
Development and Experimental Validation of a Novel Prognostic Signature for Gastric Cancer

 , , , ,
, , , , 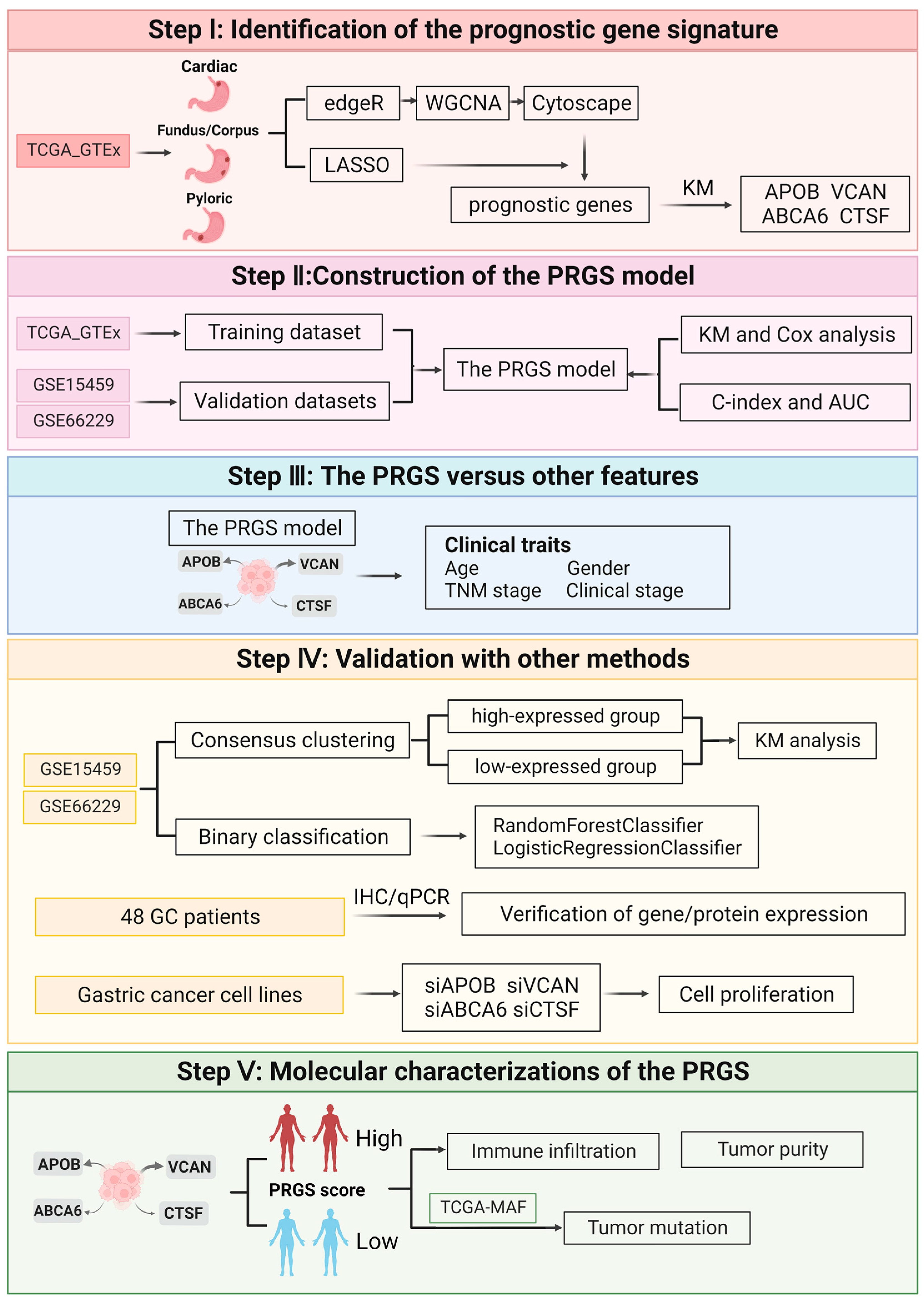{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Acquisition and Processing
2.2. Human Tissue Specimens
2.3. Differentially Expressed Analysis and Weighted Correlation Network Analysis
2.4. Construction of the Co-Expression Network
2.5. PRGS Signature Generation
2.6. Chromatin Accessibility Analysis
2.7. Consensus Clustering
2.8. Binary Classification
- (1)
- We pre-processed the GSE66229 data and selected the top 5 genes from the three generated co-expression networks of the three zones’ expression spectrum matrices, then divided the data into normal and tumor samples. We also generated the GSE66229 data and generated PRGS, CEA [21], and GCscore [6] expression spectrum matrices.
- (2)
- We performed 5-fold cross validation using the logistic regression classifier (LR) and the random forest classifier (RF) on the GSE66229 and computed the ROC curve for each fold. We then calculated the means of every fitting curve to generate the plot. We then divided the data into five subsets based on the sample tags “Tumor/Normal”, “Stage 1/Normal”, “Stage 2/Normal”, “Stage 3/Normal”, and “Stage 4/Normal”. Details of the data are shown in Table S4.
2.9. Haematoxylin–Eosin (HE) and Immunohistochemistry (IHC)
2.10. Cell Culture, Transfection, and Immunostaining
2.11. Flow Cytometric Analysis
2.12. Quantitative Real-Time PCR
2.13. Cell Infiltration Estimation
2.14. Tumor Mutation Status Analysis
2.15. Functional Enrichment Analysis
3. Results
3.1. Construction of the Gene Modules
3.2. Construction and Cross-Validation of the PRGS Model in Gastric Cancer Cohorts
3.3. PRGS Are Significantly Related to Clinical Outcomes
3.4. Experimental Validation of the PRGS in the Clinical Samples and Cell Lines
3.5. The Immune Cell Infiltration between the High- and Low-PRGS Patients
3.6. Mutation Status in GC Patients in the High- and Low-PRGS Groups
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Fong, C.; Johnston, E.; Starling, N. Neoadjuvant and Adjuvant Therapy Approaches to Gastric Cancer. Curr. Treat. Options Oncol. 2022, 23, 1247–1268. [Google Scholar] [CrossRef] [PubMed]
- Wittekind, C. The development of the TNM classification of gastric cancer. Pathol. Int. 2015, 65, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Egner, J.R. AJCC cancer staging manual. JAMA 2010, 304, 1726–1727. [Google Scholar] [CrossRef]
- Shimada, H.; Noie, T.; Ohashi, M.; Oba, K.; Takahashi, Y. Clinical significance of serum tumor markers for gastric cancer: A systematic review of literature by the Task Force of the Japanese Gastric Cancer Association. Gastric Cancer 2014, 17, 26–33. [Google Scholar] [CrossRef]
- Wei, C.; Chen, M.; Deng, W.; Bie, L.; Ma, Y.; Zhang, C.; Liu, K.; Shen, W.; Wang, S.; Yang, C.; et al. Characterization of gastric cancer stem-like molecular features, immune and pharmacogenomic landscapes. Briefings Bioinform. 2022, 23, bbab386. [Google Scholar] [CrossRef]
- Cui, K.; Yao, S.; Liu, B.; Sun, S.; Gong, L.; Li, Q.; Fei, B.; Huang, Z. A novel high-risk subpopulation identified by CTSL and ZBTB7B in gastric cancer. Br. J. Cancer 2022, 127, 1450–1460. [Google Scholar] [CrossRef] [PubMed]
- Cheong, J.H.; Wang, S.C.; Park, S.; Porembka, M.R.; Christie, A.L.; Kim, H.; Kim, H.S.; Zhu, H.; Hyung, W.J.; Noh, S.H.; et al. Development and validation of a prognostic and predictive 32-gene signature for gastric cancer. Nat. Commun. 2022, 13, 774. [Google Scholar] [CrossRef]
- Helander, H.F. The cells of the gastric mucosa. Int. Rev. Cytol. 1981, 70, 217–289. [Google Scholar] [PubMed]
- Hoffmann, W. Regeneration of the gastric mucosa and its glands from stem cells. Curr. Med. Chem. 2008, 15, 3133–3144. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, W. Current Status on Stem Cells and Cancers of the Gastric Epithelium. Int. J. Mol. Sci. 2015, 16, 19153–19169. [Google Scholar] [CrossRef] [PubMed]
- Lai, S. A human mode of intestinal type gastric carcinoma. Med. Hypotheses 2019, 123, 27–29. [Google Scholar] [CrossRef] [PubMed]
- Vivian, J.; Rao, A.A.; Nothaft, F.A.; Ketchum, C.; Armstrong, J.; Novak, A.; Pfeil, J.; Narkizian, J.; Deran, A.D.; Musselman-Brown, A.; et al. Toil enables reproducible, open source, big biomedical data analyses. Nat. Biotechnol. 2017, 35, 314–316. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.M.; MacDonald, J.A. Network analysis of TCGA and GTEx gene expression datasets for identification of trait-associated biomarkers in human cancer. STAR Protoc. 2022, 3, 101168. [Google Scholar] [CrossRef] [PubMed]
- Law, C.W.; Alhamdoosh, M.; Su, S.; Dong, X.; Tian, L.; Smyth, G.K.; Ritchie, M.E. RNA-seq analysis is easy as 1-2-3 with limma, Glimma and edgeR. F1000Research 2016, 5. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Chin, C.H.; Chen, S.H.; Wu, H.H.; Ho, C.-W.; Ko, M.-T.; Lin, C.-Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8, S11. [Google Scholar] [CrossRef] [PubMed]
- Tibshirani, R. The lasso method for variable selection in the Cox model. Stat. Med. 1997, 16, 385–395. [Google Scholar] [CrossRef]
- Wilkerson, M.D.; Hayes, D.N. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics 2010, 26, 1572–1573. [Google Scholar] [CrossRef]
- Beauchemin, N.; Arabzadeh, A. Carcinoembryonic antigen-related cell adhesion molecules (CEACAMs) in cancer progression and metastasis. Cancer Metastasis Rev. 2013, 32, 643–671. [Google Scholar] [CrossRef]
- Shen, Y.; Vignali, P.; Wang, R. Rapid profiling cell cycle by flow cytometry using concurrent staining of DNA and mitotic markers. Bio-Protocol 2017, 7, e2517. [Google Scholar] [CrossRef]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Trevino, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 1–11. [Google Scholar] [CrossRef]
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D.; et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019, 37, 773–782. [Google Scholar] [CrossRef]
- Mayakonda, A.; Lin, D.-C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-Seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef]
- Friedman, J.; Hastie, T.; Tibshirani, R. glmnet: Lasso and elastic-net regularized generalized linear models. R Package Version 2009, 1, 1–24. [Google Scholar]
- Kumar, V.; Ramnarayanan, K.; Sundar, R.; Padmanabhan, N.; Srivastava, S.; Koiwa, M.; Yasuda, T.; Koh, V.; Huang, K.K.; Tay, S.T.; et al. Single-Cell Atlas of Lineage States, Tumor Microenvironment, and Subtype-Specific Expression Programs in Gastric Cancer. Cancer Discov. 2022, 12, 670–691. [Google Scholar] [CrossRef]
- Kassambara, A.; Kosinski, M.; Biecek, P.; Fabian, S. Package ‘Survminer’. Drawing Survival Curves Using ‘ggplot2’(R Package Version 03 1). 2017. Available online: https://cran.r-project.org/web/packages/survminer/survminer.pdf (accessed on 11 November 2022).
- Geng, H.; Dong, Z.; Zhang, L.; Yang, C.; Li, T.; Lin, Y.; Ke, S.; Xia, X.; Zhang, Z.; Zhao, G.; et al. An immune signature for risk stratification and therapeutic prediction in helicobacter pylori-infected gastric cancer. Cancers 2022, 14, 3276. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Lin, R.; Liao, X.; Li, D.; Qin, Y. Identification and verification of the molecular mechanisms and prognostic values of the cadherin gene family in gastric cancer. Sci. Rep. 2021, 11, 23674. [Google Scholar] [CrossRef]
- Hu, B.-L.; Xie, M.-Z.; Li, K.-Z.; Li, J.-L.; Gui, Y.-C.; Xu, J.-W. Genome-wide analysis to identify a novel distant metastasis-related gene signature predicting survival in patients with gastric cancer. Biomed. Pharmacother. 2019, 117, 109159. [Google Scholar] [CrossRef]
- Li, Y.; Sun, R.; Zhang, Y.; Yuan, Y.; Miao, Y. A methylation-based mRNA signature predicts survival in patients with gastric cancer. Cancer Cell Int. 2020, 20, 284. [Google Scholar] [CrossRef] [PubMed]
- Șenbabaoğlu, Y.; Michailidis, G.; Li, J.Z. Critical limitations of consensus clustering in class discovery. Sci. Rep. 2014, 4, 6207. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wu, J.; Huang, W.; Weng, S.; Wang, B.; Chen, Y.; Wang, H. Development and validation of a hypoxia-immune-based microenvironment gene signature for risk stratification in gastric cancer. J. Transl. Med. 2020, 18, 201. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ou, Y.; Luo, R.; Wang, J.; Wang, D.; Guan, J.; Li, Y.; Xia, P.; Chen, P.R.; Liu, Y. SAR1B senses leucine levels to regulate mTORC1 signalling. Nature 2021, 596, 281–284. [Google Scholar] [CrossRef]
- Zhang, C.; Cheng, W.; Ren, X.; Wang, Z.; Liu, X.; Li, G.; Han, S.; Jiang, T.; Wu, A. Tumor Purity as an Underlying Key Factor in GliomaTumor Purity in Glioma. Clin. Cancer Res. 2017, 23, 6279–6291. [Google Scholar] [CrossRef]
- Boutilier, A.J.; Elsawa, S.F. Macrophage polarization states in the tumor microenvironment. Int. J. Mol. Sci. 2021, 22, 6995. [Google Scholar] [CrossRef]
- Sarvaria, A.; Madrigal, A.; Saudemont, A. B cell regulation in cancer and anti-tumor immunity. Cell. Mol. Immunol. 2017, 14, 662–674. [Google Scholar] [CrossRef]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Pasche, B.; Zhang, W.; Chen, K. Association of MUC16 Mutation With Tumor Mutation Load and Outcomes in Patients With Gastric Cancer. JAMA Oncol. 2018, 4, 1691–1698. [Google Scholar] [CrossRef]
- Huang, Y.J.; Cao, Z.F.; Wang, J.; Yang, J.; Wei, Y.-H.; Tang, Y.-C.; Cheng, Y.-X.; Zhou, J.; Zhang, Z.X. Why MUC16 mutations lead to a better prognosis: A study based on The Cancer Genome Atlas gastric cancer cohort. World J. Clin. Cases 2021, 9, 4143. [Google Scholar] [CrossRef] [PubMed]
- Myung, J.K.; Cho, H.J.; Park, C.K.; Kim, S.K.; Phi, J.H.; Park, S.H. IDH1 mutation of gliomas with long-term survival analysis. Oncol. Rep. 2012, 28, 1639–1644. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.; Cai, G.; Yu, Q.; Shen, J.; Gu, Z.; Chen, J.; Shi, W.; Shi, J. IDH1 mutation diminishes aggressive phenotype in glioma stem cells. Int. J. Oncol. 2018, 52, 270–278. [Google Scholar] [CrossRef]
- Zhu, Z.; Fu, H.; Wang, S.; Yu, X.; You, Q.; Shi, M.; Dai, C.; Wang, G.; Cha, W.; Wang, W. Whole-exome sequencing identifies prognostic mutational signatures in gastric cancer. Ann. Transl. Med. 2020, 8, 1484. [Google Scholar] [CrossRef]
- Wang, K.; Yuen, S.T.; Xu, J.; Lee, S.P.; Yan, H.H.N.; Shi, S.T.; Siu, H.C.; Deng, S.; Chu, K.M.; Law, S.; et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat. Genet. 2014, 46, 573–582. [Google Scholar] [CrossRef]
- Reske, J.J.; Wilson, M.R.; Holladay, J.; Siwicki, R.A.; Skalski, H.; Harkins, S.; Adams, M.; Risinger, J.I.; Hostetter, G.; Lin, K.; et al. Co-existing TP53 and ARID1A mutations promote aggressive endometrial tumorigenesis. PLoS Genet. 2021, 17, e1009986. [Google Scholar] [CrossRef]
- Zang, Z.J.; Cutcutache, I.; Poon, S.L.; Zhang, S.L.; McPherson, J.R.; Tao, J.; Rajasegaran, V.; Heng, H.L.; Deng, N.; Gan, A.; et al. Exome sequencing of gastric adenocarcinoma identifies recurrent somatic mutations in cell adhesion and chromatin remodeling genes. Nat. Genet. 2012, 44, 570–574. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; Li, T.; Zheng, S.; Wang, H. The Hippo pathway as a drug target in gastric cancer. Cancer Lett. 2018, 420, 14–25. [Google Scholar] [CrossRef]
- Magnelli, L.; Schiavone, N.; Staderini, F.; Biagioni, A.; Papucci, L. MAP kinases pathways in gastric cancer. Int. J. Mol. Sci. 2020, 21, 2893. [Google Scholar] [CrossRef] [PubMed]
- Cohen, D.A.; Dabbs, D.J.; Cooper, K.L.; Amin, M.; Jones, T.E.; Jones, M.W.; Chivukula, M.; Trucco, G.A.; Bhargava, R. Interobserver agreement among pathologists for semiquantitative hormone receptor scoring in breast carcinoma. Am. J. Clin. Pathol. 2012, 138, 796–802. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, C.; Huo, Y.; Zhang, Y.; Yin, F.; Chen, T.; Wang, Z.; Gao, J.; Jin, P.; Li, X.; Shi, M.; et al. Development and Experimental Validation of a Novel Prognostic Signature for Gastric Cancer. Cancers 2023, 15, 1610. https://doi.org/10.3390/cancers15051610
Liu C, Huo Y, Zhang Y, Yin F, Chen T, Wang Z, Gao J, Jin P, Li X, Shi M, et al. Development and Experimental Validation of a Novel Prognostic Signature for Gastric Cancer. Cancers. 2023; 15(5):1610. https://doi.org/10.3390/cancers15051610
Chicago/Turabian StyleLiu, Chengcheng, Yuying Huo, Yansong Zhang, Fumei Yin, Taoyu Chen, Zhenyi Wang, Juntao Gao, Peng Jin, Xiangyu Li, Minglei Shi, and et al. 2023. "Development and Experimental Validation of a Novel Prognostic Signature for Gastric Cancer" Cancers 15, no. 5: 1610. https://doi.org/10.3390/cancers15051610
APA StyleLiu, C., Huo, Y., Zhang, Y., Yin, F., Chen, T., Wang, Z., Gao, J., Jin, P., Li, X., Shi, M., & Zhang, M. Q. (2023). Development and Experimental Validation of a Novel Prognostic Signature for Gastric Cancer. Cancers, 15(5), 1610. https://doi.org/10.3390/cancers15051610