lesions | Toxicity % |
|---|---|---|---|---|
| Sunitinib [22] | VEGFR inhibitor | 15 (7) | RCC 10/18 SD, 6/18 PR, 2/18 PD PNEN 5/5 SD CNS HB 19/21 SD, 2/21 PD Retinal HB 7/7 SD | Stopped treatment—40% |
| Vandetanib [23] ongoing, partial data | VEGFR inhibitor | 37 (2) | PNEN 2/2 DS CNS HB 2/2 SD | Stopped treatment—19% (11% due to side effects) |
| Pazopanib [24] | VEGFR inhibitor | 32 (17 *) | RCC 28/59 SD, 29/59 PR, 2/59 CR Pancreatic lesions 8/17 SD, 9/17 PR, 0/17 CR CNS HB 47/49 SD, 2/49 PR, 0/49 CR | Dose reduction—58%, Stopped treatment—23% |
| Belzutifan [25] | HIF 2 inhibitor | 61 (22) | RCC 30/61 SD, 30/61 PR PNEN 20/22 (91%) confirmed response, 3 CR CNS HB—15/50 confirmed response, 3 CR Retinal HB—12/12 confirmed response | Dose reduction—15%, Dose interruption—43%, Stopped treatment—11% |
| Trial Name | Intervention | No. Patients (Intervention/Control, n) | No. Patients with PNEN (Intervention/Control, n) | Grade 1/2/3 (n) | PFS Intervention vs. Control (Months) HR, 95% CI, p Value | OS Intervention vs. Control (Months) HR, 95% CI, p value | Toxicity (%) | Notes |
|---|---|---|---|---|---|---|---|---|
| PROMID [38] | Octreotide LAR vs. Placebo | 42/43 | 0/0 | 81/3/1 | 14 vs. 6 € 0.33 (0.19–0.55, p < 0.001) | 0.81 (0.3–2.18, p = 0.77) | Most frequent (% not reported)—diarrhea, flatulence, cholelithiasis. | Advanced Midgut NEN |
| CLARINET [39] | Lanreotide autogel vs. Placebo | 101/103 | 42/48 | 138/60/0 | NR vs 18 0.47 (0.3–0.73, p < 0.001) 0.58 (0.32–1.04) ¥ | NR | Serious AE: 3% Common AE: diarrhea 26%, abdominal pain 14%, cholelithiasis 10%. | Advanced GEP-NEN |
| RADIANT-3 [40] | Everolimus vs. Placebo | 207/203 | 207/203 | 341/65/na * | 11.4 vs. 5.4 0.35 (0.27–0.45, p < 0.001) | 44 vs. 37 0.94 (0.73–1.2) | Common AE: stomatitis 64%, rash 49%, diarrhea 34%, fatigue 31%, infections 23%. Grade 3/4 AE: anemia 6%, thrombocytopenia 4%, hyperglycemia 5%, stomatitis 7%, diarrhea 3%. | Advanced PNEN |
| NETTER-1 [41] | PRRT plus Octreotide LAR 30 mg vs. Octreotide LAR 60 mg | 116/113 | 0/0 | 157/72/0 | NR vs. 8.4 0.21 (0.13–0.33, p < 0.001) | NR 14 vs. 26 deaths, p < 0.01 # | Common AE: nausea 59%, vomiting 47%, fatigue 40%. Grade 3/4 AE: lymphopenia 9%, vomiting 7%, nausea 4%, thrombocytopenia 2%. | Advanced midgut NEN |
| ECOG-ACRIN E221 [42] | CAPTEM vs. Temozolomide | 72/72 | 72/72 | 50/61/na | 23 vs. 14 0.58 (0.36–0.93, p = 0.022) # | 59 vs. 54 0.41 (0.21–0.82, p = 0.012) # | Common AE: CAPTEM—nausea 65%, fatigue 56%, constipation 48%, anemia 37%. Temozolomide—fatigue 63%, nausea 60%, constipation 31%, anemia 31%, thrombocytopenia 31%. | Advanced PNEN |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Halperin, R.; Tirosh, A. Non-Interventional Management of Advanced Pancreatic Neuroendocrine Neoplasms in Patients with von Hippel-Lindau Disease. Cancers 2023, 15, 1739. https://doi.org/10.3390/cancers15061739
Halperin R, Tirosh A. Non-Interventional Management of Advanced Pancreatic Neuroendocrine Neoplasms in Patients with von Hippel-Lindau Disease. Cancers. 2023; 15(6):1739. https://doi.org/10.3390/cancers15061739
Chicago/Turabian StyleHalperin, Reut, and Amit Tirosh. 2023. "Non-Interventional Management of Advanced Pancreatic Neuroendocrine Neoplasms in Patients with von Hippel-Lindau Disease" Cancers 15, no. 6: 1739. https://doi.org/10.3390/cancers15061739
APA StyleHalperin, R., & Tirosh, A. (2023). Non-Interventional Management of Advanced Pancreatic Neuroendocrine Neoplasms in Patients with von Hippel-Lindau Disease. Cancers, 15(6), 1739. https://doi.org/10.3390/cancers15061739