

Molecular Mechanisms of Gastrointestinal Stromal Tumors and Their Impact on Systemic Therapy Decision

Abstract
Simple Summary
Abstract
1. Introduction
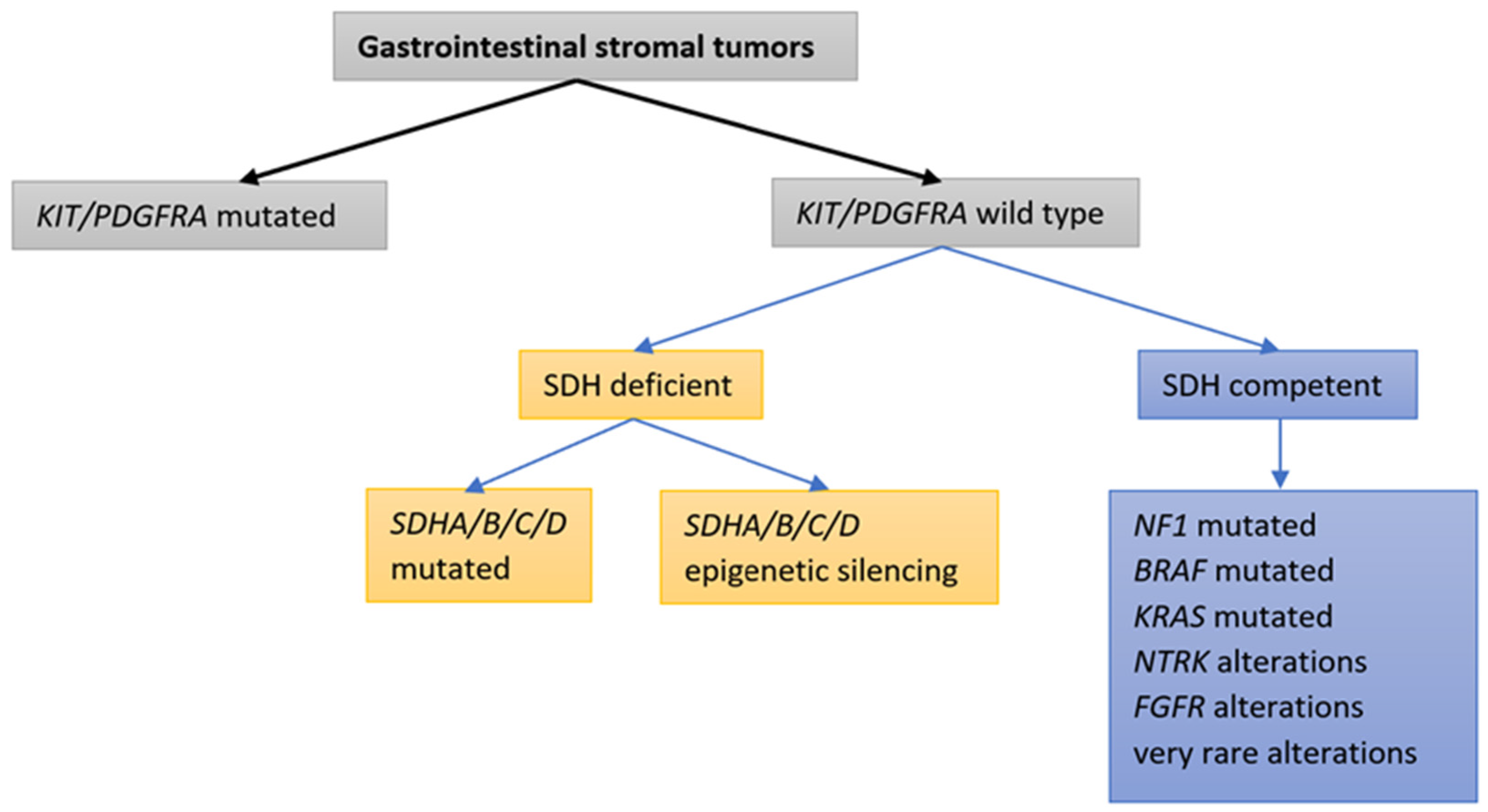
2. Molecular Classification of GISTs
3. Identifying the Molecular Mechanisms of GIST
3.1. GISTs with KIT or PDGFRA Mutations
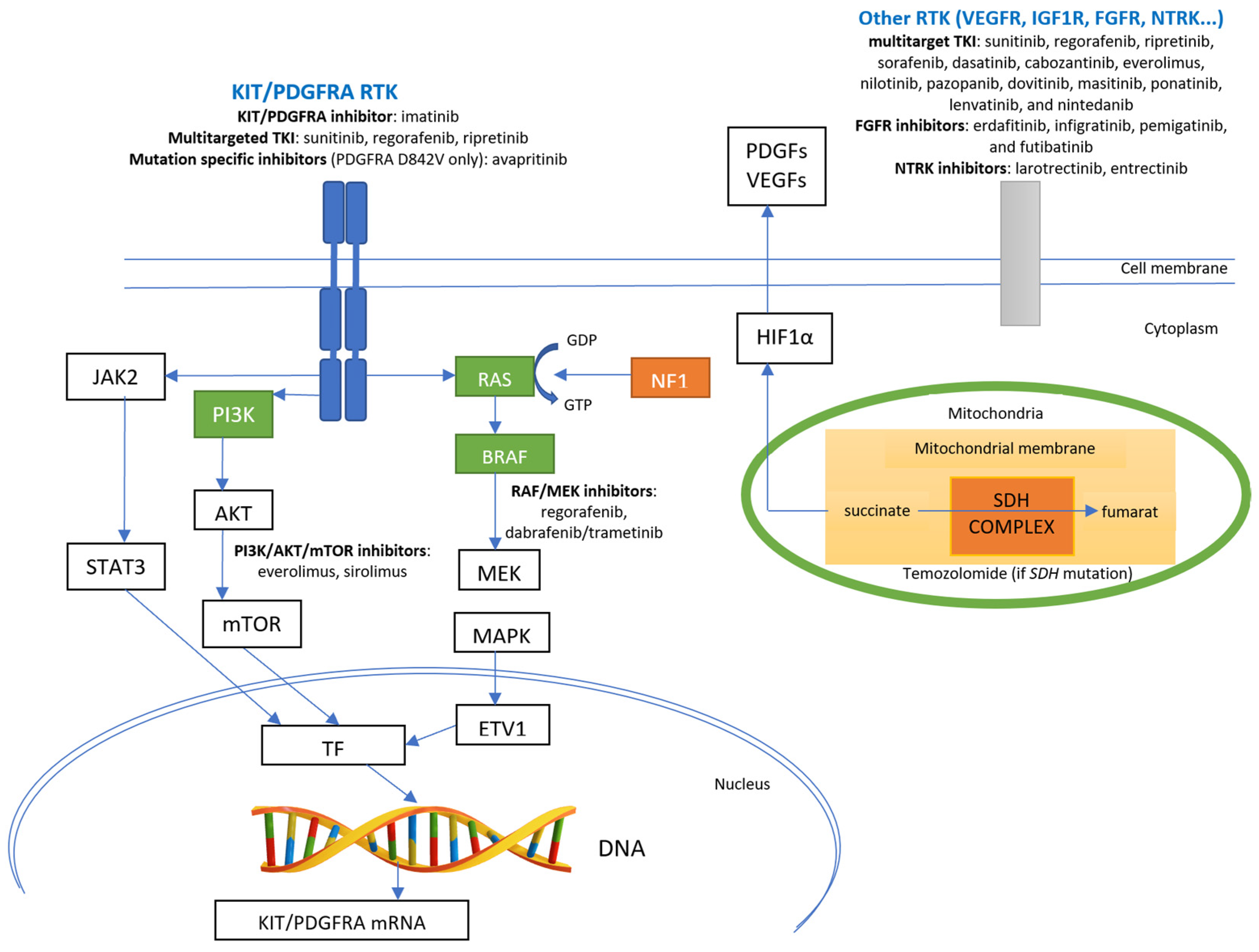
3.1.1. Targeted Therapy for GISTs with KIT or PDGFRA Mutations

{kind=link}
{kind=link}
| Study First Author/Publication Information | Number of Patients, Patient Population | Clinical Phase | Intervention | Molecular Analysis | Primary Endpoint | Results |
|---|---|---|---|---|---|---|
| Dematteo et al., Lancet 2009 [70] | 713 Patients resected for ≥3 cm GIST | 3 | 1 y adjuvant imatinib 400 mg/d vs. placebo | No | RFS (primary endpoint changed from OS to RFS) | 1 y RFS: 98% (imatinib) vs. 83% (placebo), HR 0.35. No OS benefits |
| Casali et al., JCO 2015 [71] | 908 Patients after R0-1 surgery for localized high- or intermediate-risk GISTs according to NIH criteria [72] | 3 | 2 y adjuvant imatinib 400 mg/d vs. no treatment | No | IFFS (primary endpoint changed from OS to IFFS) | 5 y IFFS: 87.0% (imatinib) vs. 84.1% (control), p = 0.21, HR 0.79. No OS benefits |
| Joensuu et al., JAMA 2020 [25] | 397 Patients resected for high-risk GISTs according to modified NIH criteria [62] | 3 | 1 y vs. 3 y adjuvant imatinib 400 mg/d | Yes (366/397) | RFS | 10 y RFS: 52.5% (3 y treatment) and 41.8% (1 y treatment), HR 0.66, p = 0.003 10 year OS: 79.0% (3 y treatment) and 65.3% (1 y treatment), p = 0.004, HR: 0.55. |
3.1.2. Progression of GISTs with KIT or PDGFRA Mutations after the First-Line Targeted Therapy
3.2. GISTs without KIT/PDGFRA Mutations
3.2.1. GISTs without KIT/PDGFRA Mutations and with Deficient SDH Complex
Targeted Therapy for GISTs without KIT/PDGFRA Mutations and with a Deficient SDH Complex
3.2.2. GISTs without KIT/PDGFRA Mutations and with a Mutation in Neurofibromin 1 (NF1)
3.2.3. GISTs without KIT/PDGFRA Mutations and with Mutations in BRAF
Targeted Therapy of GISTs without KIT/PDGFRA Mutations and with Mutations in BRAF
3.2.4. GISTs without KIT/PDGFRA Mutations and with Mutations in KRAS
3.2.5. GISTs without KIT/PDGFRA Mutations and with NTRK Alterations
Targeted Therapy of GISTs without KIT/PDGFRA Mutations and with NTRK Alterations
3.2.6. GISTs without KIT/PDGFRA Mutations and with FGFR Alterations
Targeted Therapy for GISTs without KIT/PDGFRA Mutations and with FGFR Alterations
3.2.7. GISTs without KIT/PDGFRA Mutations and with Very Rare Mutations of Unknown Clinical Significance
3.3. Future Directions
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wang, Z.H.; Liang, X.B.; Wang, Y.; Ma, G.L.; Qu, Y.Q.; Tian, X.W. Epidemiology survey of gastrointestinal stromal tumor in Shanxi Province in 2011. Zhonghua Yi Xue Za Zhi 2013, 93, 2541–2544. [Google Scholar] [PubMed]
- Chan, K.H.; Chan, C.W.; Chow, W.H.; Kwan, W.K.; Kong, C.K.; Mak, K.F.; Leung, M.Y.; Lau, L.K. Gastrointestinal stromal tumors in a cohort of Chinese patients in Hong Kong. World J. Gastroenterol. 2006, 12, 2223–2228. [Google Scholar] [CrossRef] [PubMed]
- Lv, M.; Wu, C.; Zheng, Y.; Zhao, N. Incidence and survival analysis of gastrointestinal stromal tumors in shanghai: A population-based study from 2001 to 2010. Gastroenterol. Res. Pract. 2014, 2014, 834136. [Google Scholar] [CrossRef]
- Cho, M.Y.; Sohn, J.H.; Kim, J.M.; Kim, K.M.; Park, Y.S.; Kim, W.H.; Jung, J.S.; Jung, E.S.; Jin, S.Y.; Kang, D.Y.; et al. Current trends in the epidemiological and pathological characteristics of gastrointestinal stromal tumors in Korea, 2003–2004. J. Korean Med. Sci. 2010, 25, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Chiang, N.J.; Chen, L.T.; Tsai, C.R.; Chang, J.S. The epidemiology of gastrointestinal stromal tumors in Taiwan, 1998–2008: A nation-wide cancer registry-based study. BMC Cancer 2014, 14, 102. [Google Scholar] [CrossRef]
- Steigen, S.E.; Eide, T.J. Trends in incidence and survival of mesenchymal neoplasm of the digestive tract within a defined population of northern Norway. Apmis 2006, 114, 192–200. [Google Scholar] [CrossRef]
- Søreide, K.; Sandvik, O.M.; Søreide, J.A.; Giljaca, V.; Jureckova, A.; Bulusu, V.R. Global epidemiology of gastrointestinal stromal tumours (GIST): A systematic review of population-based cohort studies. Cancer Epidemiol. 2016, 40, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Agaimy, A.; Wünsch, P.H.; Hofstaedter, F.; Blaszyk, H.; Rümmele, P.; Gaumann, A.; Dietmaier, W.; Hartmann, A. Minute gastric sclerosing stromal tumors (GIST tumorlets) are common in adults and frequently show c-KIT mutations. Am. J. Surg. Pathol. 2007, 31, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Kawanowa, K.; Sakuma, Y.; Sakurai, S.; Hishima, T.; Iwasaki, Y.; Saito, K.; Hosoya, Y.; Nakajima, T.; Funata, N. High incidence of microscopic gastrointestinal stromal tumors in the stomach. Hum. Pathol. 2006, 37, 1527–1535. [Google Scholar] [CrossRef] [PubMed]
- Corless, C.L.; Barnett, C.M.; Heinrich, M.C. Gastrointestinal stromal tumours: Origin and molecular oncology. Nat. Rev. Cancer 2011, 11, 865–878. [Google Scholar] [CrossRef]
- Ricci, R.; Giustiniani, M.C.; Gessi, M.; Lanza, P.; Castri, F.; Biondi, A.; Persiani, R.; Vecchio, F.M.; Risio, M. Telocytes are the physiological counterpart of inflammatory fibroid polyps and PDGFRA-mutant GISTs. J. Cell. Mol. Med. 2018, 22, 4856–4862. [Google Scholar] [CrossRef] [PubMed]
- Kondo, J.; Huh, W.J.; Franklin, J.L.; Heinrich, M.C.; Rubin, B.P.; Coffey, R.J. A smooth muscle-derived, Braf-driven mouse model of gastrointestinal stromal tumor (GIST): Evidence for an alternative GIST cell-of-origin. J. Pathol. 2020, 252, 441–450. [Google Scholar] [CrossRef] [PubMed]
- De Pinieux, G.; Karanian, M.; Le Loarer, F.; Le Guellec, S.; Chabaud, S.; Terrier, P.; Bouvier, C.; Batistella, M.; Neuville, A.; Robin, Y.M.; et al. Nationwide incidence of sarcomas and connective tissue tumors of intermediate malignancy over four years using an expert pathology review network. PLoS ONE 2021, 16, e0246958. [Google Scholar] [CrossRef] [PubMed]
- Hirota, S.; Isozaki, K.; Moriyama, Y.; Hashimoto, K.; Nishida, T.; Ishiguro, S.; Kawano, K.; Hanada, M.; Kurata, A.; Takeda, M.; et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998, 279, 577–580. [Google Scholar] [CrossRef]
- Ricci, R. Syndromic gastrointestinal stromal tumors. Hered. Cancer Clin. Pract. 2016, 14, 15. [Google Scholar] [CrossRef]
- Miettinen, M.; Killian, J.K.; Wang, Z.F.; Lasota, J.; Lau, C.; Jones, L.; Walker, R.; Pineda, M.; Zhu, Y.J.; Kim, S.Y.; et al. Immunohistochemical loss of succinate dehydrogenase subunit A (SDHA) in gastrointestinal stromal tumors (GISTs) signals SDHA germline mutation. Am. J. Surg. Pathol. 2013, 37, 234–240. [Google Scholar] [CrossRef]
- Stratakis, C.A.; Carney, J.A. The triad of paragangliomas, gastric stromal tumours and pulmonary chondromas (Carney triad), and the dyad of paragangliomas and gastric stromal sarcomas (Carney-Stratakis syndrome): Molecular genetics and clinical implications. J. Intern. Med. 2009, 266, 43–52. [Google Scholar] [CrossRef]
- Belinsky, M.G.; Rink, L.; von Mehren, M. Succinate dehydrogenase deficiency in pediatric and adult gastrointestinal stromal tumors. Front. Oncol. 2013, 3, 117. [Google Scholar] [CrossRef]
- Italiano, A.; Chen, C.-L.; Sung, Y.-S.; Singer, S.; DeMatteo, R.P.; LaQuaglia, M.P.; Besmer, P.; Socci, N.; Antonescu, C.R. SDHA loss of function mutations in a subset of young adult wild-type gastrointestinal stromal tumors. BMC Cancer 2012, 12, 408. [Google Scholar] [CrossRef]
- Janeway, K.A.; Liegl, B.; Harlow, A.; Le, C.; Perez-Atayde, A.; Kozakewich, H.; Corless, C.L.; Heinrich, M.C.; Fletcher, J.A. Pediatric KIT wild-type and platelet-derived growth factor receptor alpha-wild-type gastrointestinal stromal tumors share KIT activation but not mechanisms of genetic progression with adult gastrointestinal stromal tumors. Cancer Res. 2007, 67, 9084–9088. [Google Scholar] [CrossRef]
- Pantaleo, M.A.; Astolfi, A.; Urbini, M.; Nannini, M.; Paterini, P.; Indio, V.; Saponara, M.; Formica, S.; Ceccarelli, C.; Casadio, R.; et al. Analysis of all subunits, SDHA, SDHB, SDHC, SDHD, of the succinate dehydrogenase complex in KIT/PDGFRA wild-type GIST. Eur. J. Hum. Genet. 2014, 22, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Casali, P.G.; Blay, J.Y.; Abecassis, N.; Bajpai, J.; Bauer, S.; Biagini, R.; Bielack, S.; Bonvalot, S.; Boukovinas, I.; Bovee, J.V.M.G.; et al. Gastrointestinal stromal tumours: ESMO-EURACAN-GENTURIS Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2022, 33, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Network, N.C.C. NCCN Guidelines. Available online: https://www.nccn.org/guidelines/guidelines-detail?category=1&id=1507 (accessed on 9 December 2022).
- Verweij, J.; Casali, P.G.; Zalcberg, J.; LeCesne, A.; Reichardt, P.; Blay, J.Y.; Issels, R.; van Oosterom, A.; Hogendoorn, P.C.; Van Glabbeke, M.; et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: Randomised trial. Lancet 2004, 364, 1127–1134. [Google Scholar] [CrossRef] [PubMed]
- Joensuu, H.; Eriksson, M.; Sundby Hall, K.; Reichardt, A.; Hermes, B.; Schütte, J.; Cameron, S.; Hohenberger, P.; Jost, P.J.; Al-Batran, S.E.; et al. Survival Outcomes Associated with 3 Years vs. 1 Year of Adjuvant Imatinib for Patients with High-Risk Gastrointestinal Stromal Tumors: An Analysis of a Randomized Clinical Trial after 10-Year Follow-up. JAMA Oncol. 2020, 6, 1241–1246. [Google Scholar] [CrossRef] [PubMed]
- Demetri, G.D.; Benjamin, R.S.; Blanke, C.D.; Blay, J.Y.; Casali, P.; Choi, H.; Corless, C.L.; Debiec-Rychter, M.; DeMatteo, R.P.; Ettinger, D.S.; et al. NCCN Task Force report: Management of patients with gastrointestinal stromal tumor (GIST)—Update of the NCCN clinical practice guidelines. J. Natl. Compr. Canc. Netw. 2007, 5 (Suppl. 2), S1-29, quiz S30. [Google Scholar] [CrossRef]
- Pierotti, M.A.; Tamborini, E.; Negri, T.; Pricl, S.; Pilotti, S. Targeted therapy in GIST: In silico modeling for prediction of resistance. Nat. Rev. Clin. Oncol. 2011, 8, 161–170. [Google Scholar] [CrossRef]
- Brčić, I.; Argyropoulos, A.; Liegl-Atzwanger, B. Update on Molecular Genetics of Gastrointestinal Stromal Tumors. Diagnostics 2021, 11, 194. [Google Scholar] [CrossRef]
- Hu, X.; Wang, Z.; Su, P.; Zhang, Q.; Kou, Y. Advances in the research of the mechanism of secondary resistance to imatinib in gastrointestinal stromal tumors. Front. Oncol. 2022, 12, 933248. [Google Scholar] [CrossRef]
- Serrano, C.; Mariño-Enríquez, A.; Tao, D.L.; Ketzer, J.; Eilers, G.; Zhu, M.; Yu, C.; Mannan, A.M.; Rubin, B.P.; Demetri, G.D.; et al. Complementary activity of tyrosine kinase inhibitors against secondary kit mutations in imatinib-resistant gastrointestinal stromal tumours. Br. J. Cancer 2019, 120, 612–620. [Google Scholar] [CrossRef]
- Heinrich, M.C.; Corless, C.L.; Duensing, A.; McGreevey, L.; Chen, C.J.; Joseph, N.; Singer, S.; Griffith, D.J.; Haley, A.; Town, A.; et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science 2003, 299, 708–710. [Google Scholar] [CrossRef]
- Klug, L.R.; Khosroyani, H.M.; Kent, J.D.; Heinrich, M.C. New treatment strategies for advanced-stage gastrointestinal stromal tumours. Nat. Rev. Clin. Oncol. 2022, 19, 328–341. [Google Scholar] [CrossRef]
- Astolfi, A.; Indio, V.; Nannini, M.; Saponara, M.; Schipani, A.; De Leo, A.; Altimari, A.; Vincenzi, B.; Comandini, D.; Grignani, G.; et al. Targeted Deep Sequencing Uncovers Cryptic KIT Mutations in KIT/PDGFRA/SDH/RAS-P Wild-Type GIST. Front. Oncol. 2020, 10, 504. [Google Scholar] [CrossRef]
- Unk, M.; Bombač, A.; Jezeršek Novaković, B.; Stegel, V.; Šetrajčič Dragoš, V.; Blatnik, O.; Klančar, G.; Novaković, S. Correlation of treatment outcome in sanger/RT-qPCR. Oncol. Rep. 2022, 48, 167. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.C.; Rankin, C.; Blanke, C.D.; Demetri, G.D.; Borden, E.C.; Ryan, C.W.; von Mehren, M.; Blackstein, M.E.; Priebat, D.A.; Tap, W.D.; et al. Correlation of Long-term Results of Imatinib in Advanced Gastrointestinal Stromal Tumors with Next-Generation Sequencing Results: Analysis of Phase 3 SWOG Intergroup Trial S0033. JAMA Oncol. 2017, 3, 944–952. [Google Scholar] [CrossRef] [PubMed]
- Vanden Bempt, I.; Vander Borght, S.; Sciot, R.; Spans, L.; Claerhout, S.; Brems, H.; Lehnert, S.; Dehaspe, L.; Fransis, S.; Neuville, B.; et al. Comprehensive targeted next-generation sequencing approach in the molecular diagnosis of gastrointestinal stromal tumor. Genes Chromosomes Cancer 2021, 60, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Bombac, A.; Zakotnik, B.; Bucic, M.; Setrajcic Dragos, V.; Gazic, B.; Stegel, V.; Klancar, G.; Novakovic, S. Mutational spectrum and classification of novel mutations in patients with metastatic gastrointestinal stromal tumours. Int. J. Oncol. 2020, 56, 1468–1478. [Google Scholar] [CrossRef] [PubMed]
- Blay, J.Y.; Kang, Y.K.; Nishida, T.; von Mehren, M. Gastrointestinal stromal tumours. Nat. Rev. Dis. Primers 2021, 7, 22. [Google Scholar] [CrossRef] [PubMed]
- Boikos, S.A.; Stratakis, C.A. The genetic landscape of gastrointestinal stromal tumor lacking KIT and PDGFRA mutations. Endocrine 2014, 47, 401–408. [Google Scholar] [CrossRef]
- Boikos, S.A.; Pappo, A.S.; Killian, J.K.; LaQuaglia, M.P.; Weldon, C.B.; George, S.; Trent, J.C.; von Mehren, M.; Wright, J.A.; Schiffman, J.D.; et al. Molecular Subtypes of KIT/PDGFRA Wild-Type Gastrointestinal Stromal Tumors: A Report from the National Institutes of Health Gastrointestinal Stromal Tumor Clinic. JAMA Oncol. 2016, 2, 922–928. [Google Scholar] [CrossRef]
- Janeway, K.A.; Kim, S.Y.; Lodish, M.; Nosé, V.; Rustin, P.; Gaal, J.; Dahia, P.L.; Liegl, B.; Ball, E.R.; Raygada, M.; et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc. Natl. Acad. Sci. USA 2011, 108, 314–318. [Google Scholar] [CrossRef]
- Lasota, J.; Xi, L.; Coates, T.; Dennis, R.; Evbuomwan, M.O.; Wang, Z.F.; Raffeld, M.; Miettinen, M. No KRAS mutations found in gastrointestinal stromal tumors (GISTs): Molecular genetic study of 514 cases. Mod. Pathol. 2013, 26, 1488–1491. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zeng, X.; Wu, X.; He, J.; Gao, J.; Shuai, X.; Wang, G.; Zhang, P.; Tao, K. Clinicopathologic study of succinate-dehydrogenase-deficient gastrointestinal stromal tumors: A single-institutional experience in China. Medicine 2017, 96, e7668. [Google Scholar] [CrossRef] [PubMed]
- Mathias-Machado, M.C.; de Jesus, V.H.F.; de Carvalho Oliveira, L.J.; Neumann, M.; Peixoto, R.D. Current Molecular Profile of Gastrointestinal Stromal Tumors and Systemic Therapeutic Implications. Cancers 2022, 14, 5330. [Google Scholar] [CrossRef] [PubMed]
- Mei, L.; Smith, S.C.; Faber, A.C.; Trent, J.; Grossman, S.R.; Stratakis, C.A.; Boikos, S.A. Gastrointestinal Stromal Tumors: The GIST of Precision Medicine. Trends Cancer 2018, 4, 74–91. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, M.; Lasota, J. Succinate dehydrogenase deficient gastrointestinal stromal tumors (GISTs)—A review. Int. J. Biochem. Cell Biol. 2014, 53, 514–519. [Google Scholar] [CrossRef]
- Astolfi, A.; Pantaleo, M.A.; Indio, V.; Urbini, M.; Nannini, M. The Emerging Role of the FGF/FGFR Pathway in Gastrointestinal Stromal Tumor. Int. J. Mol. Sci. 2020, 21, 3313. [Google Scholar] [CrossRef]
- Nannini, M.; Biasco, G.; Astolfi, A.; Pantaleo, M.A. An overview on molecular biology of KIT/PDGFRA wild type (WT) gastrointestinal stromal tumours (GIST). J. Med. Genet. 2013, 50, 653–661. [Google Scholar] [CrossRef]
- Nannini, M.; Urbini, M.; Astolfi, A.; Biasco, G.; Pantaleo, M.A. The progressive fragmentation of the KIT/PDGFRA wild-type (WT) gastrointestinal stromal tumors (GIST). J. Transl. Med. 2017, 15, 113. [Google Scholar] [CrossRef]
- Pantaleo, M.A.; Nannini, M.; Corless, C.L.; Heinrich, M.C. Quadruple wild-type (WT) GIST: Defining the subset of GIST that lacks abnormalities of KIT, PDGFRA, SDH, or RAS signaling pathways. Cancer Med. 2015, 4, 101–103. [Google Scholar] [CrossRef]
- Pantaleo, M.A.; Urbini, M.; Indio, V.; Ravegnini, G.; Nannini, M.; De Luca, M.; Tarantino, G.; Angelini, S.; Gronchi, A.; Vincenzi, B.; et al. Genome-Wide Analysis Identifies MEN1 and MAX Mutations and a Neuroendocrine-Like Molecular Heterogeneity in Quadruple WT GIST. Mol. Cancer Res. 2017, 15, 553–562. [Google Scholar] [CrossRef]
- Wozniak, A.; Rutkowski, P.; Schöffski, P.; Ray-Coquard, I.; Hostein, I.; Schildhaus, H.U.; Le Cesne, A.; Bylina, E.; Limon, J.; Blay, J.Y.; et al. Tumor genotype is an independent prognostic factor in primary gastrointestinal stromal tumors of gastric origin: A european multicenter analysis based on ConticaGIST. Clin. Cancer Res. 2014, 20, 6105–6116. [Google Scholar] [CrossRef] [PubMed]
- (MetaGIST), G.S.T.M.-A.G. Comparison of two doses of imatinib for the treatment of unresectable or metastatic gastrointestinal stromal tumors: A meta-analysis of 1640 patients. J. Clin. Oncol. 2010, 28, 1247–1253. [Google Scholar] [CrossRef]
- Szucs, Z.; Thway, K.; Fisher, C.; Bulusu, R.; Constantinidou, A.; Benson, C.; van der Graaf, W.T.; Jones, R.L. Molecular subtypes of gastrointestinal stromal tumors and their prognostic and therapeutic implications. Future Oncol. 2017, 13, 93–107. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Liu, Q. Prognostic Indicators for Gastrointestinal Stromal Tumors: A Review. Transl. Oncol. 2020, 13, 100812. [Google Scholar] [CrossRef]
- Yarden, Y.; Kuang, W.J.; Yang-Feng, T.; Coussens, L.; Munemitsu, S.; Dull, T.J.; Chen, E.; Schlessinger, J.; Francke, U.; Ullrich, A. Human proto-oncogene c-kit: A new cell surface receptor tyrosine kinase for an unidentified ligand. EMBO J. 1987, 6, 3341–3351. [Google Scholar] [CrossRef]
- Sheikh, E.; Tran, T.; Vranic, S.; Levy, A.; Bonfil, R.D. Role and significance of c-KIT receptor tyrosine kinase in cancer: A review. Bosn. J. Basic Med. Sci. 2022, 22, 683–698. [Google Scholar] [CrossRef]
- Lennartsson, J.; Rönnstrand, L. Stem cell factor receptor/c-Kit: From basic science to clinical implications. Physiol. Rev. 2012, 92, 1619–1649. [Google Scholar] [CrossRef]
- Kelly, C.M.; Gutierrez Sainz, L.; Chi, P. The management of metastatic GIST: Current standard and investigational therapeutics. J. Hematol. Oncol. 2021, 14, 2. [Google Scholar] [CrossRef]
- Joensuu, H.; Eriksson, M.; Sundby Hall, K.; Hartmann, J.T.; Pink, D.; Schütte, J.; Ramadori, G.; Hohenberger, P.; Duyster, J.; Al-Batran, S.E.; et al. One vs three years of adjuvant imatinib for operable gastrointestinal stromal tumor: A randomized trial. JAMA 2012, 307, 1265–1272. [Google Scholar] [CrossRef]
- Joensuu, H.; Wardelmann, E.; Sihto, H.; Eriksson, M.; Sundby Hall, K.; Reichardt, A.; Hartmann, J.T.; Pink, D.; Cameron, S.; Hohenberger, P.; et al. Effect of KIT and PDGFRA Mutations on Survival in Patients with Gastrointestinal Stromal Tumors Treated with Adjuvant Imatinib: An Exploratory Analysis of a Randomized Clinical Trial. JAMA Oncol. 2017, 3, 602–609. [Google Scholar] [CrossRef]
- Joensuu, H. Risk stratification of patients diagnosed with gastrointestinal stromal tumor. Hum. Pathol. 2008, 39, 1411–1419. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, M.; Lasota, J. Gastrointestinal stromal tumors: Pathology and prognosis at different sites. Semin. Diagn. Pathol. 2006, 23, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.N.; Chen, M.H.; Chen, Y.Y.; Yang, C.Y.; Yen, C.C.; Tzen, C.Y.; Chen, L.T.; Chen, J.S. A phase II trial of regorafenib in patients with metastatic and/or a unresectable gastrointestinal stromal tumor harboring secondary mutations of exon 17. Oncotarget 2017, 8, 44121–44130. [Google Scholar] [CrossRef] [PubMed]
- Vincenzi, B.; Napolitano, A.; Fiocco, M.; Mir, O.; Rutkowski, P.; Blay, J.Y.; Reichardt, P.; Joensuu, H.; Fumagalli, E.; Gennatas, S.; et al. Adjuvant Imatinib in Patients with GIST Harboring Exon 9 KIT Mutations: Results from a Multi-institutional European Retrospective Study. Clin. Cancer Res. 2022, 28, 1672–1679. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.C.; Owzar, K.; Corless, C.L.; Hollis, D.; Borden, E.C.; Fletcher, C.D.; Ryan, C.W.; von Mehren, M.; Blanke, C.D.; Rankin, C.; et al. Correlation of kinase genotype and clinical outcome in the North American Intergroup Phase III Trial of imatinib mesylate for treatment of advanced gastrointestinal stromal tumor: CALGB 150105 Study by Cancer and Leukemia Group B and Southwest Oncology Group. J. Clin. Oncol. 2008, 26, 5360–5367. [Google Scholar] [CrossRef] [PubMed]
- Debiec-Rychter, M.; Sciot, R.; Le Cesne, A.; Schlemmer, M.; Hohenberger, P.; van Oosterom, A.T.; Blay, J.Y.; Leyvraz, S.; Stul, M.; Casali, P.G.; et al. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur. J. Cancer 2006, 42, 1093–1103. [Google Scholar] [CrossRef]
- Raut, C.P.; Espat, N.J.; Maki, R.G.; Araujo, D.M.; Trent, J.; Williams, T.F.; Purkayastha, D.D.; DeMatteo, R.P. Efficacy and Tolerability of 5-Year Adjuvant Imatinib Treatment for Patients with Resected Intermediate- or High-Risk Primary Gastrointestinal Stromal Tumor: The PERSIST-5 Clinical Trial. JAMA Oncol. 2018, 4, e184060. [Google Scholar] [CrossRef]
- Patrikidou, A.; Domont, J.; Chabaud, S.; Ray-Coquard, I.; Coindre, J.M.; Bui-Nguyen, B.; Adenis, A.; Rios, M.; Bertucci, F.; Duffaud, F.; et al. Long-term outcome of molecular subgroups of GIST patients treated with standard-dose imatinib in the BFR14 trial of the French Sarcoma Group. Eur. J. Cancer 2016, 52, 173–180. [Google Scholar] [CrossRef]
- Dematteo, R.P.; Ballman, K.V.; Antonescu, C.R.; Maki, R.G.; Pisters, P.W.; Demetri, G.D.; Blackstein, M.E.; Blanke, C.D.; von Mehren, M.; Brennan, M.F.; et al. Adjuvant imatinib mesylate after resection of localised, primary gastrointestinal stromal tumour: A randomised, double-blind, placebo-controlled trial. Lancet 2009, 373, 1097–1104. [Google Scholar] [CrossRef]
- Casali, P.G.; Le Cesne, A.; Poveda Velasco, A.; Kotasek, D.; Rutkowski, P.; Hohenberger, P.; Fumagalli, E.; Judson, I.R.; Italiano, A.; Gelderblom, H.; et al. Time to Definitive Failure to the First Tyrosine Kinase Inhibitor in Localized GI Stromal Tumors Treated with Imatinib as an Adjuvant: A European Organisation for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group Intergroup Randomized Trial in Collaboration with the Australasian Gastro-Intestinal Trials Group, UNICANCER, French Sarcoma Group, Italian Sarcoma Group, and Spanish Group for Research on Sarcomas. J. Clin. Oncol. 2015, 33, 4276–4283. [Google Scholar] [CrossRef]
- Fletcher, C.D.; Berman, J.J.; Corless, C.; Gorstein, F.; Lasota, J.; Longley, B.J.; Miettinen, M.; O’Leary, T.J.; Remotti, H.; Rubin, B.P.; et al. Diagnosis of gastrointestinal stromal tumors: A consensus approach. Hum. Pathol. 2002, 33, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.C.; Corless, C.L.; Demetri, G.D.; Blanke, C.D.; von Mehren, M.; Joensuu, H.; McGreevey, L.S.; Chen, C.J.; Van den Abbeele, A.D.; Druker, B.J.; et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J. Clin. Oncol. 2003, 21, 4342–4349. [Google Scholar] [CrossRef] [PubMed]
- Blanke, C.D.; Demetri, G.D.; von Mehren, M.; Heinrich, M.C.; Eisenberg, B.; Fletcher, J.A.; Corless, C.L.; Fletcher, C.D.; Roberts, P.J.; Heinz, D.; et al. Long-term results from a randomized phase II trial of standard- versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J. Clin. Oncol. 2008, 26, 620–625. [Google Scholar] [CrossRef]
- Blanke, C.D.; Rankin, C.; Demetri, G.D.; Ryan, C.W.; von Mehren, M.; Benjamin, R.S.; Raymond, A.K.; Bramwell, V.H.; Baker, L.H.; Maki, R.G.; et al. Phase III randomized, intergroup trial assessing imatinib mesylate at two dose levels in patients with unresectable or metastatic gastrointestinal stromal tumors expressing the kit receptor tyrosine kinase: S0033. J. Clin. Oncol. 2008, 26, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Demetri, G.D.; von Mehren, M.; Blanke, C.D.; Van den Abbeele, A.D.; Eisenberg, B.; Roberts, P.J.; Heinrich, M.C.; Tuveson, D.A.; Singer, S.; Janicek, M.; et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N. Engl. J. Med. 2002, 347, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Grunewald, S.; Klug, L.R.; Mühlenberg, T.; Lategahn, J.; Falkenhorst, J.; Town, A.; Ehrt, C.; Wardelmann, E.; Hartmann, W.; Schildhaus, H.U.; et al. Resistance to Avapritinib in PDGFRA-Driven GIST Is Caused by Secondary Mutations in the PDGFRA Kinase Domain. Cancer Discov. 2021, 11, 108–125. [Google Scholar] [CrossRef]
- Ravegnini, G.; Urbini, M.; Simeon, V.; Genovese, C.; Astolfi, A.; Nannini, M.; Gatto, L.; Saponara, M.; Ianni, M.; Indio, V.; et al. An exploratory study by DMET array identifies a germline signature associated with imatinib response in gastrointestinal stromal tumor. Pharm. J. 2019, 19, 390–400. [Google Scholar] [CrossRef]
- Corless, C.L.; Schroeder, A.; Griffith, D.; Town, A.; McGreevey, L.; Harrell, P.; Shiraga, S.; Bainbridge, T.; Morich, J.; Heinrich, M.C. PDGFRA mutations in gastrointestinal stromal tumors: Frequency, spectrum and in vitro sensitivity to imatinib. J. Clin. Oncol. 2005, 23, 5357–5364. [Google Scholar] [CrossRef]
- Cassier, P.A.; Fumagalli, E.; Rutkowski, P.; Schöffski, P.; Van Glabbeke, M.; Debiec-Rychter, M.; Emile, J.F.; Duffaud, F.; Martin-Broto, J.; Landi, B.; et al. Outcome of patients with platelet-derived growth factor receptor alpha-mutated gastrointestinal stromal tumors in the tyrosine kinase inhibitor era. Clin. Cancer Res. 2012, 18, 4458–4464. [Google Scholar] [CrossRef]
- Yoo, C.; Ryu, M.H.; Jo, J.; Park, I.; Ryoo, B.Y.; Kang, Y.K. Efficacy of Imatinib in Patients with Platelet-Derived Growth Factor Receptor Alpha-Mutated Gastrointestinal Stromal Tumors. Cancer Res. Treat. 2016, 48, 546–552. [Google Scholar] [CrossRef]
- Farag, S.; Somaiah, N.; Choi, H.; Heeres, B.; Wang, W.L.; van Boven, H.; Nederlof, P.; Benjamin, R.; van der Graaf, W.; Grunhagen, D.; et al. Clinical characteristics and treatment outcome in a large multicentre observational cohort of PDGFRA exon 18 mutated gastrointestinal stromal tumour patients. Eur. J. Cancer 2017, 76, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.I.E. Enigmas in tumor resistance to kinase inhibitors and calculation of the drug resistance index for cancer (DRIC). Semin. Cancer Biol. 2017, 45, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Casali, P.G.; Zalcberg, J.; Le Cesne, A.; Reichardt, P.; Blay, J.Y.; Lindner, L.H.; Judson, I.R.; Schöffski, P.; Leyvraz, S.; Italiano, A.; et al. Ten-Year Progression-Free and Overall Survival in Patients with Unresectable or Metastatic GI Stromal Tumors: Long-Term Analysis of the European Organisation for Research and Treatment of Cancer, Italian Sarcoma Group, and Australasian Gastrointestinal Trials Group Intergroup Phase III Randomized Trial on Imatinib at Two Dose Levels. J. Clin. Oncol. 2017, 35, 1713–1720. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.C.; Corless, C.L.; Blanke, C.D.; Demetri, G.D.; Joensuu, H.; Roberts, P.J.; Eisenberg, B.L.; von Mehren, M.; Fletcher, C.D.; Sandau, K.; et al. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J. Clin. Oncol. 2006, 24, 4764–4774. [Google Scholar] [CrossRef] [PubMed]
- Demetri, G.D.; van Oosterom, A.T.; Garrett, C.R.; Blackstein, M.E.; Shah, M.H.; Verweij, J.; McArthur, G.; Judson, I.R.; Heinrich, M.C.; Morgan, J.A.; et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: A randomised controlled trial. Lancet 2006, 368, 1329–1338. [Google Scholar] [CrossRef]
- Heinrich, M.C.; vonMehren, M.; Demetri, G.D.; Fletcher, J.A.; Sun, J.; Hodgson, J.G.; Rivera, V.M.; Turner, C.D.; George, S. A phase 2 study of ponatinib in patients (pts) with advanced gastrointestinal stromal tumors (GIST) after failure of tyrosine kinase inhibitor (TKI) therapy: Initial report. J. Clin. Oncol. 2014, 32, 10506. [Google Scholar] [CrossRef]
- Lostes-Bardaji, M.J.; García-Illescas, D.; Valverde, C.; Serrano, C. Ripretinib in gastrointestinal stromal tumor: The long-awaited step forward. Ther. Adv. Med. Oncol. 2021, 13, 1758835920986498. [Google Scholar] [CrossRef]
- Blay, J.Y.; Serrano, C.; Heinrich, M.C.; Zalcberg, J.; Bauer, S.; Gelderblom, H.; Schöffski, P.; Jones, R.L.; Attia, S.; D’Amato, G.; et al. Ripretinib in patients with advanced gastrointestinal stromal tumours (INVICTUS): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2020, 21, 923–934. [Google Scholar] [CrossRef]
- Heinrich, M.C.; Jones, R.L.; von Mehren, M.; Schöffski, P.; Serrano, C.; Kang, Y.K.; Cassier, P.A.; Mir, O.; Eskens, F.; Tap, W.D.; et al. Avapritinib in advanced PDGFRA D842V-mutant gastrointestinal stromal tumour (NAVIGATOR): A multicentre, open-label, phase 1 trial. Lancet Oncol. 2020, 21, 935–946. [Google Scholar] [CrossRef]
- Kang, Y.K.; George, S.; Jones, R.L.; Rutkowski, P.; Shen, L.; Mir, O.; Patel, S.; Zhou, Y.; von Mehren, M.; Hohenberger, P.; et al. Avapritinib Versus Regorafenib in Locally Advanced Unresectable or Metastatic GI Stromal Tumor: A Randomized, Open-Label Phase III Study. J. Clin. Oncol. 2021, 39, 3128–3139. [Google Scholar] [CrossRef]
- Demetri, G.D.; Reichardt, P.; Kang, Y.K.; Blay, J.Y.; Rutkowski, P.; Gelderblom, H.; Hohenberger, P.; Leahy, M.; von Mehren, M.; Joensuu, H.; et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013, 381, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, M.C.; Marino-Enriquez, A.; Presnell, A.; Donsky, R.S.; Griffith, D.J.; McKinley, A.; Patterson, J.; Taguchi, T.; Liang, C.W.; Fletcher, J.A. Sorafenib inhibits many kinase mutations associated with drug-resistant gastrointestinal stromal tumors. Mol. Cancer Ther. 2012, 11, 1770–1780. [Google Scholar] [CrossRef] [PubMed]
- Reichardt, P.; Blay, J.Y.; Gelderblom, H.; Schlemmer, M.; Demetri, G.D.; Bui-Nguyen, B.; McArthur, G.A.; Yazji, S.; Hsu, Y.; Galetic, I.; et al. Phase III study of nilotinib versus best supportive care with or without a TKI in patients with gastrointestinal stromal tumors resistant to or intolerant of imatinib and sunitinib. Ann. Oncol. 2012, 23, 1680–1687. [Google Scholar] [CrossRef]
- Gardino, A.K.; Evans, E.K.; Kim, J.L.; Brooijmans, N.; Hodous, B.L.; Wolf, B.; Lengauer, C. Targeting kinases with precision. Mol. Cell. Oncol. 2018, 5, e1435183. [Google Scholar] [CrossRef] [PubMed]
- Serrano, C.; George, S.; Valverde, C.; Olivares, D.; García-Valverde, A.; Suárez, C.; Morales-Barrera, R.; Carles, J. Novel Insights into the Treatment of Imatinib-Resistant Gastrointestinal Stromal Tumors. Target. Oncol. 2017, 12, 277–288. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. Hazardous Substances Data Bank; National Library of Medicine: Bethesda, MD, USA, 2022.
- Javidi-Sharifi, N.; Traer, E.; Martinez, J.; Gupta, A.; Taguchi, T.; Dunlap, J.; Heinrich, M.C.; Corless, C.L.; Rubin, B.P.; Druker, B.J.; et al. Crosstalk between KIT and FGFR3 Promotes Gastrointestinal Stromal Tumor Cell Growth and Drug Resistance. Cancer Res. 2015, 75, 880–891. [Google Scholar] [CrossRef]
- Lasota, J.; Felisiak-Golabek, A.; Wasag, B.; Kowalik, A.; Zięba, S.; Chłopek, M.; Wang, Z.F.; Coates, T.; Kopczynski, J.; Gozdz, S.; et al. Frequency and clinicopathologic profile of PIK3CA mutant GISTs: Molecular genetic study of 529 cases. Mod. Pathol. 2016, 29, 275–282. [Google Scholar] [CrossRef]
- Li, K.; Cheng, H.; Li, Z.; Pang, Y.; Jia, X.; Xie, F.; Hu, G.; Cai, Q.; Wang, Y. Genetic progression in gastrointestinal stromal tumors: Mechanisms and molecular interventions. Oncotarget 2017, 8, 60589–60604. [Google Scholar] [CrossRef]
- Reichardt, P.; Demetri, G.D.; Gelderblom, H.; Rutkowski, P.; Im, S.A.; Gupta, S.; Kang, Y.K.; Schöffski, P.; Schuette, J.; Soulières, D.; et al. Correlation of KIT and PDGFRA mutational status with clinical benefit in patients with gastrointestinal stromal tumor treated with sunitinib in a worldwide treatment-use trial. BMC Cancer 2016, 16, 22. [Google Scholar] [CrossRef]
- Heinrich, M.C.; Maki, R.G.; Corless, C.L.; Antonescu, C.R.; Harlow, A.; Griffith, D.; Town, A.; McKinley, A.; Ou, W.B.; Fletcher, J.A.; et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J. Clin. Oncol. 2008, 26, 5352–5359. [Google Scholar] [CrossRef]
- Janku, F.; Abdul Razak, A.R.; Chi, P.; Heinrich, M.C.; von Mehren, M.; Jones, R.L.; Ganjoo, K.; Trent, J.; Gelderblom, H.; Somaiah, N.; et al. Switch Control Inhibition of KIT and PDGFRA in Patients with Advanced Gastrointestinal Stromal Tumor: A Phase I Study of Ripretinib. J. Clin. Oncol. 2020, 38, 3294–3303. [Google Scholar] [CrossRef] [PubMed]
- George, S.; Jones, R.L.; Bauer, S.; Kang, Y.K.; Schöffski, P.; Eskens, F.; Mir, O.; Cassier, P.A.; Serrano, C.; Tap, W.D.; et al. Avapritinib in Patients with Advanced Gastrointestinal Stromal Tumors Following at Least Three Prior Lines of Therapy. Oncologist 2021, 26, e639–e649. [Google Scholar] [CrossRef] [PubMed]
- Green, S.; Weiss, G.R. Southwest Oncology Group standard response criteria, endpoint definitions and toxicity criteria. Investig. New Drugs 1992, 10, 239–253. [Google Scholar] [CrossRef] [PubMed]
- George, S.; Blay, J.Y.; Casali, P.G.; Le Cesne, A.; Stephenson, P.; Deprimo, S.E.; Harmon, C.S.; Law, C.N.; Morgan, J.A.; Ray-Coquard, I.; et al. Clinical evaluation of continuous daily dosing of sunitinib malate in patients with advanced gastrointestinal stromal tumour after imatinib failure. Eur. J. Cancer 2009, 45, 1959–1968. [Google Scholar] [CrossRef] [PubMed]
- Nannini, M.; Astolfi, A.; Paterini, P.; Urbini, M.; Santini, D.; Catena, F.; Indio, V.; Casadio, R.; Pinna, A.D.; Biasco, G.; et al. Expression of IGF-1 receptor in KIT/PDGF receptor-α wild-type gastrointestinal stromal tumors with succinate dehydrogenase complex dysfunction. Future Oncol. 2013, 9, 121–126. [Google Scholar] [CrossRef]
- Oudijk, L.; Gaal, J.; Korpershoek, E.; van Nederveen, F.H.; Kelly, L.; Schiavon, G.; Verweij, J.; Mathijssen, R.H.J.; den Bakker, M.A.; Oldenburg, R.A.; et al. SDHA mutations in adult and pediatric wild-type gastrointestinal stromal tumors. Mod. Pathol. 2013, 26, 456–463. [Google Scholar] [CrossRef]
- Rutkowski, P.; Magnan, H.; Chou, A.J.; Benson, C. Treatment of gastrointestinal stromal tumours in paediatric and young adult patients with sunitinib: A multicentre case series. BMC Cancer 2017, 17, 717. [Google Scholar] [CrossRef]
- Andrzejewska, M.; Czarny, J.; Derwich, K. Latest Advances in the Management of Pediatric Gastrointestinal Stromal Tumors. Cancers 2022, 14, 4989. [Google Scholar] [CrossRef]
- Janeway, K.A.; Albritton, K.H.; Van Den Abbeele, A.D.; D’Amato, G.Z.; Pedrazzoli, P.; Siena, S.; Picus, J.; Butrynski, J.E.; Schlemmer, M.; Heinrich, M.C.; et al. Sunitinib treatment in pediatric patients with advanced GIST following failure of imatinib. Pediatr. Blood Cancer 2009, 52, 767–771. [Google Scholar] [CrossRef]
- Ganjoo, K.N.; Villalobos, V.M.; Kamaya, A.; Fisher, G.A.; Butrynski, J.E.; Morgan, J.A.; Wagner, A.J.; D’Adamo, D.; McMillan, A.; Demetri, G.D.; et al. A multicenter phase II study of pazopanib in patients with advanced gastrointestinal stromal tumors (GIST) following failure of at least imatinib and sunitinib. Ann. Oncol. 2014, 25, 236–240. [Google Scholar] [CrossRef]
- Von Mehren, M.; George, S.; Heinrich, M.C.; Schuetze, S.M.; Yap, J.T.; Yu, J.Q.; Abbott, A.; Litwin, S.; Crowley, J.; Belinsky, M.; et al. Linsitinib (OSI-906) for the Treatment of Adult and Pediatric Wild-Type Gastrointestinal Stromal Tumors, a SARC Phase II Study. Clin. Cancer Res. 2020, 26, 1837–1845. [Google Scholar] [CrossRef] [PubMed]
- Yebra, M.; Bhargava, S.; Kumar, A.; Burgoyne, A.M.; Tang, C.M.; Yoon, H.; Banerjee, S.; Aguilera, J.; Cordes, T.; Sheth, V.; et al. Establishment of Patient-Derived Succinate Dehydrogenase-Deficient Gastrointestinal Stromal Tumor Models for Predicting Therapeutic Response. Clin. Cancer Res. 2022, 28, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Burgoyne, A.; Heinrich, M.C.; von Mehren, M.; Trent, J.C.; Messer, E.; Messer, K.; Metallo, C.; Sicklick, J.K. An open-label, phase 2 efficacy study of temozolomide in advanced succinate dehydrogenase-mutant/deficient gastrointestinal stromal tumor. In Proceedings of the CTOS 2022 Annual Meeting, Vancouver, BC, Canada, 16–19 November 2022. [Google Scholar]
- Bergoug, M.; Doudeau, M.; Godin, F.; Mosrin, C.; Vallée, B.; Bénédetti, H. Neurofibromin Structure, Functions and Regulation. Cells 2020, 9, 2365. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wei, C.-J.; Cui, X.-W.; Li, Y.-H.; Gu, Y.-H.; Gu, B.; Li, Q.-F.; Wang, Z.-C. Impacts of NF1 Gene Mutations and Genetic Modifiers in Neurofibromatosis Type 1. Front. Neurol. 2021, 12, 704639. [Google Scholar] [CrossRef] [PubMed]
- Gasparotto, D.; Rossi, S.; Polano, M.; Tamborini, E.; Lorenzetto, E.; Sbaraglia, M.; Mondello, A.; Massani, M.; Lamon, S.; Bracci, R.; et al. Quadruple-Negative GIST Is a Sentinel for Unrecognized Neurofibromatosis Type 1 Syndrome. Clin. Cancer Res. 2017, 23, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Maertens, O.; Prenen, H.; Debiec-Rychter, M.; Wozniak, A.; Sciot, R.; Pauwels, P.; De Wever, I.; Vermeesch, J.R.; de Raedt, T.; De Paepe, A.; et al. Molecular pathogenesis of multiple gastrointestinal stromal tumors in NF1 patients. Hum. Mol. Genet. 2006, 15, 1015–1023. [Google Scholar] [CrossRef]
- Miettinen, M.; Fetsch, J.F.; Sobin, L.H.; Lasota, J. Gastrointestinal stromal tumors in patients with neurofibromatosis 1: A clinicopathologic and molecular genetic study of 45 cases. Am. J. Surg. Pathol. 2006, 30, 90–96. [Google Scholar] [CrossRef]
- Yamamoto, H.; Tobo, T.; Nakamori, M.; Imamura, M.; Kojima, A.; Oda, Y.; Nakamura, N.; Takahira, T.; Yao, T.; Tsuneyoshi, M. Neurofibromatosis type 1-related gastrointestinal stromal tumors: A special reference to loss of heterozygosity at 14q and 22q. J. Cancer Res. Clin. Oncol. 2009, 135, 791–798. [Google Scholar] [CrossRef]
- Burgoyne, A.M.; De Siena, M.; Alkhuziem, M.; Tang, C.M.; Medina, B.; Fanta, P.T.; Belinsky, M.G.; von Mehren, M.; Thorson, J.A.; Madlensky, L.; et al. Duodenal-Jejunal Flexure GI Stromal Tumor Frequently Heralds Somatic. JCO Precis. Oncol. 2017, 2017, PO.17.00014. [Google Scholar] [CrossRef]
- Mussi, C.; Schildhaus, H.U.; Gronchi, A.; Wardelmann, E.; Hohenberger, P. Therapeutic consequences from molecular biology for gastrointestinal stromal tumor patients affected by neurofibromatosis type 1. Clin. Cancer Res. 2008, 14, 4550–4555. [Google Scholar] [CrossRef]
- Śmiech, M.; Leszczyński, P.; Kono, H.; Wardell, C.; Taniguchi, H. Emerging BRAF Mutations in Cancer Progression and Their Possible Effects on Transcriptional Networks. Genes 2020, 11, 1342. [Google Scholar] [CrossRef] [PubMed]
- Hostein, I.; Faur, N.; Primois, C.; Boury, F.; Denard, J.; Emile, J.F.; Bringuier, P.P.; Scoazec, J.Y.; Coindre, J.M. BRAF mutation status in gastrointestinal stromal tumors. Am. J. Clin. Pathol. 2010, 133, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Cohn, A.L.; Day, B.M.; Abhyankar, S.; McKenna, E.; Riehl, T.; Puzanov, I. BRAF(V600) mutations in solid tumors, other than metastatic melanoma and papillary thyroid cancer, or multiple myeloma: A screening study. Onco Targets Ther. 2017, 10, 965–971. [Google Scholar] [CrossRef] [PubMed]
- Huss, S.; Pasternack, H.; Ihle, M.A.; Merkelbach-Bruse, S.; Heitkötter, B.; Hartmann, W.; Trautmann, M.; Gevensleben, H.; Büttner, R.; Schildhaus, H.U.; et al. Clinicopathological and molecular features of a large cohort of gastrointestinal stromal tumors (GISTs) and review of the literature: BRAF mutations in KIT/PDGFRA wild-type GISTs are rare events. Hum. Pathol. 2017, 62, 206–214. [Google Scholar] [CrossRef]
- Agaimy, A.; Terracciano, L.M.; Dirnhofer, S.; Tornillo, L.; Foerster, A.; Hartmann, A.; Bihl, M.P. V600E BRAF mutations are alternative early molecular events in a subset of KIT/PDGFRA wild-type gastrointestinal stromal tumours. J. Clin. Pathol. 2009, 62, 613–616. [Google Scholar] [CrossRef]
- Daniels, M.; Lurkin, I.; Pauli, R.; Erbstösser, E.; Hildebrandt, U.; Hellwig, K.; Zschille, U.; Lüders, P.; Krüger, G.; Knolle, J.; et al. Spectrum of KIT/PDGFRA/BRAF mutations and Phosphatidylinositol-3-Kinase pathway gene alterations in gastrointestinal stromal tumors (GIST). Cancer Lett. 2011, 312, 43–54. [Google Scholar] [CrossRef]
- Jašek, K.; Váňová, B.; Grendár, M.; Štanclová, A.; Szépe, P.; Hornáková, A.; Holubeková, V.; Plank, L.; Lasabová, Z. BRAF mutations in KIT/PDGFRA positive gastrointestinal stromal tumours (GISTs): Is their frequency underestimated? Pathol. Res. Pract. 2020, 216, 153171. [Google Scholar] [CrossRef]
- Agaram, N.P.; Wong, G.C.; Guo, T.; Maki, R.G.; Singer, S.; Dematteo, R.P.; Besmer, P.; Antonescu, C.R. Novel V600E BRAF mutations in imatinib-naive and imatinib-resistant gastrointestinal stromal tumors. Genes Chromosomes Cancer 2008, 47, 853–859. [Google Scholar] [CrossRef]
- Nannini, M.; Valerio, D.S.; Gruppioni, E.; Altimari, A.; Chiusole, B.; Saponara, M.; Pantaleo, M.A.; Brunello, A. Complete radiological response to first-line regorafenib in a patient with abdominal relapse of BRAF V600E mutated GIST. Therap. Adv. Gastroenterol. 2020, 13, 1756284820927305. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. Dabrafenib–Trametinib Combination Approved for Solid Tumors with BRAF Mutations. Available online: https://www.cancer.gov/newsevents/cancer-currents-blog/2022/fda-dabrafenib-trametinib-braf-solidtumors (accessed on 9 December 2022).
- Falchook, G.S.; Trent, J.C.; Heinrich, M.C.; Beadling, C.; Patterson, J.; Bastida, C.C.; Blackman, S.C.; Kurzrock, R. BRAF mutant gastrointestinal stromal tumor: First report of regression with BRAF inhibitor dabrafenib (GSK2118436) and whole exomic sequencing for analysis of acquired resistance. Oncotarget 2013, 4, 310–315. [Google Scholar] [CrossRef]
- Charo, L.M.; Burgoyne, A.M.; Fanta, P.T.; Patel, H.; Chmielecki, J.; Sicklick, J.K.; McHale, M.T. A Novel PRKAR1B-BRAF Fusion in Gastrointestinal Stromal Tumor Guides Adjuvant Treatment Decision-Making during Pregnancy. J. Natl. Compr. Canc. Netw. 2018, 16, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Torrence, D.; Xie, Z.; Zhang, L.; Chi, P.; Antonescu, C.R. Gastrointestinal stromal tumors with BRAF gene fusions. A report of two cases showing low or absent KIT expression resulting in diagnostic pitfalls. Genes Chromosomes Cancer 2021, 60, 789–795. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Guo, Z.; Wang, F.; Fu, L. KRAS mutation: From undruggable to druggable in cancer. Signal Transduct. Target. Ther. 2021, 6, 386. [Google Scholar] [CrossRef] [PubMed]
- Ricci, R.; Dei Tos, A.P.; Rindi, G. On the prevalence of KRAS mutations in GISTs. Virchows Arch. 2013, 463, 847. [Google Scholar] [CrossRef]
- Hechtman, J.F.; Zehir, A.; Mitchell, T.; Borsu, L.; Singer, S.; Tap, W.; Oultache, A.; Ladanyi, M.; Nafa, K. Novel oncogene and tumor suppressor mutations in KIT and PDGFRA wild type gastrointestinal stromal tumors revealed by next generation sequencing. Genes Chromosomes Cancer 2015, 54, 177–184. [Google Scholar] [CrossRef]
- Miranda, C.; Nucifora, M.; Molinari, F.; Conca, E.; Anania, M.C.; Bordoni, A.; Saletti, P.; Mazzucchelli, L.; Pilotti, S.; Pierotti, M.A.; et al. KRAS and BRAF mutations predict primary resistance to imatinib in gastrointestinal stromal tumors. Clin. Cancer Res. 2012, 18, 1769–1776. [Google Scholar] [CrossRef]
- Antonescu, C.R.; Romeo, S.; Zhang, L.; Nafa, K.; Hornick, J.L.; Nielsen, G.P.; Mino-Kenudson, M.; Huang, H.-Y.; Mosquera, J.-M.; Dei Tos, P.A.; et al. Dedifferentiation in gastrointestinal stromal tumor to an anaplastic KIT-negative phenotype: A diagnostic pitfall: Morphologic and molecular characterization of 8 cases occurring either de novo or after imatinib therapy. Am. J. Surg. Pathol. 2013, 37, 385–392. [Google Scholar] [CrossRef]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRASG12C Inhibition with Sotorasib in Advanced Solid Tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef]
- Amatu, A.; Sartore-Bianchi, A.; Siena, S. NTRK gene fusions as novel targets of cancer therapy across multiple tumour types. ESMO Open 2016, 1, e000023. [Google Scholar] [CrossRef]
- Manea, C.A.; Badiu, D.C.; PLoScaru, I.C.; Zgura, A.; Bacinschi, X.; Smarandache, C.G.; Serban, D.; Popescu, C.G.; Grigorean, V.T.; Botnarciuc, V. A review of NTRK fusions in cancer. Ann. Med. Surg. 2022, 79, 103893. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. FDA Approves an Oncology Drug that Targets a Key Genetic Driver of Cancer, rather than a Specific Type of Tumor. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-oncology-drug-targets-key-genetic-driver-cancer-rather-specific-type-tumor (accessed on 3 January 2023).
- U.S. Food and Drug Administration. FDA Approves Entrectinib NTRK Solid Tumors and ROS1 NSCLC. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-entrectinib-ntrk-solid-tumors-and-ros-1-nsclc (accessed on 31 December 2022).
- Brenca, M.; Rossi, S.; Polano, M.; Gasparotto, D.; Zanatta, L.; Racanelli, D.; Valori, L.; Lamon, S.; Dei Tos, A.P.; Maestro, R. Transcriptome sequencing identifies ETV6-NTRK3 as a gene fusion involved in GIST. J. Pathol. 2016, 238, 543–549. [Google Scholar] [CrossRef]
- Shi, E.; Chmielecki, J.; Tang, C.M.; Wang, K.; Heinrich, M.C.; Kang, G.; Corless, C.L.; Hong, D.; Fero, K.E.; Murphy, J.D.; et al. FGFR1 and NTRK3 actionable alterations in “Wild-Type” gastrointestinal stromal tumors. J. Transl. Med. 2016, 14, 339. [Google Scholar] [CrossRef]
- D’Alpino Peixoto, R.; Medeiros, B.A.; Cronemberger, E.H. Resected High-Risk Rectal GIST Harboring NTRK1 Fusion: A Case Report and Review of the Literature. J. Gastrointest. Cancer 2021, 52, 316–319. [Google Scholar] [CrossRef]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: Integrated analysis of three phase 1–2 trials. Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef]
- Hong, D.S.; DuBois, S.G.; Kummar, S.; Farago, A.F.; Albert, C.M.; Rohrberg, K.S.; van Tilburg, C.M.; Nagasubramanian, R.; Berlin, J.D.; Federman, N.; et al. Larotrectinib in patients with TRK fusion-positive solid tumours: A pooled analysis of three phase 1/2 clinical trials. Lancet Oncol. 2020, 21, 531–540. [Google Scholar] [CrossRef]
- Nathenson, M.; Demetri, G.; Lassen, U.; Hong, D.; Boni, V.; Deeken, J.; Dowlati, A.; Cox, M.; Ku, N.; Cruickshank, S.; et al. O-020—Activity of larotrectinib in patients with TRK fusion GI malignancies. Ann. Oncol. 2018, 29, v107. [Google Scholar] [CrossRef]
- Krook, M.A.; Reeser, J.W.; Ernst, G.; Barker, H.; Wilberding, M.; Li, G.; Chen, H.-Z.; Roychowdhury, S. Fibroblast growth factor receptors in cancer: Genetic alterations, diagnostics, therapeutic targets and mechanisms of resistance. Br. J. Cancer 2021, 124, 880–892. [Google Scholar] [CrossRef] [PubMed]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip Rev Dev Biol 2015, 4, 215–266. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M.; Nakagama, H. FGF receptors: Cancer biology and therapeutics. Med. Res. Rev. 2014, 34, 280–300. [Google Scholar] [CrossRef] [PubMed]
- Ellinghaus, P.; Neureiter, D.; Nogai, H.; Stintzing, S.; Ocker, M. Patient Selection Approaches in FGFR Inhibitor Trials-Many Paths to the Same End? Cells 2022, 11, 3180. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, I.M.; Mariño-Enríquez, A.; Fletcher, J.A. What is New in Gastrointestinal Stromal Tumor? Adv. Anat. Pathol. 2017, 24, 259–267. [Google Scholar] [CrossRef] [PubMed]
- U.S. National Library of Medicine. ClinicalTrials.gov. 2023. Available online: https://clinicaltrials.gov/ (accessed on 17 February 2023).
- Pascual, J.; Attard, G.; Bidard, F.C.; Curigliano, G.; De Mattos-Arruda, L.; Diehn, M.; Italiano, A.; Lindberg, J.; Merker, J.D.; Montagut, C.; et al. ESMO recommendations on the use of circulating tumour DNA assays for patients with cancer: A report from the ESMO Precision Medicine Working Group. Ann. Oncol. 2022, 33, 750–768. [Google Scholar] [CrossRef] [PubMed]
- Serrano, C.; Vivancos, A.; López-Pousa, A.; Matito, J.; Mancuso, F.M.; Valverde, C.; Quiroga, S.; Landolfi, S.; Castro, S.; Dopazo, C.; et al. Clinical value of next generation sequencing of plasma cell-free DNA in gastrointestinal stromal tumors. BMC Cancer 2020, 20, 99. [Google Scholar] [CrossRef] [PubMed]
- Ravegnini, G.; Sammarini, G.; Serrano, C.; Nannini, M.; Pantaleo, M.A.; Hrelia, P.; Angelini, S. Clinical relevance of circulating molecules in cancer: Focus on gastrointestinal stromal tumors. Ther. Adv. Med. Oncol. 2019, 11, 1758835919831902. [Google Scholar] [CrossRef] [PubMed]
- Bauer, S.; Jones, R.L.; George, S.; Gelderblom, H.; Schöffski, P.; von Mehren, M.; Zalcberg, J.R.; Kang, Y.-K.; Abdul Razak, A.R.; Trent, J.C.; et al. Mutational heterogeneity of imatinib resistance and efficacy of ripretinib vs sunitinib in patients with gastrointestinal stromal tumor: ctDNA analysis from INTRIGUE. J. Clin. Oncol. 2023, 41, 397784. [Google Scholar] [CrossRef]


| First Author, Publication Information | Number of Patients, Patient Population | Clinical Phase | Intervention | Molecular Analysis Performed | Primary Endpoint | Response Evaluation Criteria | Results |
|---|---|---|---|---|---|---|---|
| Demetri et al., NEJM 2002 [76] | 147 Advanced GISTs | 2 | Imatinib 400 mg/d vs. imatinib 600 mg/d | No | ORR | Southwest Oncology Group criteria [105] | ORR: 49.3% (400 mg) vs. 58.1% (600 mg) Estimated 1 y OS: 88%. |
| Verweij et al., Lancet 2004 [24] | 946 Metastatic or unresectable GISTs | 3 | Imatinib 400 mg/d vs. imatinib 800 mg/d | No | PFS | RECIST 1.0 | PFS longer in the group with 800 mg vs. 400 mg, HR 0.82, p = 0.026 1 y OS: 85% (400 mg) vs. 86% (800 mg) ORR: 50.1% (400 mg) vs. 54.3% (800 mg) |
| Blanke et al., JCO 2008 [75] | 746 Metastatic or unresectable GISTs | 3 | Imatinib 400 mg/d vs. imatinib 800 mg/d | No | PFS and OS | RECIST 1.0 | Median PFS: 18 m (400 mg) vs. 20 m (800 mg), p = 0.13 Median OS: 55 m (400 mg) vs. 51 m (800 mg), p = 0.83 No difference in RR between the two arms |
| Demetri et al., Lancet 2006 [86] | 312 Advanced GISTs resistant or intolerant to imatinib | 3 | Sunitinib 50 mg vs. placebo | No | TTF | RECIST 1.0 or WHO (WHO Handbook for Reporting Results of Cancer Treatment) | Median TTP: 6.3 m (sunitinib) vs. 1.5 m (placebo), HR 0.33, p < 0.0001 Median PFS: 5.5 m (sunitinib) vs. 1.4 m (placebo), HR 0.33, p < 0.0001 Median OS: not reached. HR 0.49, p = 0.007 ORR: 7% (sunitinib) and 0% (placebo), p = 0.006 |
| George et al., EJC 2009 [106] | 60 Patients with unresectable GISTs resistant or intolerant to imatinib | 2 | Sunitinib 37.5 mg/d | No | DCR | RECIST 1.0 | DCR: 53% Median PFS: 7.8 m Median OS: 24.6 m ORR: 13% 1 y survival rate: 70% |
| Demetri et al., Lancet 2013 [92] | 199 Metastatic or unresectable GISTs resistant to imatinib and sunitinib | 3 | Regorafenib 160 mg/d vs. placebo | Yes * | PFS | Modified RECIST 1.1 [92] | Median PFS 4.8 m (regorafenib) vs. 0.9 m (placebo), HR 0.27, p < 0.0001 HR OS: 0.77, p = 0.199 DCR 52.6% (regorafenib) vs. 9.1% (placebo) |
| Blay et al., Lancet Oncol 2020 [89] | 129 Advanced GISTs with resistance or intolerance to imatinib, sunitinib, and regorafenib | 3 | Ripretinib 150 mg/d + BSC vs. placebo + BSC | Yes (112/129) | PFS | Modified RECIST 1.1 [92] | Median PFS: 6.3 m (ripretinib) vs. 1.0 m (placebo), HR: 0.15, p < 0.0001 Median OS: 15.1 m (ripretinib) vs. 6.6 m (placebo), HR 0.36 1 y estimated OS: 65.4% (ripretinib) vs. 25.9% (placebo) ORR: 9% |
| Heinrich et al. Lancet Oncol 2020 [90] | 56 Unresectable PDGFRA D842V-GISTs, regardless of previous therapy | 1 | Avapritinib 300/400 mg/d | Yes (56/56) | ORR | Modified RECIST 1.1 [92] | ORR: 91% CBR: 98% Median DOR: 27.6 m Median PFS: 34.0 m |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Unk, M.; Jezeršek Novaković, B.; Novaković, S. Molecular Mechanisms of Gastrointestinal Stromal Tumors and Their Impact on Systemic Therapy Decision. Cancers 2023, 15, 1498. https://doi.org/10.3390/cancers15051498
Unk M, Jezeršek Novaković B, Novaković S. Molecular Mechanisms of Gastrointestinal Stromal Tumors and Their Impact on Systemic Therapy Decision. Cancers. 2023; 15(5):1498. https://doi.org/10.3390/cancers15051498
Chicago/Turabian StyleUnk, Mojca, Barbara Jezeršek Novaković, and Srdjan Novaković. 2023. "Molecular Mechanisms of Gastrointestinal Stromal Tumors and Their Impact on Systemic Therapy Decision" Cancers 15, no. 5: 1498. https://doi.org/10.3390/cancers15051498
APA StyleUnk, M., Jezeršek Novaković, B., & Novaković, S. (2023). Molecular Mechanisms of Gastrointestinal Stromal Tumors and Their Impact on Systemic Therapy Decision. Cancers, 15(5), 1498. https://doi.org/10.3390/cancers15051498