
Evolution of the Targeted Therapy Landscape for Cholangiocarcinoma: Is Cholangiocarcinoma the ‘NSCLC’ of GI Oncology?



Abstract
Simple Summary
Abstract
1. Introduction
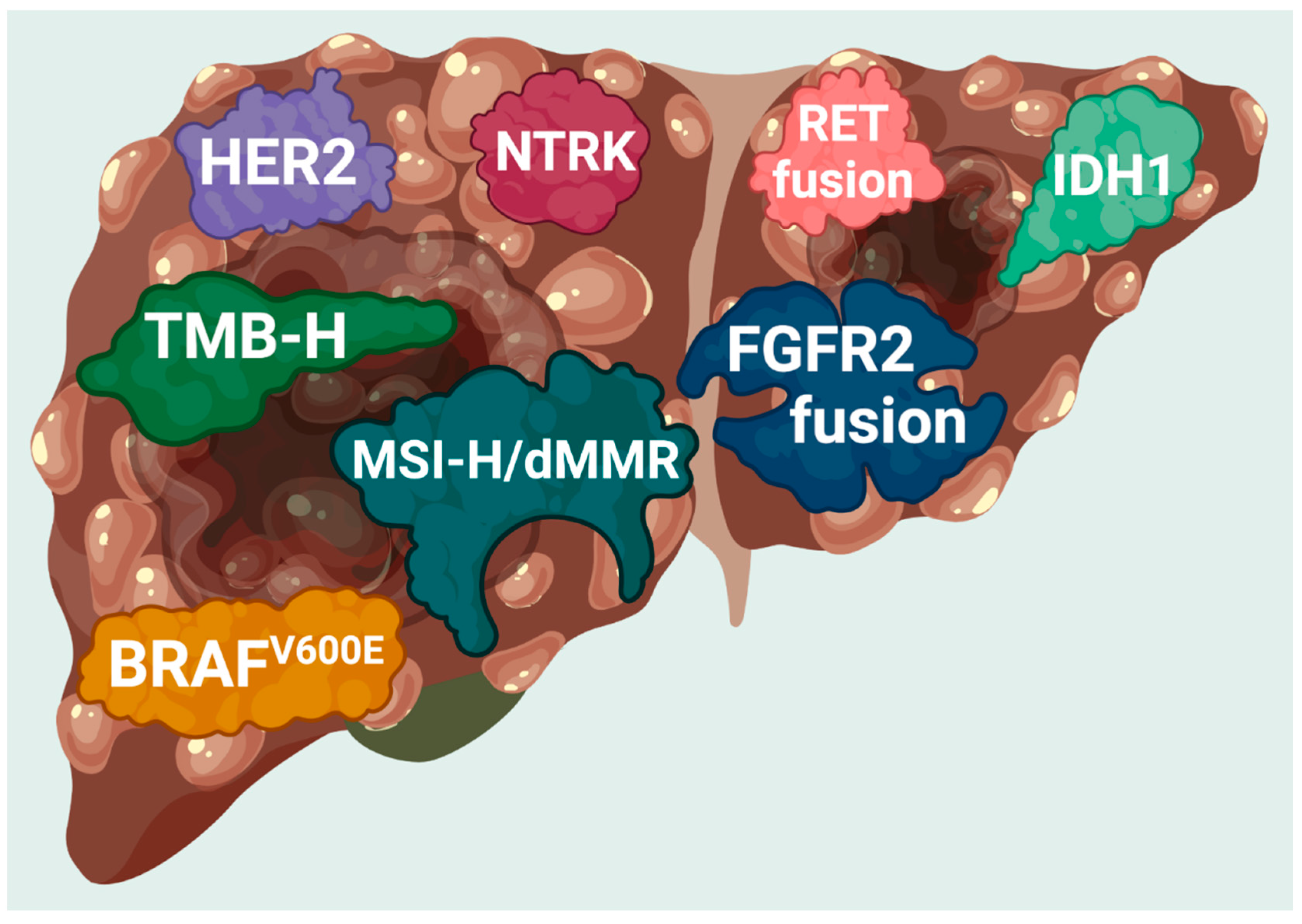
2. The Options for Molecular Targeted Therapy
2.1. Neurotrophic Tyrosine Receptor Kinase (NTRK) Gene Fusion-Positive Cholangiocarcinoma (CCA)
2.2. Cholangiocarcinoma (CCA) with BRAFV600E Mutations
Other BRAF Inhibitors Currently Undergoing Clinical Trials
2.3. Cholangiocarcinoma (CCA) with Fibroblast Growth Factor Receptor 2 (FGFR2) Gene Fusion or Rearrangement
2.4. High Tumor Mutational Burden (TMB-H) as a Predictive and Prognostic Biomarker
2.5. High Microsatellite Instability and Mismatch Repair Deficient (MSI-H/dMMR) Cholangiocarcinoma (CCA)
2.6. Isocitrate Dehydrogenase Isoenzyme (IDH1) Gene Mutations in Cholangiocarcinoma (CCA)
2.7. Erb-B2 Receptor Tyrosine Kinase 2 (ERBB2)/Human Epidermal Growth Factor Receptor 2 (HER2)-Positive Cholangiocarcinoma (CCA)
2.8. RET Gene Fusion-Positive Cholangiocarcinoma
3. Liquid Biopsy for Assessment of Circulating Tumor DNA (ctDNA) and Cholangiocarcinoma (CCA)
4. Conclusions
5. Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Hainsworth, J.D.; Meric-Bernstam, F.; Swanton, C.; Hurwitz, H.; Spigel, D.R.; Sweeney, C.; Burris, H.A.; Bose, R.; Yoo, B.; Stein, A.; et al. Targeted Therapy for Advanced Solid Tumors on the Basis of Molecular Profiles: Results from MyPathway, an Open-Label, Phase IIa Multiple Basket Study. J. Clin. Oncol. 2018, 36, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Tsimberidou, A.-M.; Iskander, N.G.; Hong, D.S.; Wheler, J.J.; Falchook, G.S.; Fu, S.; Piha-Paul, S.; Naing, A.; Janku, F.; Luthra, R.; et al. Personalized Medicine in a Phase I Clinical Trials Program: The MD Anderson Cancer Center Initiative. Clin. Cancer Res. 2012, 18, 6373–6383. [Google Scholar] [CrossRef] [PubMed]
- Janku, F.; Stewart, D.J.; Kurzrock, R. Targeted therapy in non-small-cell lung cancer--is it becoming a reality? Nat. Rev. Clin. Oncol. 2010, 7, 401–414. Available online: https://pubmed.ncbi.nlm.nih.gov/20551945/ (accessed on 1 December 2022). [CrossRef]
- Huang, R.S.P.; Severson, E.; Haberberger, J.; Duncan, D.L.; Hemmerich, A.; Edgerly, C.; Ferguson, N.L.; Frampton, G.; Owens, C.; Williams, E.; et al. Landscape of Biomarkers in Non-small Cell Lung Cancer Using Comprehensive Genomic Profiling and PD-L1 Immunohistochemistry. Pathol. Oncol. Res. 2021, 27, 592997. [Google Scholar] [CrossRef]
- Gower, A.; Wang, Y.; Giaccone, G. Oncogenic drivers, targeted therapies, and acquired resistance in non-small-cell lung cancer. J. Mol. Med. 2014, 92, 697–707. [Google Scholar] [CrossRef]
- Sepulveda, A.R.; Hamilton, S.R.; Allegra, C.J.; Grody, W.; Cushman-Vokoun, A.M.; Funkhouser, W.K.; Kopetz, S.E.; Lieu, C.; Lindor, N.M.; Minsky, B.D.; et al. Molecular Biomarkers for the Evaluation of Colorectal Cancer: Guideline from the American Society for Clinical Pathology, College of American Pathologists, Association for Molecular Pathology, and American Society of Clinical Oncology. J. Mol. Diagn. 2017, 19, 187. Available online: /pmc/articles/PMC5971222/ (accessed on 21 August 2022). [CrossRef]
- Dobashi, Y.; Goto, A.; Kimura, M.; Nakano, T. Molecularly Targeted Therapy: Past, Present and Future. Chemother. Open Access 2012, 1, 1–12. Available online: https://www.longdom.org/open-access/molecularly-targeted-therapy-past-present-and-future-19552.html (accessed on 9 September 2022).
- Majeed, U.; Manochakian, R.; Zhao, Y.; Lou, Y. Targeted therapy in advanced non-small cell lung cancer: Current advances and future trends. J. Hematol. Oncol. 2021, 14, 1–20. [Google Scholar] [CrossRef]
- De Mello, R.A.; Neves, N.M.; Tadokoro, H.; Amaral, G.A.; Castelo-Branco, P.; de Zia, V.A. New Target Therapies in Advanced Non-Small Cell Lung Cancer: A Review of the Literature and Future Perspectives. J. Clin. Med. 2020, 9, 3543. Available online: https://pubmed.ncbi.nlm.nih.gov/33153004/ (accessed on 9 September 2022). [CrossRef]
- D’Angelo, A.; Sobhani, N.; Chapman, R.; Bagby, S.; Bortoletti, C.; Traversini, M.; Ferrari, K.; Voltolini, L.; Darlow, J.; Roviello, G. Focus on ROS1-Positive Non-Small Cell Lung Cancer (NSCLC): Crizotinib, Resistance Mechanisms and the Newer Generation of Targeted Therapies. Cancers 2020, 12, 3293. [Google Scholar] [CrossRef]
- Lee, Y.T.; Tan, Y.J.; Oon, C.E. Molecular targeted therapy: Treating cancer with specificity. Eur. J. Pharmacol. 2018, 834, 188–196. [Google Scholar] [CrossRef]
- Simile, M.M.; Bagella, P.; Vidili, G.; Spanu, A.; Manetti, R.; Seddaiu, M.A.; Babudieri, S.; Madeddu, G.; Serra, P.A.; Altana, M.; et al. Targeted Therapies in Cholangiocarcinoma: Emerging Evidence from Clinical Trials. Medicina 2019, 55, 42. [Google Scholar] [CrossRef]
- Patel, T. Increasing incidence and mortality of primary intrahepatic cholangiocarcinoma in the United States. Hepatology 2001, 33, 1353–1357. [Google Scholar] [CrossRef]
- Everhart, J.E.; Ruhl, C.E. Burden of Digestive Diseases in the United States Part III: Liver, Biliary Tract, and Pancreas. Gastroenterology 2009, 136, 1134–1144. [Google Scholar] [CrossRef]
- Banales, J.M.; Cardinale, V.; Carpino, G.; Marzioni, M.; Andersen, J.B.; Invernizzi, P.; Lind, G.E.; Folseraas, T.; Forbes, S.J.; Fouassier, L.; et al. Expert consensus document: Cholangio-carcinoma: Current knowledge and future perspectives consensus statement from the European Network for the Study of Cholangiocarcinoma (ENS-CCA). Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 261–280. [Google Scholar] [CrossRef]
- Oliveira, D.V.; Zhang, S.; Chen, X.; Calvisi, D.F.; Andersen, J.B. Molecular profiling of intrahepatic cholangiocarcinoma: The search for new therapeutic targets. Expert Rev. Gastroenterol. Hepatol. 2017, 11, 349–356. [Google Scholar] [CrossRef]
- Sia, D.; Tovar, V.; Moeini, A.; Llovet, J.M. Intrahepatic cholangiocarcinoma: Pathogenesis and rationale for molecular therapies. Oncogene 2013, 32, 4861–4870. [Google Scholar] [CrossRef]
- US Food and Drug Administration. FDA Grants Accelerated Approval to Pemigatinib for Cholangiocarcinoma with an FGFR2 Rearrangement or Fusion; US Food and Drug Administration: Silver Spring, MD, USA, 2020; Volume 7, pp. 18–20. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-pemigatinib-cholangiocarcinoma-fgfr2-rearrangement-or-fusion (accessed on 30 September 2022).
- US Food and Drug Administration. FDA Grants Accelerated Approval to Infigratinib for Metastatic Cholangiocarcinoma; US Food and Drug Administration: Silver Spring, MD, USA, 2022. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-infigratinib-metastatic-cholangiocarcinoma (accessed on 22 October 2022).
- FDA. Grants Accelerated Approval to Futibatinib for Cholangiocarcinoma; US Food and Drug Administration: Silver Spring, MD, USA, 2022. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-futibatinib-cholangiocarcinoma (accessed on 1 December 2022).
- Casak, S.J.; Pradhan, S.; Fashoyin-Aje, L.A.; Ren, Y.; Shen, Y.-L.; Xu, Y.; Chow, E.C.Y.; Xiong, Y.; Zirklelbach, J.F.; Liu, J.; et al. FDA Approval Summary: Ivosidenib for the Treatment of Patients with Advanced Unresectable or Metastatic, Chemotherapy Refractory Cholangiocarcinoma with an IDH1 Mutation. Clin. Cancer Res. 2022, 28, 2733–2737. [Google Scholar] [CrossRef]
- Ambrosini, M.; Del Re, M.; Manca, P.; Hendifar, A.; Drilon, A.; Harada, G.; Ree, A.H.; Klempner, S.; Mælandsmo, G.M.; Flatmark, K.; et al. ALK Inhibitors in Patients with ALK Fusion–Positive GI Cancers: An International Data Set and a Molecular Case Series. JCO Precis. Oncol. 2022, 6, e2200015. [Google Scholar] [CrossRef]
- Lowery, M.A.; Ptashkin, R.; Jordan, E.; Berger, M.F.; Zehir, A.; Capanu, M.; Kemeny, N.E.; O’Reilly, E.M.; El-Dika, I.; Jarnagin, W.R.; et al. Comprehensive molecular profiling of intra- and extrahepatic cholangiocarcinomas: Potential targets for intervention. Clin. Cancer Res. 2018, 24, 4154. Available online: /pmc/articles/PMC6642361/ (accessed on 12 January 2023). [CrossRef]
- Javle, M.; Bekaii-Saab, T.; Jain, A.; Wang, Y.; Kelley, R.K.; Wang, K.; Kang, H.C.; Catenacci, D.; Ali, S.; Krishnan, S.; et al. Biliary cancer: Utility of next-generation sequencing for clinical management. Cancer 2016, 122, 3838–3847. [Google Scholar] [CrossRef] [PubMed]
- Montal, R.; Sia, D.; Montironi, C.; Leow, W.Q.; Esteban-Fabró, R.; Pinyol, R.; Torres-Martin, M.; Bassaganyas, L.; Moeini, A.; Peix, J.; et al. Molecular classification and therapeutic targets in extrahepatic cholangiocarcinoma. J. Hepatol. 2020. [Google Scholar] [CrossRef]
- Li, W.; Cui, Y.; Yin, F.; Peng, L.; Liu, X.; Shen, Y.; Guo, Y.; Wen, S.; Shi, J.; Lei, M.; et al. BRAF mutation in Chinese biliary tract cancer patients. J. Clin. Oncol. 2020, 38, e16678. [Google Scholar] [CrossRef]
- Spizzo, G.; Puccini, A.; Xiu, J.; Goldberg, R.M.; Grothey, A.; Shields, A.F.; Arora, S.P.; Khushmann, M.; Salem, M.E.; Battaglin, F.; et al. Molecular profile of BRCA-mutated biliary tract cancers. ESMO Open 2020, 5, e000682. [Google Scholar] [CrossRef]
- Endo, K.; Ashida, K.; Miyake, N.; Terada, T. E-Cadherin Gene Mutations in Human Intrahepatic Cholangiocarcinoma. Available online: https://onlinelibrary.wiley.com/doi/10.1002/1096-9896 (accessed on 12 January 2023).
- Lee, H.; Wang, K.; Johnson, A.; Jones, D.M.; Ali, S.M.; Elvin, J.A.; Yelensky, R.; Lipson, D.; Miller, V.A.; Stephens, P.J.; et al. Comprehensive genomic profiling of extrahepatic cholangio-carcinoma reveals a long tail of therapeutic targets. J. Clin. Pathol. 2016, 69, 403–408. Available online: https://jcp.bmj.com/content/69/5/403 (accessed on 13 January 2023). [CrossRef]
- Xue, L.; Guo, C.; Zhang, K.; Jiang, H.; Pang, F.; Dou, Y.; Liu, X.; Lin, H.; Dong, X.; Zhao, S.; et al. Comprehensive molecular profiling of extrahepatic cholangiocarcinoma in Chinese population and potential targets for clinical practice. HepatoBiliary Surg. Nutr. 2019, 8, 615–622. [Google Scholar] [CrossRef]
- Galdy, S.; Lamarca, A.; McNamara, M.G.; Hubner, R.A.; Cella, C.A.; Fazio, N.; Valle, J.W. HER2/HER3 pathway in biliary tract malignancies; systematic review and meta-analysis: A potential therapeutic target? Cancer Metastasis Rev. 2017, 36, 141–157. [Google Scholar] [CrossRef]
- Kim, H.; Kim, R.; Kim, H.R.; Jo, H.; Kim, H.; Ha, S.Y.; Park, J.O.; Park, Y.S.; Kim, S.T. HER2 Aberrations as a Novel Marker in Advanced Biliary Tract Cancer. Front Oncol. 2022, 12, 834104. [Google Scholar] [CrossRef]
- Silverman, I.M.; Hollebecque, A.; Friboulet, L.; Owens, S.; Newton, R.C.; Zhen, H.; Féliz, L.; Zecchetto, C.; Melisi, D.; Burn, T.C. Clinicogenomic Analysis of FGFR2-Rearranged Cholangiocarcinoma Identifies Correlates of Response and Mechanisms of Resistance to Pemigatinib. Cancer Discov. 2021, 11, 326–339. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Macarulla, T.; Javle, M.M.; Kelley, R.K.; Lubner, S.J.; Adeva, J.; Cleary, J.M.; Catenacci, D.V.; Borad, M.J.; Bridgewater, J.; et al. Ivosidenib in IDH1-mutant, chemothera-py-refractory cholangiocarcinoma (ClarIDHy): A multicentre, randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2020, 21, 796–807. [Google Scholar] [CrossRef]
- Naganuma, A.; Sakuda, T.; Murakami, T.; Aihara, K.; Watanuki, Y.; Suzuki, Y.; Shibasaki, E.; Masuda, T.; Uehara, S.; Yasuoka, H.; et al. Microsatellite Instability-high Intrahepatic Cholangiocarcinoma with Portal Vein Tumor Thrombosis Successfully Treated with Pembrolizumab. Intern. Med. 2020, 59, 2261–2267. [Google Scholar] [CrossRef]
- US Food and Drug Administration. FDA Grants Accelerated Approval to Pembrolizumab for First Tissue/Site Agnostic Indication. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-pembrolizumab-first-tissuesite-agnostic-indication (accessed on 4 April 2021).
- US Food and Drug Administration. FDA Approves Pembrolizumab for Adults and Children with TMB-H Solid Tumors; US Food and Drug Administration: Silver Spring, MD, USA, 2022. Available online: https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-pembrolizumab-adults-and-children-tmb-h-solid-tumors (accessed on 1 October 2022).
- US Food and Drug Administration. FDA Grants Accelerated Approval to Dostarlimab-Gxly for dMMR Advanced Solid Tumors; US Food and Drug Administration: Silver Spring, MD, USA, 2022. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-dostarlimab-gxly-dmmr-advanced-solid-tumors (accessed on 2 October 2022).
- US Food and Drug Administration. FDA Approves an Oncology Drug That Targets a Key Genetic Driver of Cancer, Rather Than a Specific Type of Tumor. Case Medical Research; US Food and Drug Administration: Silver Spring, MD, USA, 2018.
- US Food and Drug Administration. FDA Approves Entrectinib for NTRK Solid Tumors and ROS-1 NSCLC; US Food and Drug Administration: Silver Spring, MD, USA, 2022. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-entrectinib-ntrk-solid-tumors-and-ros-1-nsclc (accessed on 1 October 2022).
- US Food and Drug Administration. FDA Grants Accelerated Approval to Dabrafenib in Combination with Trametinib for Unre-Sectable or Metastatic Solid Tumors with BRAF V600E Mutation; US Food and Drug Administration: Silver Spring, MD, USA, 2022.
- Amatu, A.; Sartore-Bianchi, A.; Bencardino, K.; Pizzutilo, E.; Tosi, F.; Siena, S. Tropomyosin receptor kinase (TRK) biology and the role of NTRK gene fusions in cancer. Ann. Oncol. 2019, 30, viii5–viii15. [Google Scholar] [CrossRef]
- Cocco, E.; Scaltriti, M.; Drilon, A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat. Rev. Clin. Oncol. 2018, 15, 731–747. [Google Scholar] [CrossRef]
- Liu, F.; Wei, Y.; Zhang, H.; Jiang, J.; Zhang, P.; Chu, Q. NTRK Fusion in Non-Small Cell Lung Cancer: Diagnosis, Therapy, and TRK Inhibitor Resistance. Front. Oncol. 2022, 12, 864666. [Google Scholar] [CrossRef]
- Demols, A.; Rocq, L.; Charry, M.; De Nève, N.; Verrellen, A.; Ramadhan, A.; Van Campenhout, C.; De Clercq, S.; Salmon, I.; D’Haene, N. NTRK gene fusions in biliary tract cancers. J. Clin. Oncol. 2020, 38, 574. [Google Scholar] [CrossRef]
- Ross, J.S.; Wang, K.; Gay, L.; Al-Rohil, R.; Rand, J.V.; Jones, D.M.; Lee, H.J.; Sheehan, C.E.; Otto, G.A.; Palmer, G.; et al. New Routes to Targeted Therapy of Intrahepatic Cholangio-carcinomas Revealed by Next-Generation Sequencing. Oncologist 2014, 19, 235–242. [Google Scholar] [CrossRef]
- Drilon, A.; Siena, S.; Ou, S.-H.I.; Patel, M.; Ahn, M.J.; Lee, J.; Bauer, T.M.; Farago, A.F.; Wheler, J.J.; Liu, S.V.; et al. Safety and Antitumor Activity of the Multitargeted Pan-TRK, ROS1, and ALK Inhibitor Entrectinib: Combined Results from Two Phase I Trials (ALKA-372-001 and STARTRK-1). Cancer Discov. 2017, 7, 400–409. [Google Scholar] [CrossRef]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: Integrated analysis of three phase 1-2 trials. Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; DuBois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of Larotrectinib in TRK Fusion-Positive Cancers in Adults and Children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef]
- Subbiah, V.; Lassen, U.; Élez, E.; Italiano, A.; Curigliano, G.; Javle, M.; de Braud, F.; Prager, G.W.; Greil, R.; Stein, A.; et al. Dabrafenib plus trametinib in patients with BRAFV600E-mutated biliary tract cancer (ROAR): A phase 2, open-label, single-arm, multicentre basket trial. Lancet Oncol. 2020, 21, 1234–1243. [Google Scholar] [CrossRef]
- Salama, A.K.S.; Li, S.; Macrae, E.R.; Park, J.-I.; Mitchell, E.P.; Zwiebel, J.A.; Chen, H.X.; Gray, R.J.; McShane, L.M.; Rubinstein, L.V.; et al. Dabrafenib and Trametinib in Patients with Tumors With BRAFV600E Mutations: Results of the NCI-MATCH Trial Subprotocol H. J. Clin. Oncol. 2020, 38, 3895–3904. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Sahai, V.; Hollebecque, A.; Vaccaro, G.; Melisi, D.; Al-Rajabi, R.; Paulson, A.S.; Borad, M.J.; Gallinson, D.; Murphy, A.G.; et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: A multicentre, open-label, phase 2 study. Lancet Oncol. 2020, 21, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Javle, M.; Kelley, R.; Roychowdhury, S.; Weiss, K.; Abou-Alfa, G.; Macarulla, T.; Sadeghi, S.; Waldschmidt, D.; Zhu, A.; Goyal, L.; et al. Updated results from a phase II study of infigratinib (BGJ398), a selective pan-FGFR kinase inhibitor, in patients with previously treated advanced cholangiocarcinoma containing FGFR2 fusions. Ann. Oncol. 2018, 29, viii720. [Google Scholar] [CrossRef]
- Mohler, M.; Goyal, L.; Meric-Bernstam, F.; Hollebecque, A.; Morizane, C.; Valle, J.W.; Karasic, T.B.; Abrams, T.A.; Kelley, R.K.; Cassier, P.; et al. Abstract CT010: Primary results of phase 2 FOENIX-CCA2: The irreversible FGFR1-4 inhibitor futibatinib in intrahepatic cholangiocarcinoma (iCCA) with FGFR2 fu-sions/rearrangements. Cancer Res. 2021, 81 (Suppl. 13), CT010. [Google Scholar]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Marabelle, A.; Le, D.T.; Ascierto, P.A.; Di Giacomo, A.M.; De Jesus-Acosta, A.; Delord, J.P.; Geva, R.; Gottfried, M.; Penel, N.; Hansen, A.R.; et al. Efficacy of Pembrolizumab in Patients with Noncolorectal High Microsatellite Instability/Mismatch Repair–Deficient Cancer: Results from the Phase II KEY-NOTE-158 Study. J. Clin. Oncol. 2019, 38, 1–10. [Google Scholar] [CrossRef]
- Andre, T.; Berton, D.; Curigliano, G.; Ellard, S.; Trigo Pérez, J.M.; Arkenau, H.T.; Abdeddaim, C.; Moreno, V.; Guo, W.; Im, E.; et al. Safety and efficacy of anti–PD-1 antibody dostarlimab in patients (pts) with mismatch repair-deficient (dMMR) solid cancers: Results from GARNET study. J. Clin. Oncology. 2021, 39 (Suppl. 3), 9. [Google Scholar] [CrossRef]
- Lowery, M.A.; Burris, H.A.; Janku, F.; Shroff, R.T.; Cleary, J.M.; Azad, N.S.; Goyal, L.; Maher, E.A.; Gore, L.; Hollebecque, A.; et al. Safety and activity of ivosidenib in patients with IDH1-mutant advanced cholangiocarcinoma: A phase 1 study. Lancet Gastroenterol. Hepatol. 2019, 4, 711–720. [Google Scholar] [CrossRef]
- Javle, M.; Borad, M.J.; Azad, N.S.; Kurzrock, R.; Abou-Alfa, G.K.; George, B.; Hainsworth, J.; Meric-Bernstam, F.; Swanton, C.; Sweeney, C.J.; et al. Pertuzumab and trastuzumab for HER2-positive, metastatic biliary tract cancer (MyPathway): A multicentre, open-label, phase 2a, multiple basket study. Lancet Oncol. 2021, 22, 1290–1300. [Google Scholar] [CrossRef]
- Subbiah, V.; Cassier, P.A.; Siena, S.; Garralda, E.; Paz-Ares, L.; Garrido, P.; Nadal, E.; Vuky, J.; Lopes, G.; Kalemkerian, G.P.; et al. Pan-cancer efficacy of pralsetinib in patients with RET fusion–positive solid tumors from the phase 1/2 ARROW trial. Nat. Med. 2022, 28, 1640–1645. [Google Scholar] [CrossRef]
- Subbiah, V.; Wolf, J.; Konda, B.; Kang, H.; Spira, A.; Weiss, J.; Takeda, M.; Ohe, Y.; Khan, S.; Ohashi, K.; et al. Tumour-agnostic efficacy and safety of selpercatinib in patients with RET fusion-positive solid tumours other than lung or thyroid tumours (LIBRETTO-001): A phase 1/2, open-label, basket trial. Lancet Oncol. 2022, 23, 1261–1273. Available online: http://www.thelancet.com/article/S1470204522005411/fulltext (accessed on 23 February 2023). [CrossRef] [PubMed]
- Federman, N.; McDermott, R. Larotrectinib, a highly selective tropomyosin receptor kinase (TRK) inhibitor for the treatment of TRK fusion cancer. Expert Rev. Clin. Pharmacol. 2019, 12, 931–939. [Google Scholar] [CrossRef] [PubMed]
- Fischer, H.; Ullah, M.; de la Cruz, C.C.; Hunsaker, T.; Senn, C.; Wirz, T.; Wagner, B.; Draganov, D.; Vazvaei, F.; Donzelli, M.; et al. Entrectinib, a TRK/ROS1 inhibitor with anti-CNS tumor activity: Differentiation from other inhibitors in its class due to weak interaction with P-glycoprotein. Neuro Oncol. 2020, 22, 819–829. [Google Scholar] [CrossRef]
- Frampton, J.E. Entrectinib: A Review in NTRK+ Solid Tumours and ROS1+ NSCLC. Drugs 2021, 81, 697–708. [Google Scholar] [CrossRef]
- Dunn, D.B. Larotrectinib and Entrectinib: TRK Inhibitors for the Treatment of Pediatric and Adult Patients with NTRK Gene Fusion. J. Adv. Pr. Oncol. 2020, 11, 418–423. [Google Scholar] [CrossRef]
- US Food and Drug Administration. Rozlytrek (Entrectinib) Capsules, for Oral Use; Prescribing Information; US Food and Drug Administration: Silver Spring, MD, USA, 2021.
- European Medicines Agency. Rozlytrek (Entrectinib). Summary of Product Characteristics; European Medicines Agency: Amsterdam, The Netherlands, 2021. [Google Scholar]
- Schaider, H.; Sturm, R. The evolving universe of BRAF mutations in melanoma. Br. J. Dermatol. 2017, 177, 893. [Google Scholar] [CrossRef] [PubMed]
- Rose, A. Encorafenib and binimetinib for the treatment of BRAF V600E/K-mutated melanoma. Drugs Today 2019, 55, 247–264. [Google Scholar] [CrossRef]
- Tol, J.; Nagtegaal, I.D.; Punt, C.J.A. BRAF mutation in metastatic colorectal cancer. N. Engl. J. Med. 2009, 361, 98–99. [Google Scholar] [CrossRef]
- Sosman, J.A.; Kim, K.B.; Schuchter, L.; Gonzalez, R.; Pavlick, A.C.; Weber, J.S.; McArthur, G.A.; Hutson, T.E.; Moschos, S.J.; Flaherty, K.T.; et al. Survival in BRAF V600–Mutant Advanced Melanoma Treated with Vemurafenib. N. Engl. J. Med. 2012, 366, 707–714. [Google Scholar] [CrossRef]
- Ritterhouse, L.L.; Barletta, J.A. BRAF V600E mutation-specific antibody: A review. Semin. Diagn. Pathol. 2015, 32, 400–408. [Google Scholar] [CrossRef]
- Adashek, J.J.; Menta, A.K.; Reddy, N.K.; Desai, A.P.; Roszik, J.; Subbiah, V. Tissue-Agnostic Activity of BRAF plus MEK Inhibitor in BRAF V600–Mutant Tumors. Mol. Cancer Ther. 2022, 21, 871–878. [Google Scholar] [CrossRef] [PubMed]
- Murali, R.; Menzies, A.M.; Long, G. Dabrafenib and its potential for the treatment of metastatic melanoma. Drug Des. Dev. Ther. 2012, 6, 391–405. [Google Scholar] [CrossRef] [PubMed]
- King, A.J.; Arnone, M.R.; Bleam, M.R.; Moss, K.G.; Yang, J.; Fedorowicz, K.E.; Smitheman, K.N.; Erhardt, J.A.; Hughes-Earle, A.; Kane-Carson, L.S.; et al. Dabrafenib; Preclinical Characterization, Increased Efficacy when Combined with Trametinib, while BRAF/MEK Tool Combination Reduced Skin Lesions. PLoS ONE 2013, 8, e67583. [Google Scholar] [CrossRef]
- Kim, K.B.; Kefford, R.; Pavlick, A.C.; Infante, J.R.; Ribas, A.; Sosman, J.A.; Fecher, L.A.; Millward, M.; McArthur, G.A.; Hwu, P.; et al. Phase II study of the MEK1/MEK2 inhibitor Trametinib in patients with metastatic BRAF-mutant cutaneous melanoma previously treated with or without a BRAF inhibitor. J. Clin. Oncol. 2013, 31, 482–489. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Solit, D.B.; Rosen, N. Combination of RAF and MEK Inhibition for the Treatment of BRAF-Mutated Melanoma: Feedback Is Not Encouraged. Cancer Cell 2014, 26, 603–604. [Google Scholar] [CrossRef]
- Petitjean, A.; Achatz, M.I.W.; Borresen-Dale, A.L.; Hainaut, P.; Olivier, M. TP53 mutations in human cancers: Functional selection and impact on cancer prognosis and outcomes. Oncogene 2007, 26, 2157–2165. [Google Scholar] [CrossRef]
- Sen, S.; Meric-Bernstam, F.; Hong, D.S.; Hess, K.R.; Subbiah, V. Co-occurring Genomic Alterations and Association with Progres-sion-Free Survival in BRAFV600-Mutated Nonmelanoma Tumors. J. Natl. Cancer Inst. 2017, 109, 10. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Esposito Abate, R.; Rachiglio, A.M.; Maiello, M.R.; Esposito, C.; Schettino, C.; Izzo, F.; Nasti, G.; Normanno, N. FGFR Fusions in Cancer: From Diag-nostic Approaches to Therapeutic Intervention. Int. J. Mol. Sci. 2020, 21, 6856. [Google Scholar] [CrossRef]
- Borad, M.J.; Champion, M.D.; Egan, J.B.; Liang, W.S.; Fonseca, R.; Bryce, A.H.; McCullough, A.E.; Barrett, M.T.; Hunt, K.; Patel, M.; et al. Integrated Genomic Characterization Reveals Novel, Therapeutically Relevant Drug Targets in FGFR and EGFR Pathways in Sporadic Intrahepatic Cholangiocarcinoma. PLoS Genet. 2014, 10, e1004135. [Google Scholar] [CrossRef]
- Arai, Y.; Totoki, Y.; Hosoda, F.; Shirota, T.; Hama, N.; Nakamura, H.; Ojima, H.; Furuta, K.; Shimada, K.; Okusaka, T.; et al. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology 2013, 59, 1427–1434. [Google Scholar] [CrossRef]
- Shroff, R.T.; Yarchoan, M.; O’Connor, A.; Gallagher, D.; Zahurak, M.L.; Rosner, G.; Ohaji, C.; Sartorius-Mergenthaler, S.; Subbiah, V.; Zinner, R.; et al. The oral VEGF receptor tyrosine kinase in-hibitor pazopanib in combination with the MEK inhibitor trametinib in advanced cholangiocarcinoma. Br. J. Cancer 2017, 116, 1402–1407. [Google Scholar] [CrossRef] [PubMed]
- Ueno, M.; Ikeda, M.; Sasaki, T.; Nagashima, F.; Mizuno, N.; Shimizu, S.; Ikezawa, H.; Hayata, N.; Nakajima, R.; Morizane, C. Phase 2 study of lenvatinib monotherapy as second-line treatment in unresectable biliary tract cancer: Primary analysis results. BMC Cancer 2020, 20, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Plummer, R.; Madi, A.; Jeffels, M.; Richly, H.; Nokay, B.; Rubin, S.; Ball, H.A.; Weller, S.; Botbyl, J.; Gibson, D.M.; et al. A Phase I study of pazopanib in combination with gemcitabine in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2012, 71, 93–101. [Google Scholar] [CrossRef]
- Merz, V.; Zecchetto, C.; Melisi, D. Pemigatinib, a potent inhibitor of FGFRs for the treatment of cholangiocarcinoma. Futur. Oncol. 2021, 17, 389–402. [Google Scholar] [CrossRef]
- Botrus, G.; Raman, P.; Oliver, T.; Bekaii-Saab, T. Infigratinib (BGJ398): An investigational agent for the treatment of FGFR-altered intrahepatic cholangiocarcinoma. Expert Opin. Investig. Drugs 2021, 30, 309–316. [Google Scholar] [CrossRef]
- Nogova, L.; Sequist, L.V.; Garcia, J.M.P.; Andre, F.; Delord, J.-P.; Hidalgo, M.; Schellens, J.H.; Cassier, P.A.; Camidge, D.R.; Schuler, M.; et al. Evaluation of BGJ398, a Fibroblast Growth Factor Receptor 1-3 Kinase Inhibitor, in Patients with Advanced Solid Tumors Harboring Genetic Alterations in Fibroblast Growth Factor Receptors: Results of a Global Phase I, Dose-Escalation and Dose-Expansion Study. J. Clin. Oncol. 2017, 35, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Borbath, I.; Goyal, L.; Lamarca, A.; Macarulla, T.; Oh, D.Y.; Roychowdhury, S.; Sadeghi, S.; Shroff, R.T.; Li, A.; et al. PROOF 301: A multicenter, open-label, randomized, phase 3 trial of infigratinib versus gemcitabine plus cisplatin in patients with advanced cholangiocarcinoma with an FGFR2 gene fusion/rearrangement. J. Clin. Oncol. 2022, 40 (Suppl. 16), TPS4171. [Google Scholar] [CrossRef]
- Boichuk, S.; Dunaev, P.; Mustafin, I.; Mani, S.; Syuzov, K.; Valeeva, E.; Bikinieva, F.; Galembikova, A. Infigratinib (BGJ 398), a Pan-FGFR Inhibitor, Targets P-Glycoprotein and Increases Chemotherapeutic-Induced Mortality of Multidrug-Resistant Tumor Cells. Biomedicines 2022, 10, 601. [Google Scholar] [CrossRef]
- Sootome, H.; Fujita, H.; Ito, K.; Ochiiwa, H.; Fujioka, Y.; Ito, K.; Miura, A.; Sagara, T.; Ito, S.; Ohsawa, H.; et al. Futibatinib Is a Novel Irreversible FGFR 1–4 Inhibitor That Shows Selective Antitumor Activity against FGFR-Deregulated Tumors. Cancer Res 2020, 80, 4986–4997. [Google Scholar] [CrossRef] [PubMed]
- Meric-Bernstam, F.; Bahleda, R.; Hierro, C.; Sanson, M.; Bridgewater, J.; Arkenau, H.T.; Tran, B.; Kelley, R.K.; Park, J.O.; Javle, M.; et al. Futibatinib, an Irreversible FGFR1–4 Inhibitor, in Patients with Advanced Solid Tumors Harboring FGF/FGFR Aberrations: A Phase I Dose-Expansion Study. Cancer Discov. 2022, 12, 402–415. [Google Scholar] [CrossRef]
- Borad, M.; Javle, M.; Shaib, W.L.; Mody, K.; Bergamo, F.; Harris, W.P.; Damjanov, N.; Macarulla, T.; Brandi, G.; Masi, G.; et al. 59P Efficacy of derazantinib in intrahepatic cholangio-carcinoma (iCCA) patients with FGFR2 fusions, mutations or amplifications. Ann. Oncol. 2022, 33, S567–S568. [Google Scholar] [CrossRef]
- Hollebecque, A.; Borad, M.; Goyal, L.; Schram, A.; Park, J.O.; Cassier, P.A.; Kamath, S.D.; Meng, D.W.; Dotan, E.; Kim, R.; et al. LBA12 - Efficacy of RLY-4008, a highly selective FGFR2 inhibitor in patients (pts) with an FGFR2-fusion or rearrangement (f/r), FGFR inhibitor (FGFRi)-naïve cholangiocarcinoma (CCA): ReFocus trial. Ann. Oncol. 2022, 33, S808–S869. Available online: https://oncologypro.esmo.org/meeting-resources/esmo-congress/efficacy-of-rly-4008-a-highly-selective-fgfr2-inhibitor-in-patients-pts-with-an-fgfr2-fusion-or-rearrangement-f-r-fgfr-inhibitor-fgfri-naiv (accessed on 26 February 2023). [CrossRef]
- Hollebecque, A.; Silverman, I.; Owens, S.; Féliz, L.; Lihou, C.; Zhen, H.; Newton, R.; Burn, T.; Melisi, D. Comprehensive genomic profiling and clinical outcomes in patients (pts) with fibroblast growth factor receptor rearrangement-positive (FGFR2+) cholangiocarcinoma (CCA) treated with pemigatinib in the fight-202 trial. Ann. Oncol. 2019, 30, v276. [Google Scholar] [CrossRef]
- Javle, M.M.; Murugesan, K.; Shroff, R.T.; Borad, M.J.; Abdel-Wahab, R.; Schrock, A.B.; Chung, J.; Goyal, L.; Frampton, G.M.; Kelley, R.K.; et al. Profiling of 3,634 cholangiocarcinomas (CCA) to identify genomic alterations (GA), tumor mutational burden (TMB), and genomic loss of heterozygosity (gLOH). J. Clin. Oncol. 2019, 37, 4087. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef]
- Riaz, N.; Havel, J.J.; Makarov, V.; Desrichard, A.; Urba, W.J.; Sims, J.S.; Hodi, F.S.; Martín-Algarra, S.; Mandal, R.; Sharfman, W.H.; et al. Tumor and Microenvironment Evolution during Immu-notherapy with Nivolumab. Cell 2017, 171, 934–949.e16. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Rizvi, H.; Sanchez-Vega, F.; La, K.; Chatila, W.; Jonsson, P.; Halpenny, D.; Plodkowski, A.; Long, N.; Sauter, J.L.; Rekhtman, N.; et al. Molecular Determinants of Response to An-ti-Programmed Cell Death (PD)-1 and Anti-Programmed Death-Ligand 1 (PD-L1) Blockade in Patients with Non-Small-Cell Lung Cancer Profiled with Targeted Next-Generation Sequencing. J. Clin. Oncol. 2018, 36, 633–641. [Google Scholar] [CrossRef]
- Kim, H.; Kim, H.; Kim, R.; Jo, H.; Kim, H.R.; Hong, J.; Park, J.O.; Park, Y.S.; Kim, S.T. Tumor Mutational Burden as a Biomarker for Advanced Biliary Tract Cancer. Technol. Cancer Res. Treat. 2021, 20, 15330338211062324. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Francisco, L.M.; Sage, P.T.; Sharpe, A.H. The PD-1 pathway in tolerance and autoimmunity. Immunol. Rev. 2010, 236, 219–242. [Google Scholar] [CrossRef] [PubMed]
- Syn, N.L.; Teng, M.W.L.; Mok, T.S.K.; Soo, R.A. De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol. 2017, 18, e731–e741. [Google Scholar] [CrossRef] [PubMed]
- Marabelle, A.; Fakih, M.; Lopez, J.; Shah, M.; Shapira-Frommer, R.; Nakagawa, K.; Chung, H.C.; Kindler, H.L.; Lopez-Martin, J.A.; Miller, W.H.; et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: Prospective biomarker analysis of the mul-ticohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020, 21, 1353–1365. [Google Scholar] [CrossRef]
- Li, K.; Luo, H.; Huang, L.; Luo, H.; Zhu, X. Microsatellite instability: A review of what the oncologist should know. Cancer Cell Int. 2020, 20, 1–13. [Google Scholar] [CrossRef]
- Park, U.B.; Jeong, T.J.; Gu, N.; Lee, H.T.; Heo, Y.-S. Molecular basis of PD-1 blockade by dostarlimab, the FDA-approved antibody for cancer immunotherapy. Biochem. Biophys. Res. Commun. 2022, 599, 31–37. [Google Scholar] [CrossRef]
- Xiang, X.; Liu, Z.; Zhang, C.; Li, Z.; Gao, J.; Zhang, C.; Cao, Q.; Cheng, J.; Liu, H.; Chen, D.; et al. IDH Mutation Subgroup Status Associates with Intratumor Heterogeneity and the Tumor Microenvironment in Intrahepatic Cholangiocarcinoma. Adv. Sci. 2021, 8, e2101230. [Google Scholar] [CrossRef]
- Valle, J.W.; Kelley, R.K.; Nervi, B.; Oh, D.Y.; Zhu, A.X. Biliary tract cancer. Lancet 2021, 397, 428–444. [Google Scholar] [CrossRef]
- Boscoe, A.N.; Rolland, C.; Kelley, R.K. Frequency and prognostic significance of isocitrate dehydrogenase 1 mutations in cholan-giocarcinoma: A systematic literature review. J. Gastrointest. Oncol. 2019, 10, 751–765. [Google Scholar] [CrossRef]
- Zhang, H.; Berezov, A.; Wang, Q.; Zhang, G.; Drebin, J.; Murali, R.; Greene, M.I. ErbB receptors: From oncogenes to targeted cancer therapies. J. Clin. Investig. 2007, 117, 2051–2058. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Hurwitz, H.; Raghav, K.P.S.; McWilliams, R.R.; Fakih, M.; VanderWalde, A.; Swanton, C.; Kurzrock, R.; Burris, H.; Sweeney, C.; et al. Pertuzumab plus trastuzumab for HER2-amplified metastatic colorectal cancer (MyPathway): An updated report from a multicentre, open-label, phase 2a, multiple basket study. Lancet Oncol. 2019, 20, 518–530. [Google Scholar] [CrossRef]
- Hudis, C.A. Trastuzumab—Mechanism of Action and Use in Clinical Practice. N. Engl. J. Med. 2007, 357, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Junttila, T.T.; Akita, R.W.; Parsons, K.; Fields, C.; Phillips, G.D.; Friedman, L.S.; Sampath, D.; Sliwkowski, M.X. Ligand-independent HER2/HER3/PI3K complex is disrupted by trastuzumab and is effectively inhibited by the PI3K inhibitor GDC-0941. Cancer Cell 2009, 15, 429–440. [Google Scholar] [CrossRef]
- Tsurutani, J.; Iwata, H.; Krop, I.; Jänne, P.A.; Doi, T.; Takahashi, S.; Park, H.; Redfern, C.; Tamura, K.; Wise-Draper, T.M.; et al. Targeting her2 with trastuzumab deruxtecan: A dose-expansion, phase i study in multiple advanced solid tumors. Cancer Discov. 2020, 10, 688–701. Available online: https://aacrjournals.org/cancerdiscovery/article/10/5/688/2521/Targeting-HER2-with-Trastuzumab-Deruxtecan-A-Dose (accessed on 27 February 2023). [CrossRef] [PubMed]
- Ohba, A.; Morizane, C.; Kawamoto, Y.; Komatsu, Y.; Ueno, M.; Kobayashi, S.; Ikeda, M.; Sasaki, M.; Furuse, J.; Okano, N.; et al. Trastuzumab deruxtecan (T-DXd; DS-8201) in patients (pts) with HER2-expressing unresectable or recurrent biliary tract cancer (BTC): An investigator-initiated multicenter phase 2 study (HERB trial). J. Clin. Oncol. 2022, 40, 4006. [Google Scholar] [CrossRef]
- Paratala, B.S.; Chung, J.H.; Williams, C.B.; Yilmazel, B.; Petrosky, W.; Williams, K.; Schrock, A.B.; Gay, L.M.; Lee, E.; Dolfi, S.C.; et al. RET rearrangements are actionable alterations in breast cancer. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Drusbosky, L.M.; Rodriguez, E.; Dawar, R.; Ikpeazu, C.V. Therapeutic strategies in RET gene rearranged non-small cell lung cancer. J. Hematol. Oncol. 2021, 14, 1–8. [Google Scholar] [CrossRef]
- Adashek, J.J.; Desai, A.P.; Andreev-Drakhlin, A.Y.; Roszik, J.; Cote, G.J.; Subbiah, V. Hallmarks of RET and Co-occuring Genomic Alterations in RET-aberrant Cancers. Mol. Cancer Ther. 2021, 20, 1769–1776. [Google Scholar] [CrossRef]
- Kim, J.; Bradford, D.; Larkins, E.; Pai-Scherf, L.H.; Chatterjee, S.; Mishra-Kalyani, P.S.; Wearne, E.; Helms, W.S.; Ayyoub, A.; Bi, Y.; et al. FDA Approval Summary: Pralsetinib for the Treatment of Lung and Thyroid Cancers with RET Gene Mutations or Fusions. Clin. Cancer Res. 2021, 27, 5452–5456. [Google Scholar] [CrossRef]
- Subbiah, V.; Gainor, J.F.; Oxnard, G.R.; Tan, D.S.; Owen, D.H.; Cho, B.C.; Loong, H.H.; McCoach, C.E.; Weiss, J.; Kim, Y.J.; et al. Intracranial Efficacy of Selpercatinib in RET Fu-sion-Positive Non-Small Cell Lung Cancers on the LIBRETTO-001. Trial. Clin Cancer Res. 2021, 27, 4160–4167. Available online: https://pubmed.ncbi.nlm.nih.gov/34088726/ (accessed on 12 February 2023). [CrossRef]
- FDA Approves Selpercatinib for Locally Advanced or Metastatic RET Fusion-Positive Solid Tumors|FDA. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-selpercatinib-locally-advanced-or-metastatic-ret-fusion-positive-solid-tumors (accessed on 12 February 2023).
- Adashek, J.; Janku, F.; Kurzrock, R. Signed in Blood: Circulating Tumor DNA in Cancer Diagnosis, Treatment and Screening. Cancers 2021, 13, 3600. [Google Scholar] [CrossRef]
- Nikanjam, M.; Kato, S.; Kurzrock, R. Liquid biopsy: Current technology and clinical applications. J. Hematol. Oncol. 2022, 15, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Nygaard, A.D.; Holdgaard, P.C.; Spindler, K.-L.G.; Pallisgaard, N.; Jakobsen, A. The correlation between cell-free DNA and tumour burden was estimated by PET/CT in patients with advanced NSCLC. Br. J. Cancer 2013, 110, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Birkenkamp-Demtröder, K.; Christensen, E.; Nordentoft, I.; Knudsen, M.; Taber, A.; Høyer, S.; Lamy, P.; Agerbæk, M.; Jensen, J.B.; Dyrskjøt, L. Monitoring Treatment Response and Metastatic Relapse in Advanced Bladder Cancer by Liquid Biopsy Analysis. Eur. Urol. 2018, 73, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Ghidini, M.; Pizzo, C.; Botticelli, A.; Hahne, J.C.; Passalacqua, R.; Tomasello, G.; Petrelli, F. Biliary tract cancer: Current challenges and future prospects. Cancer Manag. Res. 2018, ume 11, 379–388. [Google Scholar] [CrossRef]
- Tamada, K.; Ushio, J.; Sugano, K. Endoscopic diagnosis of extrahepatic bile duct carcinoma: Advances and current limitations. World J. Clin. Oncol. 2011, 2, 203–216. [Google Scholar] [CrossRef]
- Zou, S.; Li, J.; Zhou, H.; Frech, C.; Jiang, X.; Chu, J.S.C.; Zhao, X.; Li, Y.; Li, Q.; Wang, H.; et al. Mutational landscape of intrahepatic cholangiocarcinoma. Nat. Commun. 2014, 5, 5696. [Google Scholar] [CrossRef]
- Zill, O.A.; Greene, C.; Sebisanovic, D.; Siew, L.M.; Leng, J.; Vu, M.; Hendifar, A.E.; Wang, Z.; Atreya, C.E.; Kelley, R.K.; et al. Cell-Free DNA Next-Generation Sequencing in Pancreatobiliary Carcinomas. Cancer Discov. 2015, 5, 1040–1048. [Google Scholar] [CrossRef]
- Mody, K.; Kasi, P.M.; Yang, J.; Surapaneni, P.K.; Bekaii-Saab, T.; Ahn, D.H.; Mahipal, A.; Sonbol, M.B.; Starr, J.S.; Roberts, A.; et al. Circulating Tumor DNA Profiling of Advanced Biliary Tract Cancers. JCO Precis. Oncol. 2019, 1–9. [Google Scholar] [CrossRef]
- Kumari, S.; Tewari, S.; Husain, N.; Agarwal, A.; Pandey, A.; Singhal, A.; Lohani, M. Quantification of Circulating Free DNA as a Diagnostic Marker in Gall Bladder Cancer. Pathol. Oncol. Res. 2016, 23, 91–97. [Google Scholar] [CrossRef]
- Eaton, J.E.; Gossard, A.A.; Talwalkar, J.A. Recall processes for biliary cytology in primary sclerosing cholangitis. Curr. Opin. Gastroenterol. 2014, 30, 287–294. [Google Scholar] [CrossRef]
- Lucci, A.; Hall, C.S.; Lodhi, A.K.; Bhattacharyya, A.; Anderson, A.E.; Xiao, L.; Bedrosian, I.; Kuerer, H.M.; Krishnamurthy, S. Circulating tumour cells in non-metastatic breast cancer: A prospective study. Lancet Oncol. 2012, 13, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Ettrich, T.J.; Schwerdel, D.; Dolnik, A.; Beuter, F.; Blätte, T.J.; Schmidt, S.A.; Stanescu-Siegmund, N.; Steinacker, J.; Marienfeld, R.; Kleger, A.; et al. Genotyping of circulating tumor DNA in cholangi-ocarcinoma reveals diagnostic and prognostic information. Sci. Rep. 2019, 9, 13261. [Google Scholar] [CrossRef] [PubMed]
- Goyal, L.; Saha, S.K.; Liu, L.Y.; Siravegna, G.; Leshchiner, I.; Ahronian, L.G.; Lennerz, J.K.; Vu, P.; Deshpande, V.; Kambadakone, A.; et al. Polyclonal Secondary FGFR2 Mutations Drive Ac-quired Resistance to FGFR Inhibition in Patients with FGFR2 Fusion–Positive Cholangiocarcinoma. Cancer Discov. 2017, 7, 252–263. [Google Scholar] [CrossRef]
- Kato, S.; Okamura, R.; Adashek, J.J.; Khalid, N.; Lee, S.; Nguyen, V.; Sicklick, J.K.; Kurzrock, R. Targeting G1/S phase cell-cycle genomic alterations and accompanying co-alterations with individualized CDK4/6 inhibitor-based regimens. JCI Insight 2021, 6, 1. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Adashek, J.J.; Shaya, J.; Okamura, R.; Jimenez, R.E.; Lee, S.; Sicklick, J.K.; Kurzrock, R. Concomitant MEK and Cyclin Gene Alterations: Implications for Response to Targeted Therapeutics. Clin. Cancer Res. 2021, 27, 2792–2797. [Google Scholar] [CrossRef]
- Kato, S.; Kim, K.H.; Lim, H.J.; Boichard, A.; Nikanjam, M.; Weihe, E.; Kuo, D.J.; Eskander, R.N.; Goodman, A.; Galanina, N.; et al. Real-world data from a molecular tumor board demonstrates improved outcomes with a precision N-of-One strategy. Nat. Commun. 2020, 11, 1–9. [Google Scholar] [CrossRef]
- Adashek, J.J.; Kato, S.; Parulkar, R.; Szeto, C.W.; Sanborn, J.Z.; Vaske, C.J.; Benz, S.C.; Reddy, S.K.; Kurzrock, R. Transcriptomic silencing as a potential mechanism of treatment resistance. J. Clin. Investig. 2020, 5. [Google Scholar] [CrossRef]
- Kato, S.; Gumas, S.; Adashek, J.J.; Okamura, R.; Lee, S.; Sicklick, J.K.; Kurzrock, R. Multi-omic analysis in carcinoma of unknown primary (CUP): Therapeutic impact of knowing the unknown. Mol. Oncol. 2022. [Google Scholar] [CrossRef]
- Adashek, J.J.; Subbiah, V.; Kurzrock, R. From Tissue-Agnostic to N-of-One Therapies: (R)Evolution of the Precision Paradigm. Trends Cancer 2020, 7, 15–28. [Google Scholar] [CrossRef]
- Wahida, A.; Buschhorn, L.; Fröhling, S.; Jost, P.J.; Schneeweiss, A.; Lichter, P.; Kurzrock, R. The coming decade in precision oncology: Six riddles. Nat. Rev. Cancer 2022, 23, 43–54. [Google Scholar] [CrossRef]
- Sicklick, J.K.; Kato, S.; Okamura, R.; Schwaederle, M.; Hahn, M.E.; Williams, C.B.; De, P.; Krie, A.; Piccioni, D.E.; Miller, V.A.; et al. Molecular profiling of cancer patients enables personalized combination therapy: The I-PREDICT study. Nat. Med. 2019, 25, 744–750. [Google Scholar] [CrossRef] [PubMed]
- Szeto, C.W.; Kurzrock, R.; Kato, S.; Goloubev, A.; Veerapaneni, S.; Preble, A.; Reddy, S.K.; Adashek, J.J. Association of differential expression of immuno-regulatory molecules and presence of targetable mutations may inform rational design of clinical trials. ESMO Open 2022, 7, 100396. [Google Scholar] [CrossRef] [PubMed]
- Adashek, J.; Subbiah, V.; Westphalen, C.; Naing, A.; Kato, S.; Kurzrock, R. Cancer: Slaying the nine-headed Hydra. Ann. Oncol. 2022, 34, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Adashek, J.J.; Goloubev, A.; Kato, S.; Kurzrock, R. Missing the target in cancer therapy. Nat. Cancer 2021, 2, 369–371. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| CCA Subtype | iCCA (Affects Bile Ducts within the Liver) | eCCA (Affects Bile Ducts Outside of the Liver) |
|---|---|---|
| ARID1A | 18–23% [23,24] | 14% [25] |
| BAP1 | 15–20% [23,24] | -- |
| BRAF V600E | 1.5% [26] | -- |
| BRCA1 | 0.4% [27] | 2% [27] |
| BRCA2 | 2.8% [27] | 2.5% [27] |
| CDH1 | 11.8% [28] | -- |
| CDKN2A/B | 9–27% [23,24] | 9–28% [25,29,30] |
| ERBB2/HER2 | 5.8% [31,32] | 1.3–20% [25,31,32] |
| FGFR2 fusion | 10–16% [23,33] | 0 [23,33] |
| IDH1/2 | 13–30% [23,34] | 4.7% [25] |
| KRAS | 7–54% [23,24,25] | 36.7–46% [25,29,30] |
| MSI-H/dMMR | 4.7–18.2% [35] | 4% [25] |
| PI3K | 7% [23,25] | 5% [23,25] |
| SMAD4 | -- | 10.7% [25] |
| TP53 | 18–27% [23,24] | 18–68% [25,29,30] |
| Target (Gene) | % in CCA | FDA-Approved Drug | Date of Approval | Trials | Total | ORR | OS | PFS | Disease-Free Survival | Duration of Response | Major Adverse Events (Grade ≥ 3) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| No. of CCA/ BTCs (%) | |||||||||||
| NTRK gene fusion-positive | 3–9% [46] | Entrectinib | 15 August 2019 | Drilon et al., 2017 [47] | 55 (100%) | 100% (95% CI: 44 to 100) | NR | NR | NR | For the three patients, the DoR was 2.6 months, 4.6 months, and 15.1 months | Fatigue/asthenia: 5 (4%) Weight increase: 2 (2%) Diarrhea: 1 (1%) Arthralgia: 1 (1%) |
| NR | NR | NR | NR | NR | NR | ||||||
| Doebele et al., 2020 [48] | 54 (100%) | 57% (95% CI: 43.2 to 70.8) | 21 months (95% CI 14.9 to NE) | 11.2 months (95% CI 8.0 to 14.9) | NR | 10.4 months (95% CI: 7.1 to NE) | Anemia: 8 (12%) Increased weight: 7 (10%) Fatigue: 5 (7%) | ||||
| 1 (2%) | NR | NR | NR | NR | NR | ||||||
| Larotrectinib | 26 November 2018 | Drilon et al., 2018 [49] | 55 (100%) | 80% (95% CI: 67 to 90) | NR | Not reached | NR | Not reached | Anemia: 114 (11%) Increased weight: 73 (7%) Decreased neutrophil count: 73 (7%) Increased ALT and AST: 73 (7%) | ||
| 2 (4%) | 50% objective tumor shrinkage | NR | NR | NR | NR | ||||||
| BRAF-V600E | 1.5 [26] | Trametinib plus dabrafenib | 22 June 2022 | Subbiah et al., 2020 [50] | 43 (100%) * | 51% (95% CI: 36 to 67) | 14 months (95% CI: 10 to 33) | 9 months (95% CI: 5 to 10) | NR | 9 months (95% CI: 6 to 14) | γ-glutamyltransferase increased: 5 (12%) Decreased WBC count: 3 (7%) Pyrexia: 3 (7%) |
| Salama et al., 2020 [51] | 29 (100%) | 38% (95% CI: 22.9% to 54.9%) | 28.6 months | 11.4 months (90% CI: 8.4 to 16.3) | NR | 25.1 months (90% CI: 12.8 to NE) | Fatigue: 4 (11.4%) Decreased neutrophil count: 3 (8.6%) Decreased WBC count: 3 (8.6%) | ||||
| 4 (13.8%) | 75% (3/4 pts), one is ongoing for 29 months | NR | NR | NR | NR | ||||||
| FGFR2 fusion or rearrangements | 10–16% [33] | Pemigatinib | 17 April 2020 | Abou-Alfa et al., 2020 [52] | 107 (100%) * | 35.5% (95% CI: 26.5 to 45.4%) | 21.1 months (95% CI: 14.8 to NE) | 6.93 months (95% CI: 6.18 to 9.59) | NR | 7.5 months (95% CI: 5.7 to 14.5) | Hypophosphataemia: 10 (7%) Stomatitis: 8 (5%) Arthralgia: 6 (4%) Palmar-plantar erythrodysesthesia: 6 (4%) |
| Infigratinib | 28 May 2021 | Javle et al., 2021 [53] | 108 (100%) * | 23.1 (95% CI 15.6 to 32.3%) | 12.5 (95% CI: 9.9 to 16.6) | 6.8 (95% CI: 5.3 to 7.6) | NR | 5.4 (95% CI: 3.7 to 7.4) | Hypophosphatemia: 10 (14.1%) Hyperphosphatemia: 9 (12.7%) Hyponatremia: 8 (11.3%) | ||
| Futibatinib | 30 September 2022 | Goyal et al. [54] | 103 (100%) * | 41.7% | 20.0 | 8.9 | NR | 9.5 | NR | ||
| MSI-H/dMMR tumors | 4.7–18.2% [35] | Pembrolizumab | 16 June 2020 | Le et al., 2017 [55] | 86 (100%) | 53% (95% CI: 42% to 64%) | Not reached 2-year OS: 64% (95% CI: 53% to 78%) | Not reached 2-year PFS: 53% (95% CI: 42% to 68%) | NR | NR | Diarrhea/colitis: 5 (6%) Pancreatitis/Hyperamylasemia: 5 (6%) Fatigue: 2 (2%) Anemia: 2 (2%) |
| 4 (4.7%) | NR | NR | NR | NR | NR | ||||||
| Marabelle et al., 2019 [56] | 233 (100%) | 34.3% (95% CI: 28.3 to 40.8) | 23.5 months (95% CI: 13.5 to NR) | 4.1 months (95% CI: 2.4 to 4.9) | NR | Not reached (range, 2.9 to 31.3+ months) | Fatigue: 2 (0.9%) Asthenia: 1 (0.4%) | ||||
| 22 (9.4%) | 40.9% (95% CI: 20.7 to 63.6) in CCA pts | 24.3 months (95% CI: 6.5 to NE) in CCA pts | 4.2 months (2.1 to NE) in CCA pts | NR | NR | ||||||
| Dostarlimab | 17 August 2021 | Andre et al., 2021 (Abstract) [57] | 106 (100%) | 38.7% (95% CI: 29.4 to 48.6) | NR | NR | NR | Not reached | Lipase increased: 2 (1.4%) | ||
| 2 (1.9%) | 100% CR | NR | NR | NR | NR | ||||||
| IDH1 | 13% [34] | Ivosidenib | 25 August 2021 | Lowery et al., 2019 [58] | 73 (100%) * | 5% (95% CI: 1.5 to 13.4) | 13.8 months (95% CI: 11.1 to 29.3) | 3.8 months (95% CI: 3.6 to 7.3) | NR | NR | Ascites: 4 (5%) Anemia: 3 (4%) Fatigue: 2 (3%) |
| Abou-Alfa et al., 2020 [34] | 185 (100%) * | 2% (95% CI: 0.5 to 6.9) | 10.8 months (95% CI: 7.7 to 17.6) | 2.7 months (95% CI: 1.6 to 4.2) | NR | NR | Ascites: 9 (7%) Aspartate aminotransferase increased: 6 (5%) Anemia: 4 (3%) Fatigue: 4 (3%) | ||||
| HER2-positive tumor | 5.8% of iCCA and 13–20% of eCCA [31,32] | Pertuzumab plus trastuzumab | Not yet approved in CCA | Javle et al., 2021 [59] | 39 (100%) * | 23% (95% CI: 11 to 39) | 10.9 months (95% CI: 5.2 to 15.6) | 4.0 months (95% CI: 1.8 to 5.7) | NR | 10.8 months (95% CI: 0.7 to 25.4) | Increased alanine aminotransferase: 5 (13%) Increased aspartate aminotransferase: 5 (13%) Blood alkaline phosphatase increased: 4 (10%) |
| RET fusion-positive | NR | Pralsetinib | Not yet approved in CCA | Subbiah et al., 2022 [60] | 23 (100%) | 57% (95% CI: 35%–77%) | 13.6 months (95% CI: 7.5 to NE) | 7.4 months (95% CI: 5.1 to 13.6) | NR | 11.7 months (95% CI: 5.5 to 19.0) | Neutropenia: 9 (31%) Anemia: 4 (14%) Increased AST: 3 (10%) |
| 3 (13%) | 66.7% | NR | NR | NR | NR | ||||||
| Selpercatinib | 21 September 2022 | Subbiah et al., 2022 [61] | 45 (41 evaluated for efficacy) | 43.9% (95% CI 28.5–60.3) | 18.0 months (95% CI: 10.7– Not estimated) *** | 13.2 months (95% CI: 7.4–26.2) | NR | 24.5 (95% CI: 9.2–Not estimated) | Hypertension (22%) Increased alanine aminotransferase (16%) Increased aspartate aminotransferase (13%). | ||
| 2 (1 evaluated for efficacy) | 100% | 5.6 months |
| Target | Phase | Clinical Trial Identifier | Treated Cancer Group | Experimental Arm | Control Arm | Primary Outcome | Secondary Outcome (Main) |
|---|---|---|---|---|---|---|---|
| First Line | |||||||
| FGFR2 fusion/rearrangement | III | NCT03656536 | CCA | Pemigatinib | Gemcitabine/Cisplatin | PFS | OS, OR, DOR, DCR |
| III | NCT03773302 | CCA | Infigratinib | Gemcitabine/Cisplatin | PFS | OS, DCR, DOR, BOR | |
| III | NCT04093362 | iCCA | Futibatinib | Gemcitabine/Cisplatin | PFS | OS, safety, ORR, DCR | |
| II | NCT03230318 | iCCA | Derazantinib | None | ORR, PFS | OS, safety, DCR | |
| I/II | NCT04526106 | iCCA and other advanced tumors | RLY-4008 | None | ORR, MTD, safety | DOR, DCR, pharmacokinetics | |
| HER 2 mutations | II | NCT03613168 | BTCs | Trastuzumab plus gemcitabine/cisplatin | None | BOR, safety | PFS, OS |
| I/II | NCT02992340 | BTCs | Varlitinib plus gemcitabine/cisplatin | None | MTD, safety, PFS, ORR | OS, DOR, DCR, PK | |
| Subsequent lines | |||||||
| NTRK gene fusion- | II | NCT04879121 | Advanced solid tumors | Larotrectinib | None | ORR | PFS, OS, safety, DOR, GMI, CBR |
| II | NCT03213704 | Advanced solid tumors | Larotrectinib | None | ORR | PFS, safety, PK, changes in tumor genomics | |
| Non-V600E BRAF mutations | II | NCT03839342 | Advanced solid tumors | Bimimetinib + Encorafenib | None | ORR | PFS, safety, DCR |
| I | NCT04190628 | Advanced solid tumors | ABM-1310 | None | MTD | PFS, OS, safety, PK, ORR, DCR, DOR | |
| I | NCT04249843 | Advanced solid tumors | BGB-3245 | None | Safety, MTD | PFS, OS, PK, ORR, DCR, DORƒ | |
| I | NCT04418167 | Advanced solid tumors | JSI-1187 monotherapy or in combination with dabrafenib | None | Safety | PFS, OS, ORR, DOR, time to response, DCR, PK | |
| IDH1/2 mutations | II | NCT02428855 | iCCA | Dasatinib | None | ORR | PFS, OS, safety |
| II | NCT03212274 | CCA | Olaparib | None | ORR | PFS, OS, safety | |
| II | NCT03878095 | CCA | Ceralasertib + Olaparib | None | ORR | PFS, OS, safety, DOR | |
| I/II | NCT02273739 | Advanced solid tumors | Enasidenib | None | DLT, ECOG | Plasma concentration metrics | |
| I | NCT04521686 | CCA | LY3410738 LY3410738 + Gemcitabine/Cisplatin | MTD | ORR, safety and tolerability, efficacy, PK | ||
| dMMR/MSI-H | I/II | NCT04800627 | Advanced solid tumors | Pevonedistat in combination with Pembrolizumab | None | Recommended phase 2 dose, ORR | PFS, OS, safety, changes in protein misfolding |
| HER 2 mutations | II/III | NCT03093870 | BTCs | Varlitinib with Capecitabine | Capecitabine | ORR, PFS | OS, safety, DOR, DCR, tumor size, ECOG |
| II | NCT03185988 | Metastatic carcinoma of digestive system including BTCs | Trastuzumab plus 5-FU or IRI or Capecitabine | None | RR | OS, PFS, DCR, DOR, time of response, ECOG | |
| II | jRCT2031180150 | Advanced solid tumors | Trastuzumab and Pertuzumab | None | ORR | PFS, OS, safety, DOR | |
| II | NCT02999672 | CCA | Trastuzumab emtansine | None | BOR | PFS, OS, safety, PK | |
| II | NCT02675829 | Advanced solid tumors | Ado-Trastuzumab emtansine | None | ORR | None | |
| II | NCT04482309 | Advanced solid tumors | Trastuzumab Deruxtecan | None | ORR | OS, PFS, safety, DOR, DCR, PK, immunogenicity | |
| I/II | NCT03410927 | Advanced solid tumors | TAS0728 | None | Safety, ORR | OS, DOR, PK, DCR | |
| I | NCT04764084 | CCA | Niraparib + Anlotinib | None | DLT, MTD | PFS, ORR | |
| I | NCT02892123 | Advanced solid tumors | Zanidatamab plus chemotherapy | None | MTD, Safety | PFS, ORR, PK, antidrug antibodies | |
| I | NCT02564900 | Non-breast/non-gastric solid tumors | Trastuzumab Deruxtecan | None | ORR | DCR, BOR, DOR, PFS, OS, pharmacokinetics, safety | |
| BAP1 and other DDR genes | II | NCT03207347 | CCA | Niraparib | None | ORR | PFS, OS, safety |
| DNA repair gene mutation | II | NCT03207347 | CCA | Niraparib | None | ORR | PFS, OS, safety |
| Matched molecular therapy | |||||||
| Matched molecular therapy | N/A | NCT04504604 | Rare tumors | FoundationOne CDx and FoundationOne Liquid CDx | None | % who receive a molecularly targeted matched, PFS | Tumor molecular profiles correlation to treatment outcome. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gupta, A.; Kurzrock, R.; Adashek, J.J. Evolution of the Targeted Therapy Landscape for Cholangiocarcinoma: Is Cholangiocarcinoma the ‘NSCLC’ of GI Oncology? Cancers 2023, 15, 1578. https://doi.org/10.3390/cancers15051578
Gupta A, Kurzrock R, Adashek JJ. Evolution of the Targeted Therapy Landscape for Cholangiocarcinoma: Is Cholangiocarcinoma the ‘NSCLC’ of GI Oncology? Cancers. 2023; 15(5):1578. https://doi.org/10.3390/cancers15051578
Chicago/Turabian StyleGupta, Amol, Razelle Kurzrock, and Jacob J. Adashek. 2023. "Evolution of the Targeted Therapy Landscape for Cholangiocarcinoma: Is Cholangiocarcinoma the ‘NSCLC’ of GI Oncology?" Cancers 15, no. 5: 1578. https://doi.org/10.3390/cancers15051578
APA StyleGupta, A., Kurzrock, R., & Adashek, J. J. (2023). Evolution of the Targeted Therapy Landscape for Cholangiocarcinoma: Is Cholangiocarcinoma the ‘NSCLC’ of GI Oncology? Cancers, 15(5), 1578. https://doi.org/10.3390/cancers15051578