
2-Arachidonoylglycerol Modulates CXCL12-Mediated Chemotaxis in Mantle Cell Lymphoma and Chronic Lymphocytic Leukemia

 ,
,
Abstract
Simple Summary
Abstract
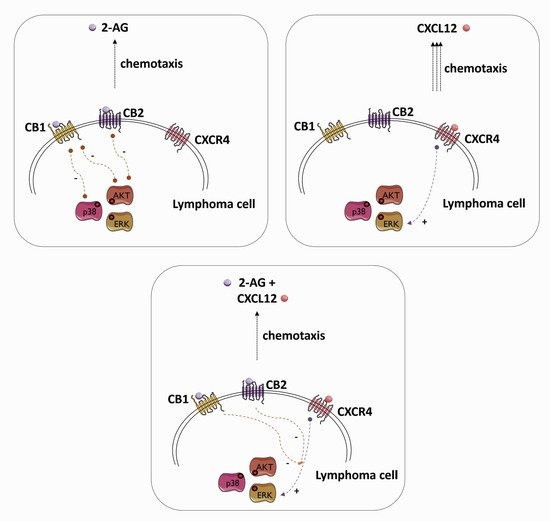
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Primary Samples
2.2. Lymphoma Cell Isolation from Peripheral Blood Samples
2.3. Antibodies and Reagents
2.4. siRNA Introduction by Electroporation
2.5. RNA Isolation and cDNA Synthesis
2.6. qPCR
2.7. Activation of p38, AKT and p44/42 Signaling Pathway and Western Blot
2.8. Flow Cytometric Analysis of CXCR4 Surface Expression
2.9. Immunofluorescence Staining
2.10. Chemotaxis
2.11. Statistics
3. Results
3.1. Chemotaxis of Primary MCL and CLL Cells towards 2-AG
3.2. Cannabinoid Receptors Are Differentially Expressed in MCL and CLL
3.3. Both CB1 and CB2 Receptors Contribute to the 2-AG Mediated Chemotaxis in MCL Cell Line
3.4. Chemotaxis towards 2-AG and CXCL12 in Combination
3.5. Effect of 2-AG on CXCL12-Mediated Chemotaxis in MCL Cell Lines
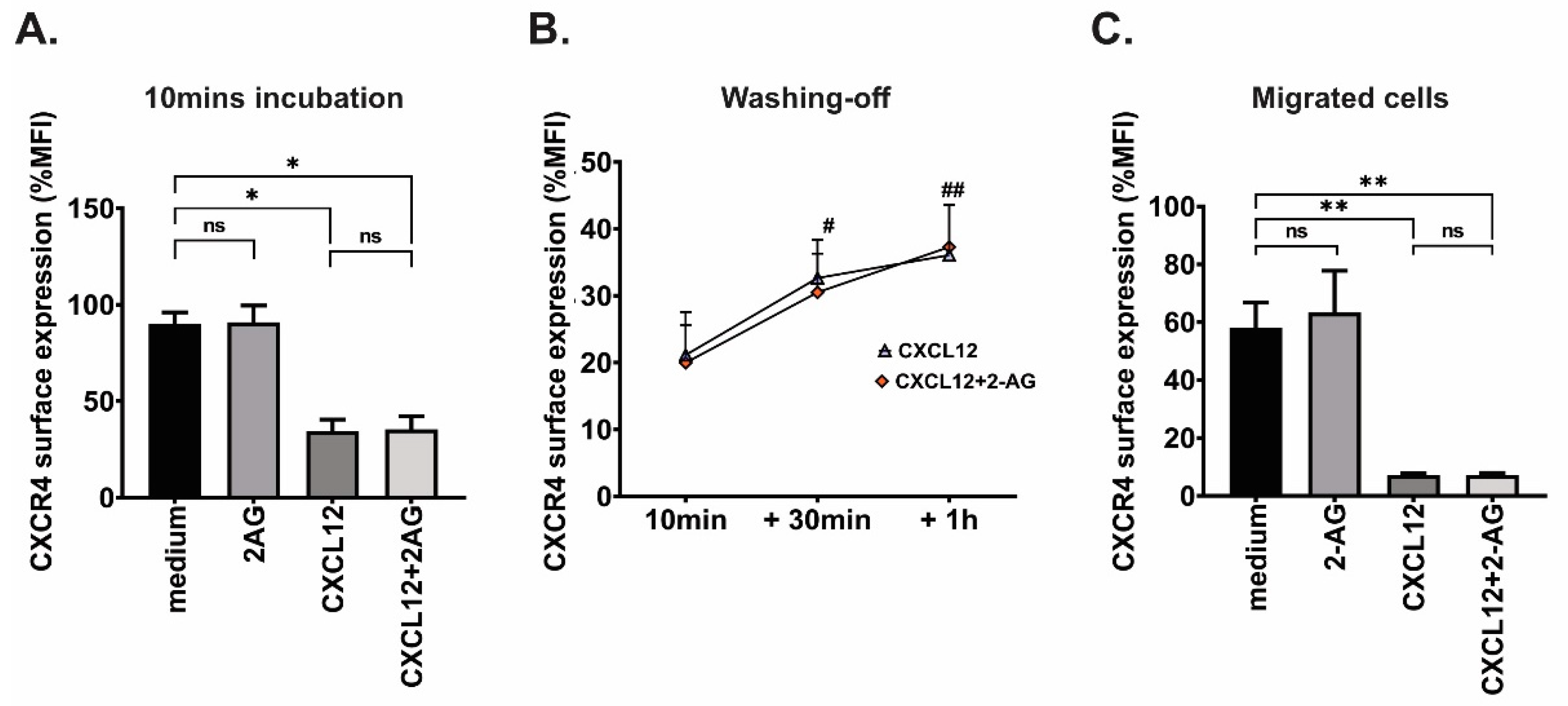
3.6. Effect of 2-AG on CXCR4 Surface Expression
3.7. 2-AG and CXCL12 Combination Impact on the p38, Akt and p44/42 Signaling Pathways
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kurtova, A.V.; Tamayo, A.T.; Ford, R.J.; Burger, J.A. Mantle cell lymphoma cells express high levels of CXCR4, CXCR5, and VLA-4 (CD49d): Importance for interactions with the stromal microenvironment and specific targeting. Blood 2009, 113, 4604–4613. [Google Scholar] [CrossRef] [PubMed]
- Medina, D.J.; Goodell, L.; Glod, J.; Gelinas, C.; Rabson, A.B.; Strair, R.K. Mesenchymal stromal cells protect mantle cell lymphoma cells from spontaneous and drug-induced apoptosis through secretion of B-cell activating factor and activation of the canonical and non-canonical nuclear factor kappaB pathways. Haematologica 2012, 97, 1255–1263. [Google Scholar] [CrossRef] [PubMed]
- Puente, X.S.; Jares, P.; Campo, E. Chronic lymphocytic leukemia and mantle cell lymphoma: Crossroads of genetic and microenvironment interactions. Blood 2018, 131, 2283–2296. [Google Scholar] [CrossRef] [PubMed]
- Shain, K.H.; Tao, J. The B-cell receptor orchestrates environment-mediated lymphoma survival and drug resistance in B-cell malignancies. Oncogene 2014, 33, 4107–4113. [Google Scholar] [CrossRef] [PubMed]
- Bond, D.A.; Woyach, J.A. Targeting BTK in CLL: Beyond Ibrutinib. Curr. Hematol. Malig. Rep. 2019, 14, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Becker, M.; Hobeika, E.; Jumaa, H.; Reth, M.; Maity, P.C. CXCR4 signaling and function require the expression of the IgD-class B-cell antigen receptor. Proc. Natl. Acad. Sci. USA 2017, 114, 5231–5236. [Google Scholar] [CrossRef]
- Haerzschel, A.; Catusse, J.; Hutterer, E.; Paunovic, M.; Zirlik, K.; Eibel, H.; Krenn, P.W.; Hartmann, T.N.; Burger, M. BCR and chemokine responses upon anti-IgM and anti-IgD stimulation in chronic lymphocytic leukaemia. Ann. Hematol. 2016, 95, 1979–1988. [Google Scholar] [CrossRef]
- Montresor, A.; Toffali, L.; Rigo, A.; Ferrarini, I.; Vinante, F.; Laudanna, C. CXCR4- and BCR-triggered integrin activation in B-cell chronic lymphocytic leukemia cells depends on JAK2-activated Bruton’s tyrosine kinase. Oncotarget 2018, 9, 35123–35140. [Google Scholar] [CrossRef]
- Pavlasova, G.; Borsky, M.; Seda, V.; Cerna, K.; Osickova, J.; Doubek, M.; Mayer, J.; Calogero, R.; Trbusek, M.; Pospisilova, S.; et al. Ibrutinib inhibits CD20 upregulation on CLL B cells mediated by the CXCR4/SDF-1 axis. Blood 2016, 128, 1609–1613. [Google Scholar] [CrossRef]
- Saint-Georges, S.; Quettier, M.; Bouyaba, M.; Le Coquil, S.; Lauriente, V.; Guittat, L.; Levy, V.; Ajchenbaum-Cymbalista, F.; Varin-Blank, N.; Le Roy, C.; et al. Protein kinase D-dependent CXCR4 down-regulation upon BCR triggering is linked to lymphadenopathy in chronic lymphocytic leukaemia. Oncotarget 2016, 7, 41031–41046. [Google Scholar] [CrossRef]
- Mohle, R.; Failenschmid, C.; Bautz, F.; Kanz, L. Overexpression of the chemokine receptor CXCR4 in B cell chronic lymphocytic leukemia is associated with increased functional response to stromal cell-derived factor-1 (SDF-1). Leukemia 1999, 13, 1954–1959. [Google Scholar] [CrossRef] [PubMed]
- Arai, J.; Yasukawa, M.; Yakushijin, Y.; Miyazaki, T.; Fujita, S. Stromal cells in lymph nodes attract B-lymphoma cells via production of stromal cell-derived factor-1. Eur. J. Haematol. 2000, 64, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Corcione, A.; Ottonello, L.; Tortolina, G.; Facchetti, P.; Airoldi, I.; Guglielmino, R.; Dadati, P.; Truini, M.; Sozzani, S.; Dallegri, F.; et al. Stromal cell-derived factor-1 as a chemoattractant for follicular center lymphoma B cells. J. Natl. Cancer Inst. 2000, 92, 628–635. [Google Scholar] [CrossRef]
- Okada, T.; Ngo, V.N.; Ekland, E.H.; Forster, R.; Lipp, M.; Littman, D.R.; Cyster, J.G. Chemokine requirements for B cell entry to lymph nodes and Peyer’s patches. J. Exp. Med. 2002, 196, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Peled, A.; Klein, S.; Beider, K.; Burger, J.A.; Abraham, M. Role of CXCL12 and CXCR4 in the pathogenesis of hematological malignancies. Cytokine 2018, 109, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Ek, S.; Hogerkorp, C.M.; Dictor, M.; Ehinger, M.; Borrebaeck, C.A. Mantle cell lymphomas express a distinct genetic signature affecting lymphocyte trafficking and growth regulation as compared with subpopulations of normal human B cells. Cancer Res. 2002, 62, 4398–4405. [Google Scholar]
- Islam, T.C.; Asplund, A.C.; Lindvall, J.M.; Nygren, L.; Liden, J.; Kimby, E.; Christensson, B.; Smith, C.I.; Sander, B. High level of cannabinoid receptor 1, absence of regulator of G protein signalling 13 and differential expression of Cyclin D1 in mantle cell lymphoma. Leukemia 2003, 17, 1880–1890. [Google Scholar] [CrossRef]
- Freund, P.; Porpaczy, E.A.; Le, T.; Gruber, M.; Pausz, C.; Staber, P.; Jager, U.; Vanura, K. Cannabinoid Receptors Are Overexpressed in CLL but of Limited Potential for Therapeutic Exploitation. PLoS ONE 2016, 11, e0156693. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, K.; Wang, X.; Severa, D.; Eriksson, M.; Kimby, E.; Merup, M.; Christensson, B.; Flygare, J.; Sander, B. Expression of cannabinoid receptors type 1 and type 2 in non-Hodgkin lymphoma: Growth inhibition by receptor activation. Int. J. Cancer 2008, 123, 1025–1033. [Google Scholar] [CrossRef]
- Basu, S.; Ray, A.; Dittel, B.N. Cannabinoid receptor 2 is critical for the homing and retention of marginal zone B lineage cells and for efficient T-independent immune responses. J. Immunol. 2011, 187, 5720–5732. [Google Scholar] [CrossRef]
- Muppidi, J.R.; Arnon, T.I.; Bronevetsky, Y.; Veerapen, N.; Tanaka, M.; Besra, G.S.; Cyster, J.G. Cannabinoid receptor 2 positions and retains marginal zone B cells within the splenic marginal zone. J. Exp. Med. 2011, 208, 1941–1948. [Google Scholar] [CrossRef]
- Pereira, J.P.; An, J.; Xu, Y.; Huang, Y.; Cyster, J.G. Cannabinoid receptor 2 mediates the retention of immature B cells in bone marrow sinusoids. Nat. Immunol. 2009, 10, 403–411. [Google Scholar] [CrossRef]
- Chan, L.N.; Chen, Z.; Braas, D.; Lee, J.W.; Xiao, G.; Geng, H.; Cosgun, K.N.; Hurtz, C.; Shojaee, S.; Cazzaniga, V.; et al. Metabolic gatekeeper function of B-lymphoid transcription factors. Nature 2017, 542, 479–483. [Google Scholar] [CrossRef]
- Pisanti, S.; Picardi, P.; D’Alessandro, A.; Laezza, C.; Bifulco, M. The endocannabinoid signaling system in cancer. Trends Pharmacol. Sci. 2013, 34, 273–282. [Google Scholar] [CrossRef]
- Sailler, S.; Schmitz, K.; Jager, E.; Ferreiros, N.; Wicker, S.; Zschiebsch, K.; Pickert, G.; Geisslinger, G.; Walter, C.; Tegeder, I.; et al. Regulation of circulating endocannabinoids associated with cancer and metastases in mice and humans. Oncoscience 2014, 1, 272–282. [Google Scholar] [CrossRef]
- Zhang, J.; Medina-Cleghorn, D.; Bernal-Mizrachi, L.; Bracci, P.M.; Hubbard, A.; Conde, L.; Riby, J.; Nomura, D.K.; Skibola, C.F. The potential relevance of the endocannabinoid, 2-arachidonoylglycerol, in diffuse large B-cell lymphoma. Oncoscience 2016, 3, 31–41. [Google Scholar] [CrossRef]
- Kose, S.; Aerts-Kaya, F.; Kopru, C.Z.; Nemutlu, E.; Kuskonmaz, B.; Karaosmanoglu, B.; Taskiran, E.Z.; Altun, B.; Uckan Cetinkaya, D.; Korkusuz, P. Human bone marrow mesenchymal stem cells secrete endocannabinoids that stimulate in vitro hematopoietic stem cell migration effectively comparable to beta-adrenergic stimulation. Exp. Hematol. 2018, 57, 30–41.e31. [Google Scholar] [CrossRef] [PubMed]
- Wasik, A.M.; Nygren, L.; Almestrand, S.; Zong, F.; Flygare, J.; Wennerholm, S.B.; Saft, L.; Andersson, P.; Kimby, E.; Wahlin, B.E.; et al. Perturbations of the endocannabinoid system in mantle cell lymphoma: Correlations to clinical and pathological features. Oncoscience 2014, 1, 550–557. [Google Scholar] [CrossRef]
- Flygare, J.; Sander, B. The endocannabinoid system in cancer-potential therapeutic target? Semin. Cancer Biol. 2008, 18, 176–189. [Google Scholar] [CrossRef] [PubMed]
- Scott, K.A.; Dalgleish, A.G.; Liu, W.M. Anticancer effects of phytocannabinoids used with chemotherapy in leukaemia cells can be improved by altering the sequence of their administration. Int. J. Oncol. 2017, 51, 369–377. [Google Scholar] [CrossRef]
- Melén, C.M.; Merrien, M.; Wasik, A.M.; Panagiotidis, G.; Beck, O.; Sonnevi, K.; Junlén, H.R.; Christensson, B.; Sander, B.; Wahlin, B.E. Clinical effects of a single dose of cannabinoids to patients with chronic lymphocytic leukemia. Leuk. Lymphoma 2022, 63, 1387–1397. [Google Scholar] [CrossRef] [PubMed]
- Clot, G.; Jares, P.; Giné, E.; Navarro, A.; Royo, C.; Pinyol, M.; Martín-Garcia, D.; Demajo, S.; Espinet, B.; Salar, A.; et al. A gene signature that distinguishes conventional and leukemic nonnodal mantle cell lymphoma helps predict outcome. Blood 2018, 132, 413–422. [Google Scholar] [CrossRef]
- Holmes, K.; Williams, C.M.; Chapman, E.A.; Cross, M.J. Detection of siRNA induced mRNA silencing by RT-qPCR: Considerations for experimental design. BMC Res. Notes 2010, 3, 53. [Google Scholar] [CrossRef]
- Fanelli, F.; Di Lallo, V.D.; Belluomo, I.; De Iasio, R.; Baccini, M.; Casadio, E.; Gasparini, D.I.; Colavita, M.; Gambineri, A.; Grossi, G.; et al. Estimation of reference intervals of five endocannabinoids and endocannabinoid related compounds in human plasma by two dimensional-LC/MS/MS. J. Lipid Res. 2012, 53, 481–493. [Google Scholar] [CrossRef]
- Wasik, A.M.; Almestrand, S.; Wang, X.; Hultenby, K.; Dackland, A.L.; Andersson, P.; Kimby, E.; Christensson, B.; Sander, B. WIN55,212-2 induces cytoplasmic vacuolation in apoptosis-resistant MCL cells. Cell Death Dis. 2011, 2, e225. [Google Scholar] [CrossRef] [PubMed]
- Wasik, A.M.; Sander, B. Cannabinoid receptors in mantle cell lymphoma. Cell Cycle 2015, 14, 291–292. [Google Scholar] [CrossRef]
- Coke, C.J.; Scarlett, K.A.; Chetram, M.A.; Jones, K.J.; Sandifer, B.J.; Davis, A.S.; Marcus, A.I.; Hinton, C.V. Simultaneous Activation of Induced Heterodimerization between CXCR4 Chemokine Receptor and Cannabinoid Receptor 2 (CB2) Reveals a Mechanism for Regulation of Tumor Progression. J. Biol. Chem. 2016, 291, 9991–10005. [Google Scholar] [CrossRef]
- Freret, M.; Gouel, F.; Buquet, C.; Legrand, E.; Vannier, J.P.; Vasse, M.; Dubus, I. Rac-1 GTPase controls the capacity of human leukaemic lymphoblasts to migrate on fibronectin in response to SDF-1α (CXCL12). Leuk. Res. 2011, 35, 971–973. [Google Scholar] [CrossRef]
- Haribabu, B.; Richardson, R.M.; Fisher, I.; Sozzani, S.; Peiper, S.C.; Horuk, R.; Ali, H.; Snyderman, R. Regulation of human chemokine receptors CXCR4. Role of phosphorylation in desensitization and internalization. J. Biol. Chem. 1997, 272, 28726–28731. [Google Scholar] [CrossRef]
- Messmer, D.; Fecteau, J.F.; O’Hayre, M.; Bharati, I.S.; Handel, T.M.; Kipps, T.J. Chronic lymphocytic leukemia cells receive RAF-dependent survival signals in response to CXCL12 that are sensitive to inhibition by sorafenib. Blood 2011, 117, 882–889. [Google Scholar] [CrossRef]
- Chatterjee, S.; Behnam Azad, B.; Nimmagadda, S. The intricate role of CXCR4 in cancer. Adv. Cancer Res. 2014, 124, 31–82. [Google Scholar] [CrossRef] [PubMed]
- Maccarrone, M.; Bab, I.; Bíró, T.; Cabral, G.A.; Dey, S.K.; Di Marzo, V.; Konje, J.C.; Kunos, G.; Mechoulam, R.; Pacher, P.; et al. Endocannabinoid signaling at the periphery: 50 years after THC. Trends Pharmacol. Sci. 2015, 36, 277–296. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, S.; Muramatsu, M.; Gokoh, M.; Oka, S.; Waku, K.; Sugiura, T. Endogenous cannabinoid receptor ligand induces the migration of human natural killer cells. J. Biochem. 2005, 137, 217–223. [Google Scholar] [CrossRef]
- Kishimoto, S.; Gokoh, M.; Oka, S.; Muramatsu, M.; Kajiwara, T.; Waku, K.; Sugiura, T. 2-arachidonoylglycerol induces the migration of HL-60 cells differentiated into macrophage-like cells and human peripheral blood monocytes through the cannabinoid CB2 receptor-dependent mechanism. J. Biol. Chem. 2003, 278, 24469–24475. [Google Scholar] [CrossRef]
- Kishimoto, S.; Oka, S.; Gokoh, M.; Sugiura, T. Chemotaxis of human peripheral blood eosinophils to 2-arachidonoylglycerol: Comparison with other eosinophil chemoattractants. Int. Arch. Allergy Immunol. 2006, 140, 3–7. [Google Scholar] [CrossRef]
- Rossi, F.; Bernardo, M.E.; Bellini, G.; Luongo, L.; Conforti, A.; Manzo, I.; Guida, F.; Cristino, L.; Imperatore, R.; Petrosino, S.; et al. The cannabinoid receptor type 2 as mediator of mesenchymal stromal cell immunosuppressive properties. PLoS ONE 2013, 8, e80022. [Google Scholar] [CrossRef]
- Gasperi, V.; Evangelista, D.; Chiurchiu, V.; Florenzano, F.; Savini, I.; Oddi, S.; Avigliano, L.; Catani, M.V.; Maccarrone, M. 2-Arachidonoylglycerol modulates human endothelial cell/leukocyte interactions by controlling selectin expression through CB1 and CB2 receptors. Int. J. Biochem. Cell Biol. 2014, 51, 79–88. [Google Scholar] [CrossRef]
- Maestroni, G.J. The endogenous cannabinoid 2-arachidonoyl glycerol as in vivo chemoattractant for dendritic cells and adjuvant for Th1 response to a soluble protein. FASEB J. 2004, 18, 1914–1916. [Google Scholar] [CrossRef]
- Tanikawa, T.; Kurohane, K.; Imai, Y. Induction of preferential chemotaxis of unstimulated B-lymphocytes by 2-arachidonoylglycerol in immunized mice. Microbiol. Immunol. 2007, 51, 1013–1019. [Google Scholar] [CrossRef]
- Ten Hacken, E.; Burger, J.A. Microenvironment interactions and B-cell receptor signaling in Chronic Lymphocytic Leukemia: Implications for disease pathogenesis and treatment. Biochim. Biophys. Acta 2016, 1863, 401–413. [Google Scholar] [CrossRef]
- Xie, J.; Xiao, D.; Xu, Y.; Zhao, J.; Jiang, L.; Hu, X.; Zhang, Y.; Yu, L. Up-regulation of immunomodulatory effects of mouse bone-marrow derived mesenchymal stem cells by tetrahydrocannabinol pre-treatment involving cannabinoid receptor CB2. Oncotarget 2016, 7, 6436–6447. [Google Scholar] [CrossRef]
- Jorda, M.A.; Verbakel, S.E.; Valk, P.J.; Vankan-Berkhoudt, Y.V.; Maccarrone, M.; Finazzi-Agro, A.; Lowenberg, B.; Delwel, R. Hematopoietic cells expressing the peripheral cannabinoid receptor migrate in response to the endocannabinoid 2-arachidonoylglycerol. Blood 2002, 99, 2786–2793. [Google Scholar] [CrossRef]
- Larose, M.C.; Turcotte, C.; Chouinard, F.; Ferland, C.; Martin, C.; Provost, V.; Laviolette, M.; Flamand, N. Mechanisms of human eosinophil migration induced by the combination of IL-5 and the endocannabinoid 2-arachidonoyl-glycerol. J. Allergy Clin. Immunol. 2014, 133, 1480–1482.e3. [Google Scholar] [CrossRef]
- Rayman, N.; Lam, K.H.; Laman, J.D.; Simons, P.J.; Löwenberg, B.; Sonneveld, P.; Delwel, R. Distinct expression profiles of the peripheral cannabinoid receptor in lymphoid tissues depending on receptor activation status. J. Immunol. 2004, 172, 2111–2117. [Google Scholar] [CrossRef] [PubMed]
- Coopman, K.; Smith, L.D.; Wright, K.L.; Ward, S.G. Temporal variation in CB2R levels following T lymphocyte activation: Evidence that cannabinoids modulate CXCL12-induced chemotaxis. Int. Immunopharmacol. 2007, 7, 360–371. [Google Scholar] [CrossRef] [PubMed]
- Shoemaker, J.L.; Ruckle, M.B.; Mayeux, P.R.; Prather, P.L. Agonist-directed trafficking of response by endocannabinoids acting at CB2 receptors. J. Pharmacol. Exp. Ther. 2005, 315, 828–838. [Google Scholar] [CrossRef] [PubMed]
- Walter, L.; Franklin, A.; Witting, A.; Wade, C.; Xie, Y.; Kunos, G.; Mackie, K.; Stella, N. Nonpsychotropic cannabinoid receptors regulate microglial cell migration. J. Neurosci. 2003, 23, 1398–1405. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhang, H.; Geng, B.; Xie, Q.; Li, W.; Deng, Y.; Shi, W.; Pan, Y.; Kang, X.; Wang, J. 2-arachidonyl glycerol modulates astrocytic glutamine synthetase via p38 and ERK1/2 pathways. J. Neuroinflamm. 2018, 15, 220. [Google Scholar] [CrossRef]
- Punzo, F.; Argenziano, M.; Tortora, C.; Di Paola, A.; Mutarelli, M.; Pota, E.; Di Martino, M.; Di Pinto, D.; Marrapodi, M.M.; Roberti, D.; et al. Effect of CB2 Stimulation on Gene Expression in Pediatric B-Acute Lymphoblastic Leukemia: New Possible Targets. Int. J. Mol. Sci. 2022, 23, 8651. [Google Scholar] [CrossRef]
- Callén, L.; Moreno, E.; Barroso-Chinea, P.; Moreno-Delgado, D.; Cortés, A.; Mallol, J.; Casadó, V.; Lanciego, J.L.; Franco, R.; Lluis, C.; et al. Cannabinoid receptors CB1 and CB2 form functional heteromers in brain. J. Biol. Chem. 2012, 287, 20851–20865. [Google Scholar] [CrossRef]
- Kleyer, J.; Nicolussi, S.; Taylor, P.; Simonelli, D.; Furger, E.; Anderle, P.; Gertsch, J. Cannabinoid receptor trafficking in peripheral cells is dynamically regulated by a binary biochemical switch. Biochem. Pharmacol. 2012, 83, 1393–1412. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Age (y) | Sex (F Female; M Male) | Diagnosis | Lymphocyte Count (×109/L) | Other Malignancy/Immune Disease | Treated (1 Yes, 0 No) | Time to Treatment from Diagnosis (Months) |
|---|---|---|---|---|---|---|---|
| 1 | 72 | M | MCL | 20 | - | 0 | - |
| 2 | 64 | M | CLL | 19.5 | - | 0 | - |
| 3 | 69 | M | CLL | 66.4 | - | 0 | - |
| 4 | 75 | F | CLL | 42.6 | - | 0 | - |
| 6 | 48 | M | CLL | 144.9 | - | 0 | - |
| 10 | 73 | F | CLL | 21.3 | - | 0 | - |
| 11 | 77 | M | CLL | 27.7 | - | 0 | - |
| 12 | 56 | M | CLL | 129.9 | - | 0 | - |
| 13 | 73 | M | CLL | 103.8 | - | 1 | 1 |
| 14 | 77 | F | CLL | 66 | - | 0 | - |
| 15 | 81 | F | CLL | 22.5 | - | 1 | 84 |
| 16 | 47 | F | CLL | 11 | - | 0 | - |
| 17 | 69 | M | CLL | 17.3 | - | 0 | - |
| 18 | 72 | M | MCL | 50.5 | - | 0 | - |
| 19 | 55 | M | CLL | 54 | - | 0 | - |
| 20 | 81 | M | CLL | 93.3 | - | 1 | 7 |
| 21 | 80 | M | CLL | 11.1 | 1 | 0 | - |
| 22 | 38 | F | CLL | 216.6 | - | 0 | - |
| 25 | 87 | M | MCL | 60.6 | polymyalgia rheumatica | 0 | - |
| 26 | 71 | M | CLL | 10.4 | - | 0 | - |
| 27 | 59 | F | CLL | 84 | - | 0 | - |
| 30 | 78 | M | MCL | not known | - | not known | - |
| 31 | 77 | F | CLL | 141 | - | 0 | - |
| 32 | 81 | F | MCL | 89 | - | 0 | - |
| 40 | 58 | F | CLL | 100 | - | 0 | - |
| 41 | 44 | M | CLL | 335 | - | 0 | - |
| 42 | 78 | F | CLL | 114 | - | 1 | 66 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merrien, M.; Wasik, A.M.; Melén, C.M.; Morsy, M.H.A.; Sonnevi, K.; Junlén, H.-R.; Christensson, B.; Wahlin, B.E.; Sander, B. 2-Arachidonoylglycerol Modulates CXCL12-Mediated Chemotaxis in Mantle Cell Lymphoma and Chronic Lymphocytic Leukemia. Cancers 2023, 15, 1585. https://doi.org/10.3390/cancers15051585
Merrien M, Wasik AM, Melén CM, Morsy MHA, Sonnevi K, Junlén H-R, Christensson B, Wahlin BE, Sander B. 2-Arachidonoylglycerol Modulates CXCL12-Mediated Chemotaxis in Mantle Cell Lymphoma and Chronic Lymphocytic Leukemia. Cancers. 2023; 15(5):1585. https://doi.org/10.3390/cancers15051585
Chicago/Turabian StyleMerrien, Magali, Agata M. Wasik, Christopher M. Melén, Mohammad Hamdy Abdelrazak Morsy, Kristina Sonnevi, Henna-Riikka Junlén, Birger Christensson, Björn E. Wahlin, and Birgitta Sander. 2023. "2-Arachidonoylglycerol Modulates CXCL12-Mediated Chemotaxis in Mantle Cell Lymphoma and Chronic Lymphocytic Leukemia" Cancers 15, no. 5: 1585. https://doi.org/10.3390/cancers15051585
APA StyleMerrien, M., Wasik, A. M., Melén, C. M., Morsy, M. H. A., Sonnevi, K., Junlén, H.-R., Christensson, B., Wahlin, B. E., & Sander, B. (2023). 2-Arachidonoylglycerol Modulates CXCL12-Mediated Chemotaxis in Mantle Cell Lymphoma and Chronic Lymphocytic Leukemia. Cancers, 15(5), 1585. https://doi.org/10.3390/cancers15051585