
The Clinical Spectrum, Diagnosis, and Management of GATA2 Deficiency

 ,
, {kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
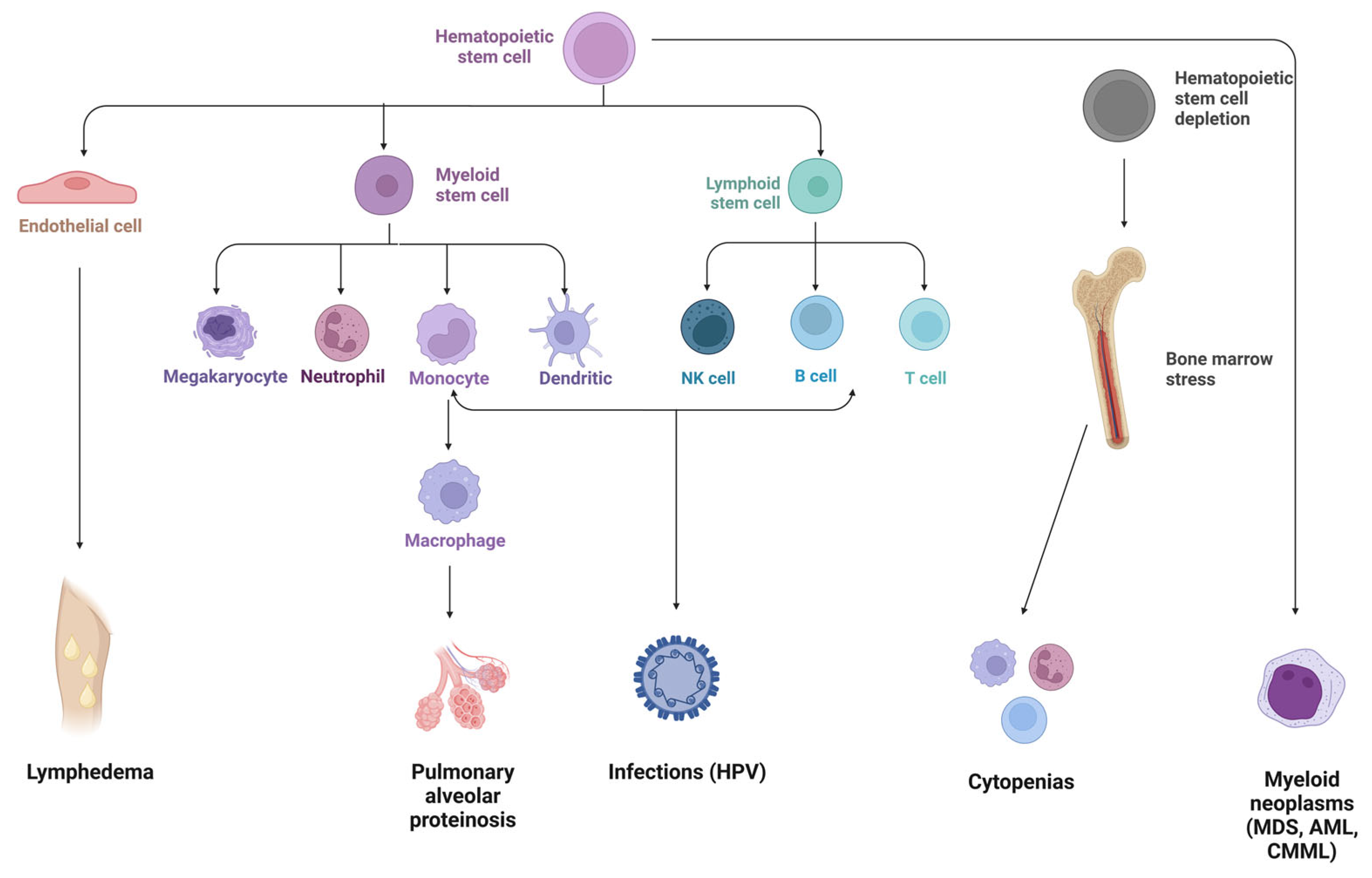
1. Background
2. GATA2 Molecular Insights
3. GATA2 Phenotypic Spectrum
4. Hematological Presentation
4.1. Bone Marrow Failure
4.2. Myeloid Neoplasms
5. Immunodeficiency Disorder
6. Non-Hemato-/Immunologic Manifestations
6.1. Pulmonary Involvement
6.2. Emberger Syndrome: Dysmorphic Features
6.3. Other Dysmorphic Features
7. Management and Surveillance
7.1. Allogeneic-HSCT
7.1.1. Indications for and Timing of allo-HSCT
7.1.2. Conditioning, Graft Source, and Donors
7.1.3. HSCT-Derived Complications
7.2. Antibiotic Prophylaxis, Immunoglobulins, and Vaccination
7.3. Surveillance
7.4. Family Monitoring
7.5. Genetic Counseling
8. Conclusions
9. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Song, W.J.; Sullivan, M.G.; Legare, R.D.; Hutchings, S.; Tan, X.; Kufrin, D.; Ratajczak, J.; Resende, I.C.; Haworth, C.; Hock, R.; et al. Haploinsufficiency of CBFA2 Causes Familial Thrombocytopenia with Propensity to Develop Acute Myelogenous Leukaemia. Nat. Genet. 1999, 23, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Pippucci, T.; Savoia, A.; Perrotta, S.; Pujol-Moix, N.; Noris, P.; Castegnaro, G.; Pecci, A.; Gnan, C.; Punzo, F.; Marconi, C.; et al. Mutations in the 5’ UTR of ANKRD26, the Ankirin Repeat Domain 26 Gene, Cause an Autosomal-Dominant Form of Inherited Thrombocytopenia, THC2. Am. J. Hum. Genet. 2011, 88, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Noris, P.; Perrotta, S.; Seri, M.; Pecci, A.; Gnan, C.; Loffredo, G.; Pujol-Moix, N.; Zecca, M.; Scognamiglio, F.; de Rocco, D.; et al. Mutations in ANKRD26 Are Responsible for a Frequent Form of Inherited Thrombocytopenia: Analysis of 78 Patients from 21 Families. Blood 2011, 117, 6673–6680. [Google Scholar] [CrossRef] [PubMed]
- Noris, P.; Favier, R.; Alessi, M.C.; Geddis, A.E.; Kunishima, S.; Heller, P.G.; Giordano, P.; Niederhoffer, K.Y.; Bussel, J.B.; Podda, G.M.; et al. ANKRD26-Related Thrombocytopenia and Myeloid Malignancies. Blood 2013, 122, 1987–1989. [Google Scholar] [CrossRef]
- Zhang, M.Y.; Churpek, J.E.; Keel, S.B.; Walsh, T.; Lee, M.K.; Loeb, K.R.; Gulsuner, S.; Pritchard, C.C.; Sanchez-Bonilla, M.; Delrow, J.J.; et al. Germline ETV6 Mutations in Familial Thrombocytopenia and Hematologic Malignancy. Nat. Genet. 2015, 47, 180. [Google Scholar] [CrossRef]
- Noetzli, L.; Lo, R.W.; Lee-Sherick, A.B.; Callaghan, M.; Noris, P.; Savoia, A.; Rajpurkar, M.; Jones, K.; Gowan, K.; Balduini, C.L.; et al. Germline Mutations in ETV6 Are Associated with Thrombocytopenia, Red Cell Macrocytosis and Predisposition to Lymphoblastic Leukemia. Nat. Genet. 2015, 47, 535–538. [Google Scholar] [CrossRef]
- Topka, S.; Vijai, J.; Walsh, M.F.; Jacobs, L.; Maria, A.; Villano, D.; Gaddam, P.; Wu, G.; McGee, R.B.; Quinn, E.; et al. Germline ETV6 Mutations Confer Susceptibility to Acute Lymphoblastic Leukemia and Thrombocytopenia. PLoS Genet. 2015, 11, e1005262. [Google Scholar] [CrossRef]
- Smith, M.L.; Cavenagh, J.D.; Lister, T.A.; Fitzgibbon, J. Mutation of CEBPA in Familial Acute Myeloid Leukemia. N. Engl. J. Med. 2004, 351, 2403–2407. [Google Scholar] [CrossRef]
- Polprasert, C.; Schulze, I.; Sekeres, M.A.; Makishima, H.; Przychodzen, B.; Hosono, N.; Singh, J.; Padgett, R.A.; Gu, X.; Phillips, J.G.; et al. Inherited and Somatic Defects in DDX41 in Myeloid Neoplasms. Cancer Cell 2015, 27, 658–670. [Google Scholar] [CrossRef]
- Hahn, C.N.; Chong, C.E.; Carmichael, C.L.; Wilkins, E.J.; Brautigan, P.J.; Li, X.C.; Babic, M.; Lin, M.; Carmagnac, A.; Lee, Y.K.; et al. Heritable GATA2 Mutations Associated with Familial Myelodysplastic Syndrome and Acute Myeloid Leukemia. Nat. Genet. 2011, 43, 1012. [Google Scholar] [CrossRef]
- Harutyunyan, A.S.; Giambruno, R.; Krendl, C.; Stukalov, A.; Klampfl, T.; Berg, T.; Chen, D.; Feenstra, J.D.M.; Jäger, R.; Gisslinger, B.; et al. Germline RBBP6 Mutations in Familial Myeloproliferative Neoplasms. Blood 2016, 127, 362–365. [Google Scholar] [CrossRef]
- Kirwan, M.; Vulliamy, T.; Marrone, A.; Walne, A.J.; Beswick, R.; Hillmen, P.; Kelly, R.; Stewart, A.; Bowen, D.; Schonland, S.O.; et al. Defining the Pathogenic Role of Telomerase Mutations in Myelodysplastic Syndrome and Acute Myeloid Leukemia. Hum. Mutat. 2009, 30, 1567–1573. [Google Scholar] [CrossRef]
- Narumi, S.; Amano, N.; Ishii, T.; Katsumata, N.; Muroya, K.; Adachi, M.; Toyoshima, K.; Tanaka, Y.; Fukuzawa, R.; Miyako, K.; et al. SAMD9 Mutations Cause a Novel Multisystem Disorder, MIRAGE Syndrome, and Are Associated with Loss of Chromosome 7. Nat. Genet. 2016, 48, 792–797. [Google Scholar] [CrossRef]
- Chen, D.H.; Below, J.E.; Shimamura, A.; Keel, S.B.; Matsushita, M.; Wolff, J.; Sul, Y.; Bonkowski, E.; Castella, M.; Taniguchi, T.; et al. Ataxia-Pancytopenia Syndrome Is Caused by Missense Mutations in SAMD9L. Am. J. Hum. Genet. 2016, 98, 1146–1158. [Google Scholar] [CrossRef]
- Tesi, B.; Davidsson, J.; Voss, M.; Rahikkala, E.; Holmes, T.D.; Chiang, S.C.C.; Komulainen-Ebrahim, J.; Gorcenco, S.; Nilsson, A.R.; Ripperger, T.; et al. Gain-of-Function SAMD9L Mutations Cause a Syndrome of Cytopenia, Immunodeficiency, MDS, and Neurological Symptoms. Blood 2017, 129, 2266–2279. [Google Scholar] [CrossRef]
- Akpan, I.J.; Osman, A.E.G.; Drazer, M.W.; Godley, L.A. Hereditary Myelodysplastic Syndrome and Acute Myeloid Leukemia: Diagnosis, Questions, and Controversies. Curr. Hematol. Malig. Rep. 2018, 13, 426–434. [Google Scholar] [CrossRef]
- Huang, K.L.; Mashl, R.J.; Wu, Y.; Ritter, D.I.; Wang, J.; Oh, C.; Paczkowska, M.; Reynolds, S.; Wyczalkowski, M.A.; Oak, N.; et al. Pathogenic Germline Variants in 10,389 Adult Cancers. Cell 2018, 173, 355–370.e14. [Google Scholar] [CrossRef]
- Lu, C.; Xie, M.; Wendl, M.C.; Wang, J.; McLellan, M.D.; Leiserson, M.D.M.; Huang, K.L.; Wyczalkowski, M.A.; Jayasinghe, R.; Banerjee, T.; et al. Patterns and Functional Implications of Rare Germline Variants across 12 Cancer Types. Nat. Commun. 2015, 6, 10086. [Google Scholar] [CrossRef]
- Wartiovaara-Kautto, U.; Hirvonen, E.A.M.; Pitkänen, E.; Heckman, C.; Saarela, J.; Kettunen, K.; Porkka, K.; Kilpivaara, O. Germline Alterations in a Consecutive Series of Acute Myeloid Leukemia. Leukemia 2018, 32, 2282–2285. [Google Scholar] [CrossRef]
- Yang, F.; Long, N.; Anekpuritanang, T.; Bottomly, D.; Savage, J.C.; Lee, T.; Solis-Ruiz, J.; Borate, U.; Wilmot, B.; Tognon, C.; et al. Identification and Prioritization of Myeloid Malignancy Germline Variants in a Large Cohort of Adult Patients with AML. Blood 2022, 139, 1208–1221. [Google Scholar] [CrossRef]
- Drazer, M.W.; Kadri, S.; Sukhanova, M.; Patil, S.A.; West, A.H.; Feurstein, S.; Calderon, D.A.; Jones, M.F.; Weipert, C.M.; Daugherty, C.K.; et al. Prognostic Tumor Sequencing Panels Frequently Identify Germ Line Variants Associated with Hereditary Hematopoietic Malignancies. Blood Adv. 2018, 2, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 Revision to the World Health Organization Classification of Myeloid Neoplasms and Acute Leukemia. Blood. 2016, 127, 2391–2405, Erratum in Blood 2016, 128, 462–463. [Google Scholar] [CrossRef]
- Baliakas, P.; Tesi, B.; Wartiovaara-Kautto, U.; Stray-Pedersen, A.; Friis, L.S.; Dybedal, I.; Hovland, R.; Jahnukainen, K.; Raaschou-Jensen, K.; Ljungman, P.; et al. Nordic Guidelines for Germline Predisposition to Myeloid Neoplasms in Adults: Recommendations for Genetic Diagnosis, Clinical Management and Follow-Up. Hemasphere 2019, 3, e321. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and Management of AML in Adults: 2022 Recommendations from an International Expert Panel on Behalf of the ELN. Blood 2022, 140, 1345–1377. [Google Scholar] [CrossRef] [PubMed]
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th Edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating Morphologic, Clinical, and Genomic Data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef]
- Wlodarski, M.W.; Collin, M.; Horwitz, M.S. GATA2 Deficiency and Related Myeloid Neoplasms. Semin Hematol. 2017, 54, 81–86. [Google Scholar] [CrossRef]
- McGowan-Jordan, J.; Simon, A.; Schmid, M. An International System for Human Cytogenetic Nomenclature; Karger: Basel, Switzerland; New York, NY, USA, 2016. [Google Scholar]
- Hsu, A.P.; Sampaio, E.P.; Khan, J.; Calvo, K.R.; Lemieux, J.E.; Patel, S.Y.; Frucht, D.M.; Vinh, D.C.; Auth, R.D.; Freeman, A.F.; et al. Mutations in GATA2 Are Associated with the Autosomal Dominant and Sporadic Monocytopenia and Mycobacterial Infection (MonoMAC) Syndrome. Blood 2011, 118, 2653–2655. [Google Scholar] [CrossRef]
- Rodrigues, N.P.; Tipping, A.J.; Wang, Z.; Enver, T. GATA-2 Mediated Regulation of Normal Hematopoietic Stem/Progenitor Cell Function, Myelodysplasia and Myeloid Leukemia. Int. J. Biochem. Cell Biol. 2012, 44, 457–460. [Google Scholar] [CrossRef]
- Rodrigues, N.P.; Janzen, V.; Forkert, R.; Dombkowski, D.M.; Boyd, A.S.; Orkin, S.H.; Enver, T.; Vyas, P.; Scadden, D.T. Haploinsufficiency of GATA-2 Perturbs Adult Hematopoietic Stem-Cell Homeostasis. Blood 2005, 106, 477–484. [Google Scholar] [CrossRef]
- Rodrigues, N.P.; Boyd, A.S.; Fugazza, C.; May, G.E.; Guo, Y.P.; Tipping, A.J.; Scadden, D.T.; Vyas, P.; Enver, T. GATA-2 Regulates Granulocyte-Macrophage Progenitor Cell Function. Blood 2008, 112, 4862–4873. [Google Scholar] [CrossRef]
- Nandakumar, S.K.; Johnson, K.; Throm, S.L.; Pestina, T.I.; Neale, G.; Persons, D.A. Low-Level GATA2 Overexpression Promotes Myeloid Progenitor Self-Renewal and Blocks Lymphoid Differentiation in Mice. Exp. Hematol. 2015, 43, 565–577. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, Y.; Chen, B.; Dong, Y.; Zhang, Y.; Mao, B.; Pan, X.; Lai, M.; Chen, Y.; Bian, G.; et al. Overexpression of GATA2 Enhances Development and Maintenance of Human Embryonic Stem Cell-Derived Hematopoietic Stem Cell-like Progenitors. Stem Cell Rep. 2019, 13, 31–47. [Google Scholar] [CrossRef]
- Menendez-Gonzalez, J.B.; Vukovic, M.; Abdelfattah, A.; Saleh, L.; Almotiri, A.; Thomas, L.A.; Agirre-Lizaso, A.; Azevedo, A.; Menezes, A.C.; Tornillo, G.; et al. Gata2 as a Crucial Regulator of Stem Cells in Adult Hematopoiesis and Acute Myeloid Leukemia. Stem Cell Rep. 2019, 13, 291–306. [Google Scholar] [CrossRef]
- Beck, D.; Thoms, J.A.I.; Perera, D.; Schütte, J.; Unnikrishnan, A.; Knezevic, K.; Kinston, S.J.; Wilson, N.K.; O’Brien, T.A.; Göttgens, B.; et al. Genome-Wide Analysis of Transcriptional Regulators in Human HSPCs Reveals a Densely Interconnected Network of Coding and Noncoding Genes. Blood 2013, 122, e12–e22. [Google Scholar] [CrossRef]
- May, G.; Soneji, S.; Tipping, A.J.; Teles, J.; McGowan, S.J.; Wu, M.; Guo, Y.; Fugazza, C.; Brown, J.; Karlsson, G.; et al. Dynamic Analysis of Gene Expression and Genome-Wide Transcription Factor Binding during Lineage Specification of Multipotent Progenitors. Cell Stem Cell 2013, 13, 754–768. [Google Scholar] [CrossRef]
- Homan, C.C.; Venugopal, P.; Arts, P.; Shahrin, N.H.; Feurstein, S.; Rawlings, L.; Lawrence, D.M.; Andrews, J.; King-Smith, S.L.; Harvey, N.L.; et al. GATA2 Deficiency Syndrome: A Decade of Discovery. Hum. Mutat. 2021, 42, 1399–1421. [Google Scholar] [CrossRef]
- Bruzzese, A.; Leardini, D.; Masetti, R.; Strocchio, L.; Girardi, K.; Algeri, M.; del Baldo, G.; Locatelli, F.; Mastronuzzi, A. GATA2 Related Conditions and Predisposition to Pediatric Myelodysplastic Syndromes. Cancers 2020, 12, 2962. [Google Scholar] [CrossRef] [PubMed]
- Collin, M.; Dickinson, R.; Bigley, V. Haematopoietic and Immune Defects Associated with GATA2 Mutation. Br. J. Haematol. 2015, 169, 173–187. [Google Scholar] [CrossRef]
- Spinner, M.A.; Sanchez, L.A.; Hsu, A.P.; Shaw, P.A.; Zerbe, C.S.; Calvo, K.R.; Arthur, D.C.; Gu, W.; Gould, C.M.; Brewer, C.C.; et al. GATA2 Deficiency: A Protean Disorder of Hematopoiesis, Lymphatics, and Immunity. Blood 2014, 123, 809–821. [Google Scholar] [CrossRef]
- Kozyra, E.J.; Pastor, V.B.; Lefkopoulos, S.; Sahoo, S.S.; Busch, H.; Voss, R.K.; Erlacher, M.; Lebrecht, D.; Szvetnik, E.A.; Hirabayashi, S.; et al. Synonymous GATA2 Mutations Result in Selective Loss of Mutated RNA and Are Common in Patients with GATA2 Deficiency. Leukemia 2020, 34, 2673–2687. [Google Scholar] [CrossRef] [PubMed]
- Wehr, C.; Grotius, K.; Casadei, S.; Bleckmann, D.; Bode, S.F.N.; Frye, B.C.; Seidl, M.; Gulsuner, S.; King, M.C.; Percival, M.B.; et al. A Novel Disease-Causing Synonymous Exonic Mutation in GATA2 Affecting RNA Splicing. Blood 2018, 132, 1211–1215. [Google Scholar] [CrossRef] [PubMed]
- Bresnick, E.H.; Jung, M.M.; Katsumura, K.R. Human GATA2 Mutations and Hematologic Disease: How Many Paths to Pathogenesis? Blood Adv. 2020, 4, 4584–4592. [Google Scholar] [CrossRef] [PubMed]
- Hsu, A.P.; Johnson, K.D.; Falcone, E.L.; Sanalkumar, R.; Sanchez, L.; Hickstein, D.D.; Cuellar-Rodriguez, J.; Lemieux, J.E.; Zerbe, C.S.; Bresnick, E.H.; et al. GATA2 Haploinsufficiency Caused by Mutations in a Conserved Intronic Element Leads to MonoMAC Syndrome. Blood 2013, 121, 3830–3837. [Google Scholar] [CrossRef] [PubMed]
- You, X.; Zhou, Y.; Chang, Y.I.; Kong, G.; Ranheim, E.A.; Johnson, K.D.; Soukup, A.A.; Bresnick, E.H.; Zhang, J. Gata2 +9.5 Enhancer Regulates Adult Hematopoietic Stem Cell Self-Renewal and T-Cell Development. Blood Adv. 2022, 6, 1095–1099. [Google Scholar] [CrossRef]
- al Seraihi, A.F.; Rio-Machin, A.; Tawana, K.; Bödör, C.; Wang, J.; Nagano, A.; Heward, J.A.; Iqbal, S.; Best, S.; Lea, N.; et al. GATA2 Monoallelic Expression Underlies Reduced Penetrance in Inherited GATA2-Mutated MDS/AML. Leukemia 2018, 32, 2502–2507. [Google Scholar] [CrossRef]
- Zhang, S.J.; Ma, L.Y.; Huang, Q.H.; Li, G.; Gu, B.W.; Gao, X.D.; Shi, J.Y.; Wang, Y.Y.; Gao, L.; Cai, X.; et al. Gain-of-Function Mutation of GATA-2 in Acute Myeloid Transformation of Chronic Myeloid Leukemia. Proc. Natl. Acad. Sci. USA 2008, 105, 2076–2081. [Google Scholar] [CrossRef]
- Wlodarski, M.W.; Hirabayashi, S.; Pastor, V.; Starý, J.; Hasle, H.; Masetti, R.; Dworzak, M.; Schmugge, M.; van den Heuvel-Eibrink, M.; Ussowicz, M.; et al. Prevalence, Clinical Characteristics, and Prognosis of GATA2-Related Myelodysplastic Syndromes in Children and Adolescents. Blood 2016, 127, 1387–1397. [Google Scholar] [CrossRef]
- West, R.R.; Calvo, K.R.; Embree, L.J.; Wang, W.; Tuschong, L.M.; Bauer, T.R.; Tillo, D.; Lack, J.; Droll, S.; Hsu, A.P.; et al. ASXL1 and STAG2 Are Common Mutations in GATA2 Deficiency Patients with Bone Marrow Disease and Myelodysplastic Syndrome. Blood Adv. 2022, 6, 793–807. [Google Scholar] [CrossRef]
- McReynolds, L.J.; Calvo, K.R.; Holland, S.M. Germline GATA2 Mutation and Bone Marrow Failure. Hematol. Oncol. Clin. N. Am. 2018, 32, 713–728. [Google Scholar] [CrossRef]
- Bödör, C.; Renneville, A.; Smith, M.; Charazac, A.; Iqbal, S.; Étancelin, P.; Cavenagh, J.; Barnett, M.J.; Kramarzová, K.; Krishnan, B.; et al. Germ-Line GATA2 p.THR354MET Mutation in Familial Myelodysplastic Syndrome with Acquired Monosomy 7 and ASXL1 Mutation Demonstrating Rapid Onset and Poor Survival. Haematologica 2012, 97, 890–894. [Google Scholar] [CrossRef]
- McReynolds, L.J.; Zhang, Y.; Yang, Y.; Tang, J.; Mulé, M.; Hsu, A.P.; Townsley, D.M.; West, R.R.; Zhu, J.; Hickstein, D.D.; et al. Rapid Progression to AML in a Patient with Germline GATA2 Mutation and Acquired NRAS Q61K Mutation. Leuk Res. Rep. 2019, 12, 100176. [Google Scholar] [CrossRef]
- Fisher, K.E.; Hsu, A.P.; Williams, C.L.; Sayeed, H.; Merritt, B.Y.; Tarek Elghetany, M.; Holland, S.M.; Bertuch, A.A.; Gramatges, M.M. Somatic Mutations in Children with GATA2-Associated Myelodysplastic Syndrome Who Lack Other Features of GATA2 Deficiency. Blood Adv. 2017, 1, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Pastor Loyola, V.B.; Hirabayashi, S.; Pohl, S.; Kozyra, E.J.; Catala, A.; de Moerloose, B.; Dworzak, M.; Hasle, H.; Masetti, R.; Schmugge, M.; et al. Somatic Genetic and Epigenetic Architecture of Myelodysplastic Syndromes Arising from GATA2 Deficiency. Blood 2015, 126, 299. [Google Scholar] [CrossRef]
- Churpek, J.E.; Pyrtel, K.; Kanchi, K.L.; Shao, J.; Koboldt, D.; Miller, C.A.; Shen, D.; Fulton, R.; O’Laughlin, M.; Fronick, C.; et al. Genomic Analysis of Germ Line and Somatic Variants in Familial Myelodysplasia/Acute Myeloid Leukemia. Blood 2015, 126, 2484–2490. [Google Scholar] [CrossRef]
- Zhang, M.Y.; Keel, S.B.; Walsh, T.; Lee, M.K.; Gulsuner, S.; Watts, A.C.; Pritchard, C.C.; Salipante, S.J.; Jeng, M.R.; Hofmann, I.; et al. Genomic Analysis of Bone Marrow Failure and Myelodysplastic Syndromes Reveals Phenotypic and Diagnostic Complexity. Haematologica 2015, 100, 42–48. [Google Scholar] [CrossRef]
- Keel, S.B.; Scott, A.; Bonilla, M.S.; Ho, P.A.; Gulsuner, S.; Pritchard, C.C.; Abkowitz, J.L.; King, M.C.; Walsh, T.; Shimamura, A. Genetic Features of Myelodysplastic Syndrome and Aplastic Anemia in Pediatric and Young Adult Patients. Haematologica 2016, 101, 1343–1350. [Google Scholar] [CrossRef]
- Wang, X.; Muramatsu, H.; Okuno, Y.; Sakaguchi, H.; Yoshida, K.; Kawashima, N.; Xu, Y.; Shiraishi, Y.; Chiba, K.; Tanaka, H.; et al. GATA2 and Secondary Mutations in Familial Myelodysplastic Syndromes and Pediatric Myeloid Malignancies. Haematologica 2015, 100, e398–e401. [Google Scholar] [CrossRef]
- Kotmayer, L.; Romero-Moya, D.; Marin-Bejar, O.; Kozyra, E.; Català, A.; Bigas, A.; Wlodarski, M.W.; Bödör, C.; Giorgetti, A. GATA2 Deficiency and MDS/AML: Experimental Strategies for Disease Modelling and Future Therapeutic Prospects. Br. J. Haematol. 2022, 199, 482–495. [Google Scholar] [CrossRef]
- Martignoles, J.A.; Delhommeau, F.; Hirsch, P. Genetic Hierarchy of Acute Myeloid Leukemia: From Clonal Hematopoiesis to Molecular Residual Disease. Int. J. Mol. Sci. 2018, 19, 3850. [Google Scholar] [CrossRef]
- Fasan, A.; Eder, C.; Haferlach, C.; Grossmann, V.; Kohlmann, A.; Dicker, F.; Kern, W.; Haferlach, T.; Schnittger, S. GATA2 Mutations Are Frequent in Intermediate-Risk Karyotype AML with Biallelic CEBPA Mutations and Are Associated with Favorable Prognosis. Leukemia 2013, 27, 482–485. [Google Scholar] [CrossRef] [PubMed]
- Greif, P.A.; Dufour, A.; Konstandin, N.P.; Ksienzyk, B.; Zellmeier, E.; Tizazu, B.; Sturm, J.; Benthaus, T.; Herold, T.; Yaghmaie, M.; et al. GATA2 Zinc Finger 1 Mutations Associated with Biallelic CEBPA Mutations Define a Unique Genetic Entity of Acute Myeloid Leukemia. Blood 2012, 120, 395–403. [Google Scholar] [CrossRef]
- Theis, F.; Corbacioglu, A.; Gaidzik, V.I.; Paschka, P.; Weber, D.; Bullinger, L.; Heuser, M.; Ganser, A.; Thol, F.; Schlegelberger, B.; et al. Clinical Impact of GATA2 Mutations in Acute Myeloid Leukemia Patients Harboring CEBPA Mutations: A Study of the AML Study Group. Leukemia 2016, 30, 2248–2250. [Google Scholar] [CrossRef]
- Alfayez, M.; Wang, S.A.; Bannon, S.A.; Kontoyiannis, D.P.; Kornblau, S.M.; Orange, J.S.; Mace, E.M.; DiNardo, C.D. Myeloid Malignancies with Somatic GATA2 Mutations Can Be Associated with an Immunodeficiency Phenotype. Leuk Lymphoma 2019, 60, 2025–2033. [Google Scholar] [CrossRef] [PubMed]
- Sekhar, M.; Pocock, R.; Lowe, D.; Mitchell, C.; Marafioti, T.; Dickinson, R.; Collin, M.; Lipman, M. Can Somatic GATA2 Mutation Mimic Germ Line GATA2 Mutation? Blood Adv. 2018, 2, 904. [Google Scholar] [CrossRef]
- Saettini, F.; Coliva, T.; Vendemini, F.; Moratto, D.; Savoldi, G.; Borlenghi, E.; Masetti, R.; Niemeyer, C.M.; Biondi, A.; Balduzzi, A.; et al. When to Suspect GATA2 Deficiency in Pediatric Patients: From Complete Blood Count to Diagnosis. Pediatr. Hematol. Oncol. 2021, 38, 510–514. [Google Scholar] [CrossRef]
- Ostergaard, P.; Simpson, M.A.; Connell, F.C.; Steward, C.G.; Brice, G.; Woollard, W.J.; Dafou, D.; Kilo, T.; Smithson, S.; Lunt, P.; et al. Mutations in GATA2 Cause Primary Lymphedema Associated with a Predisposition to Acute Myeloid Leukemia (Emberger Syndrome). Nat. Genet. 2011, 43, 929–931. [Google Scholar] [CrossRef]
- Dickinson, R.E.; Griffin, H.; Bigley, V.; Reynard, L.N.; Hussain, R.; Haniffa, M.; Lakey, J.H.; Rahman, T.; Wang, X.N.; McGovern, N.; et al. Exome Sequencing Identifies GATA-2 Mutation as the Cause of Dendritic Cell, Monocyte, B and NK Lymphoid Deficiency. Blood 2011, 118, 2656–2658. [Google Scholar] [CrossRef]
- Oleaga-Quintas, C.; de Oliveira-Júnior, E.B.; Rosain, J.; Rapaport, F.; Deswarte, C.; Guérin, A.; Sajjath, S.M.; Zhou, Y.J.; Marot, S.; Lozano, C.; et al. Inherited GATA2 Deficiency Is Dominant by Haploinsufficiency and Displays Incomplete Clinical Penetrance. J. Clin. Immunol. 2021, 41, 639–657. [Google Scholar] [CrossRef]
- Donadieu, J.; Lamant, M.; Fieschi, C.; de Fontbrune, F.S.; Caye, A.; Ouachee, M.; Beaupain, B.; Bustamante, J.; Poirel, H.A.; Isidor, B.; et al. Natural History of GATA2 Deficiency in a Survey of 79 French and Belgian Patients. Haematologica 2018, 103, 1278–1287. [Google Scholar] [CrossRef]
- Kazenwadel, J.; Secker, G.A.; Liu, Y.J.; Rosenfeld, J.A.; Wildin, R.S.; Cuellar-Rodriguez, J.; Hsu, A.P.; Dyack, S.; Fernandez, C.V.; Chong, C.E.; et al. Loss-of-Function Germline GATA2 Mutations in Patients with MDS/AML or MonoMAC Syndrome and Primary Lymphedema Reveal a Key Role for GATA2 in the Lymphatic Vasculature. Blood 2012, 119, 1283–1291. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Harada, Y.; Huang, G.; Harada, H. Myeloid Neoplasms with Germ Line RUNX1 Mutation. Int. J. Hematol. 2017, 106, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Marconi, C.; Canobbio, I.; Bozzi, V.; Pippucci, T.; Simonetti, G.; Melazzini, F.; Angori, S.; Martinelli, G.; Saglio, G.; Torti, M.; et al. 5’UTR Point Substitutions and N-Terminal Truncating Mutations of ANKRD26 in Acute Myeloid Leukemia. J. Hematol. Oncol. 2017, 10, 18. [Google Scholar] [CrossRef]
- Feurstein, S.; Godley, L.A. Germline ETV6 Mutations and Predisposition to Hematological Malignancies. Int. J. Hematol. 2017, 106, 189–195. [Google Scholar] [CrossRef]
- Fabozzi, F.; Strocchio, L.; Mastronuzzi, A.; Merli, P. GATA2 and Marrow Failure. Best Pract. Res. Clin. Haematol. 2021, 34, 101278. [Google Scholar] [CrossRef]
- Rütsche, C.V.; Haralambieva, E.; Lysenko, V.; Balabanov, S.; Theocharides, A.P.A. A Patient with a Germline GATA2 Mutation and Primary Myelofibrosis. Blood Adv. 2021, 5, 791–795. [Google Scholar] [CrossRef]
- Sahoo, S.S.; Pastor, V.B.; Goodings, C.; Voss, R.K.; Kozyra, E.J.; Szvetnik, A.; Noellke, P.; Dworzak, M.; Starý, J.; Locatelli, F.; et al. Clinical Evolution, Genetic Landscape and Trajectories of Clonal Hematopoiesis in SAMD9/SAMD9L Syndromes. Nat. Med. 2021, 27, 1806–1817. [Google Scholar] [CrossRef]
- Sahoo, S.S.; Kozyra, E.J.; Wlodarski, M.W. Germline Predisposition in Myeloid Neoplasms: Unique Genetic and Clinical Features of GATA2 Deficiency and SAMD9/SAMD9L Syndromes. Best Pract. Res. Clin. Haematol. 2020, 33, 101197. [Google Scholar] [CrossRef]
- Shamriz, O.; Zahalka, N.; Simon, A.J.; Lev, A.; Barel, O.; Mor, N.; Tal, Y.; Segel, M.J.; Somech, R.; Yonath, H.; et al. GATA2 Deficiency in Adult Life Is Characterized by Phenotypic Diversity and Delayed Diagnosis. Front. Immunol. 2022, 13, 886117. [Google Scholar] [CrossRef]
- Mace, E.M.; Hsu, A.P.; Monaco-Shawver, L.; Makedonas, G.; Rosen, J.B.; Dropulic, L.; Cohen, J.I.; Frenkel, E.P.; Bagwell, J.C.; Sullivan, J.L.; et al. Mutations in GATA2 Cause Human NK Cell Deficiency with Specific Loss of the CD56(Bright) Subset. Blood 2013, 121, 2669–2677. [Google Scholar] [CrossRef]
- Maciejewski-Duval, A.; Meuris, F.; Bignon, A.; Aknin, M.-L.; Balabanian, K.; Faivre, L.; Pasquet, M.; Barlogis, V.; Fieschi, C.; Bellanné-Chantelot, C.; et al. Altered Chemotactic Response to CXCL12 in Patients Carrying GATA2 Mutations. J. Leukoc Biol. 2016, 99, 1065–1076. [Google Scholar] [CrossRef] [PubMed]
- Ganapathi, K.A.; Townsley, D.M.; Hsu, A.P.; Arthur, D.C.; Zerbe, C.S.; Cuellar-Rodriguez, J.; Hickstein, D.D.; Rosenzweig, S.D.; Braylan, R.C.; Young, N.S.; et al. GATA2 Deficiency-Associated Bone Marrow Disorder Differs from Idiopathic Aplastic Anemia. Blood 2015, 125, 56–70. [Google Scholar] [CrossRef] [PubMed]
- Nováková, M.; Žaliová, M.; Suková, M.; Wlodarski, M.; Janda, A.; Froňková, E.; Campr, V.; Lejhancová, K.; Zapletal, O.; Pospíšilová, D.; et al. Loss of B Cells and Their Precursors Is the Most Constant Feature of GATA-2 Deficiency in Childhood Myelodysplastic Syndrome. Haematologica 2016, 101, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, R.E.; Milne, P.; Jardine, L.; Zandi, S.; Swierczek, S.I.; McGovern, N.; Cookson, S.; Ferozepurwalla, Z.; Langridge, A.; Pagan, S.; et al. The Evolution of Cellular Deficiency in GATA2 Mutation. Blood 2014, 123, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Vinh, D.C.; Patel, S.Y.; Uzel, G.; Anderson, V.L.; Freeman, A.F.; Olivier, K.N.; Spalding, C.; Hughes, S.; Pittaluga, S.; Raffeld, M.; et al. Autosomal Dominant and Sporadic Monocytopenia with Susceptibility to Mycobacteria, Fungi, Papillomaviruses, and Myelodysplasia. Blood 2010, 115, 1519–1529. [Google Scholar] [CrossRef]
- Chou, J.; Lutskiy, M.; Tsitsikov, E.; Notarangelo, L.D.; Geha, R.S.; Dioun, A. Presence of Hypogammaglobulinemia and Abnormal Antibody Responses in GATA2 Deficiency. J. Allergy Clin. Immunol. 2014, 134, 223–226. [Google Scholar] [CrossRef]
- Toboni, M.D.; Bevis, K.S. Vulvar Cancer as a Result of GATA2 Deficiency, a Rare Genetic Immunodeficiency Syndrome. Obstet. Gynecol. 2018, 132, 1112–1115. [Google Scholar] [CrossRef]
- Amarnani, A.A.; Poladian, K.R.; Marciano, B.E.; Daub, J.R.; Williams, S.G.; Livinski, A.A.; Hsu, A.P.; Palmer, C.L.; Kenney, C.M.; Avila, D.N.; et al. A Panoply of Rheumatological Manifestations in Patients with GATA2 Deficiency. Sci. Rep. 2020, 10, 8305. [Google Scholar] [CrossRef]
- Marciano, B.E.; Olivier, K.N.; Folio, L.R.; Zerbe, C.S.; Hsu, A.P.; Freeman, A.F.; Filie, A.C.; Spinner, M.A.; Sanchez, L.A.; Lovell, J.P.; et al. Pulmonary Manifestations of GATA2 Deficiency. Chest 2021, 160, 1350–1359. [Google Scholar] [CrossRef]
- Raffáč, Š.; Aljubouri, M.A.S.; Gabzdilová, J. Alveolar Proteinosis, Infectious Complications and Monocytopenia Associated with GATA2 Deficiency. Neuro Endocrinol. Lett. 2021, 41, 290–295. [Google Scholar]
- Cuellar-Rodriguez, J.; Gea-Banacloche, J.; Freeman, A.F.; Hsu, A.P.; Zerbe, C.S.; Calvo, K.R.; Wilder, J.; Kurlander, R.; Olivier, K.N.; Holland, S.M.; et al. Successful Allogeneic Hematopoietic Stem Cell Transplantation for GATA2 Deficiency. Blood 2011, 118, 3715–3720. [Google Scholar] [CrossRef] [PubMed]
- Simonis, A.; Fux, M.; Nair, G.; Mueller, N.J.; Haralambieva, E.; Pabst, T.; Pachlopnik Schmid, J.; Schmidt, A.; Schanz, U.; Manz, M.G.; et al. Allogeneic Hematopoietic Cell Transplantation in Patients with GATA2 Deficiency-a Case Report and Comprehensive Review of the Literature. Ann. Hematol. 2018, 97, 1961–1973. [Google Scholar] [CrossRef] [PubMed]
- Kazenwadel, J.; Betterman, K.L.; Chong, C.E.; Stokes, P.H.; Lee, Y.K.; Secker, G.A.; Agalarov, Y.; Demir, C.S.; Lawrence, D.M.; Sutton, D.L.; et al. GATA2 Is Required for Lymphatic Vessel Valve Development and Maintenance. J. Clin. Investig. 2015, 125, 2879–2994. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, H.; Yamaguchi, T.; Sakai, H.; Maruyama, M.; Kawakami, T.; Kawakami, F.; Nishina, S.; Ishikawa, M.; Kosho, T.; Ishida, F. A Novel Germline GATA2 Frameshift Mutation with a Premature Stop Codon in a Family with Congenital Sensory Hearing Loss and Myelodysplastic Syndrome. Int. J. Hematol. 2021, 114, 286–291. [Google Scholar] [CrossRef]
- Saida, S.; Umeda, K.; Yasumi, T.; Matsumoto, A.; Kato, I.; Hiramatsu, H.; Ohara, O.; Heike, T.; Adachi, S. Successful Reduced-Intensity Stem Cell Transplantation for GATA2 Deficiency before Progression of Advanced MDS. Pediatr Transplant. 2016, 20, 333–336. [Google Scholar] [CrossRef]
- Rio-Machin, A.; Vulliamy, T.; Hug, N.; Walne, A.; Tawana, K.; Cardoso, S.; Ellison, A.; Pontikos, N.; Wang, J.; Tummala, H.; et al. The Complex Genetic Landscape of Familial MDS and AML Reveals Pathogenic Germline Variants. Nat. Commun. 2020, 11, 1044. [Google Scholar] [CrossRef]
- Bortnick, R.; Wlodarski, M.; de Haas, V.; de Moerloose, B.; Dworzak, M.; Hasle, H.; Masetti, R.; Starý, J.; Turkiewicz, D.; Ussowicz, M.; et al. Hematopoietic Stem Cell Transplantation in Children and Adolescents with GATA2-Related Myelodysplastic Syndrome. Bone Marrow Transplant. 2021, 56, 2732–2741. [Google Scholar] [CrossRef]
- Jørgensen, S.F.; Buechner, J.; Myhre, A.E.; Galteland, E.; Spetalen, S.; Kulseth, M.A.; Sorte, H.S.; Holla, Ø.L.; Lundman, E.; Alme, C.; et al. A Nationwide Study of GATA2 Deficiency in Norway-the Majority of Patients Have Undergone Allo-HSCT. J. Clin. Immunol. 2022, 42, 404–420. [Google Scholar] [CrossRef]
- Nichols-Vinueza, D.X.; Parta, M.; Shah, N.N.; Cuellar-Rodriguez, J.M.; Bauer, T.R.; West, R.R.; Hsu, A.P.; Calvo, K.R.; Steinberg, S.M.; Notarangelo, L.D.; et al. Donor Source and Post-Transplantation Cyclophosphamide Influence Outcome in Allogeneic Stem Cell Transplantation for GATA2 Deficiency. Br. J. Haematol. 2022, 196, 169–178. [Google Scholar] [CrossRef]
- Hofmann, I.; Avagyan, S.; Stetson, A.; Guo, D.; Al-Sayegh, H.; London, W.B.; Lehmann, L. Comparison of Outcomes of Myeloablative Allogeneic Stem Cell Transplantation for Pediatric Patients with Bone Marrow Failure, Myelodysplastic Syndrome and Acute Myeloid Leukemia with and without Germline GATA2 Mutations. Biol. Blood Marrow Transplant. 2020, 26, 1124–1130. [Google Scholar] [CrossRef]
- Grossman, J.; Cuellar-Rodriguez, J.; Gea-Banacloche, J.; Zerbe, C.; Calvo, K.; Hughes, T.; Hakim, F.; Cole, K.; Parta, M.; Freeman, A.; et al. Nonmyeloablative Allogeneic Hematopoietic Stem Cell Transplantation for GATA2 Deficiency. Biol. Blood Marrow Transplant. 2014, 20, 1940–1948. [Google Scholar] [CrossRef]
- Parta, M.; Shah, N.N.; Baird, K.; Rafei, H.; Calvo, K.R.; Hughes, T.; Cole, K.; Kenyon, M.; Schuver, B.B.; Cuellar-Rodriguez, J.; et al. Allogeneic Hematopoietic Stem Cell Transplantation for GATA2 Deficiency Using a Busulfan-Based Regimen. Biol Blood Marrow Transplant. 2018, 24, 1250–1259. [Google Scholar] [CrossRef]
- Parta, M.; Cole, K.; Avila, D.; Duncan, L.; Baird, K.; Schuver, B.B.; Wilder, J.; Palmer, C.; Daub, J.; Hsu, A.P.; et al. Hematopoietic Cell Transplantation and Outcomes Related to Human Papillomavirus Disease in GATA2 Deficiency. Transplant. Cell Ther. 2021, 27, 435.e1–435.e11. [Google Scholar] [CrossRef]
- Bogaert, D.J.; Laureys, G.; Naesens, L.; Mazure, D.; de Bruyne, M.; Hsu, A.P.; Bordon, V.; Wouters, E.; Tavernier, S.J.; Lambrecht, B.N.; et al. GATA2 Deficiency and Haematopoietic Stem Cell Transplantation: Challenges for the Clinical Practitioner. Br. J. Haematol. 2020, 188, 768–773. [Google Scholar] [CrossRef]
- Khan, N.E.; Rosenberg, P.S.; Alter, B.P. Preemptive Bone Marrow Transplantation and Event-Free Survival in Fanconi Anemia. Biol. Blood Marrow Transplant. 2016, 22, 1888–1892. [Google Scholar] [CrossRef]
- Godley, L.A.; Shimamura, A. Genetic Predisposition to Hematologic Malignancies: Management and Surveillance. Blood 2017, 130, 424–432. [Google Scholar] [CrossRef]
- Hamilton, K.V.; Maese, L.; Marron, J.M.; Pulsipher, M.A.; Porter, C.C.; Nichols, K.E. Stopping Leukemia in Its Tracks: Should Preemptive Hematopoietic Stem-Cell Transplantation Be Offered to Patients at Increased Genetic Risk for Acute Myeloid Leukemia? J. Clin. Oncol. 2019, 37, 2098–2104. [Google Scholar] [CrossRef]
- Connelly, J.A.; Savani, B.N. Finding the Best Haematopoietic Stem Cell Transplant Regimen for GATA2 Haploinsufficiency: How Close Are We? Br. J. Haematol. 2022, 196, 13–14. [Google Scholar] [CrossRef]
- Hickstein, D. HSCT for GATA2 Deficiency across the Pond. Blood 2018, 131, 1272–1274. [Google Scholar] [CrossRef]
- Linnemann, A.K.; O’Geen, H.; Keles, S.; Farnham, P.J.; Bresnick, E.H. Genetic Framework for GATA Factor Function in Vascular Biology. Proc. Natl. Acad. Sci. USA 2011, 108, 13641–13646. [Google Scholar] [CrossRef]
- Johnson, K.D.; Hsu, A.P.; Ryu, M.J.; Wang, J.; Gao, X.; Boyer, M.E.; Liu, Y.; Lee, Y.; Calvo, K.R.; Keles, S.; et al. Cis-Element Mutated in GATA2-Dependent Immunodeficiency Governs Hematopoiesis and Vascular Integrity. J. Clin. Investig. 2012, 122, 3692–3704. [Google Scholar] [CrossRef] [PubMed]
- Portich, J.P.; Condino Neto, A.; Faulhaber, G.A.M. Humoral Deficiency in a Novel GATA2 Mutation: A New Clinical Presentation Successfully Treated with Hematopoietic Stem Cell Transplantation. Pediatr. Blood Cancer 2020, 67, e28374. [Google Scholar] [CrossRef] [PubMed]
- Yüksel, H.; Zafer, E. Gynecologic Manifestations in Emberger Syndrome. Turk. J. Obstet Gynecol. 2021, 18, 65–67. [Google Scholar] [CrossRef] [PubMed]
- Feurstein, S.; Drazer, M.W.; Godley, L.A. Genetic Predisposition to Leukemia and Other Hematologic Malignancies. Semin Oncol. 2016, 43, 598–608. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santiago, M.; Liquori, A.; Such, E.; Zúñiga, Á.; Cervera, J. The Clinical Spectrum, Diagnosis, and Management of GATA2 Deficiency. Cancers 2023, 15, 1590. https://doi.org/10.3390/cancers15051590
Santiago M, Liquori A, Such E, Zúñiga Á, Cervera J. The Clinical Spectrum, Diagnosis, and Management of GATA2 Deficiency. Cancers. 2023; 15(5):1590. https://doi.org/10.3390/cancers15051590
Chicago/Turabian StyleSantiago, Marta, Alessandro Liquori, Esperanza Such, Ángel Zúñiga, and José Cervera. 2023. "The Clinical Spectrum, Diagnosis, and Management of GATA2 Deficiency" Cancers 15, no. 5: 1590. https://doi.org/10.3390/cancers15051590
APA StyleSantiago, M., Liquori, A., Such, E., Zúñiga, Á., & Cervera, J. (2023). The Clinical Spectrum, Diagnosis, and Management of GATA2 Deficiency. Cancers, 15(5), 1590. https://doi.org/10.3390/cancers15051590