
Prognostic and Predictive Biomarkers in Familial Breast Cancer

{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Prognostic and Predictive Biomarkers
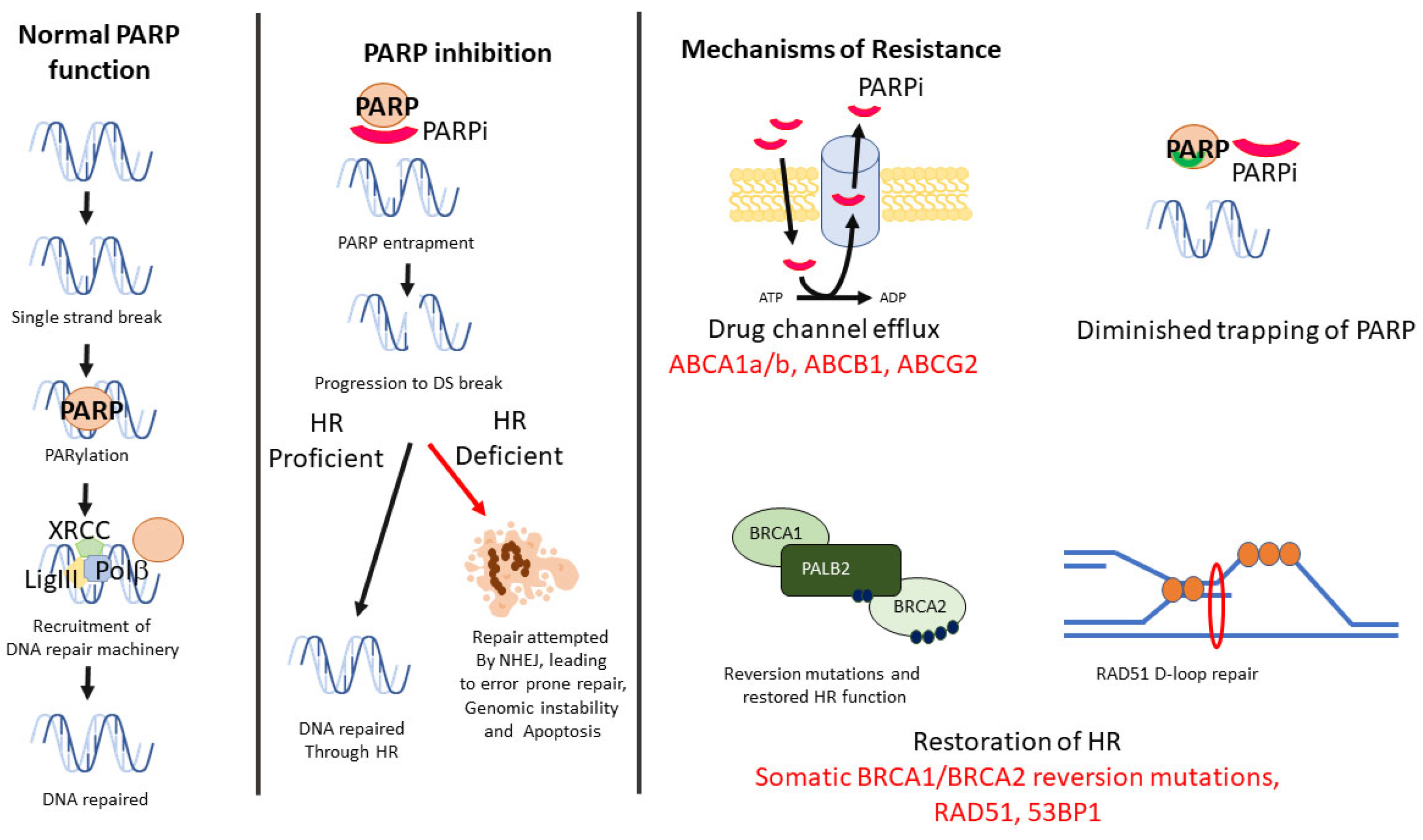
2.1. BRCA-Associated Homologous Recombination
2.2. Non-BRCA-Associated Homologous Recombination
2.3. HER2
2.4. Hypoxia
2.5. Vascular Endothelial Growth Factor (VEGF)
2.6. Cell Cycle Regulation
2.7. Androgen Receptor
2.8. Epidermal Growth Factor Receptor
2.9. MicroRNA (miR)
2.10. Single-Nucleotide Polymorphisms (SNPs)/Single-Nucleotide Variants (SNVs)
2.11. Commercial Expression Profile Assays
2.12. PIK3CA
2.13. Immunotherapy Biomarkers
2.14. Tumour-Infiltrating Lymphocytes
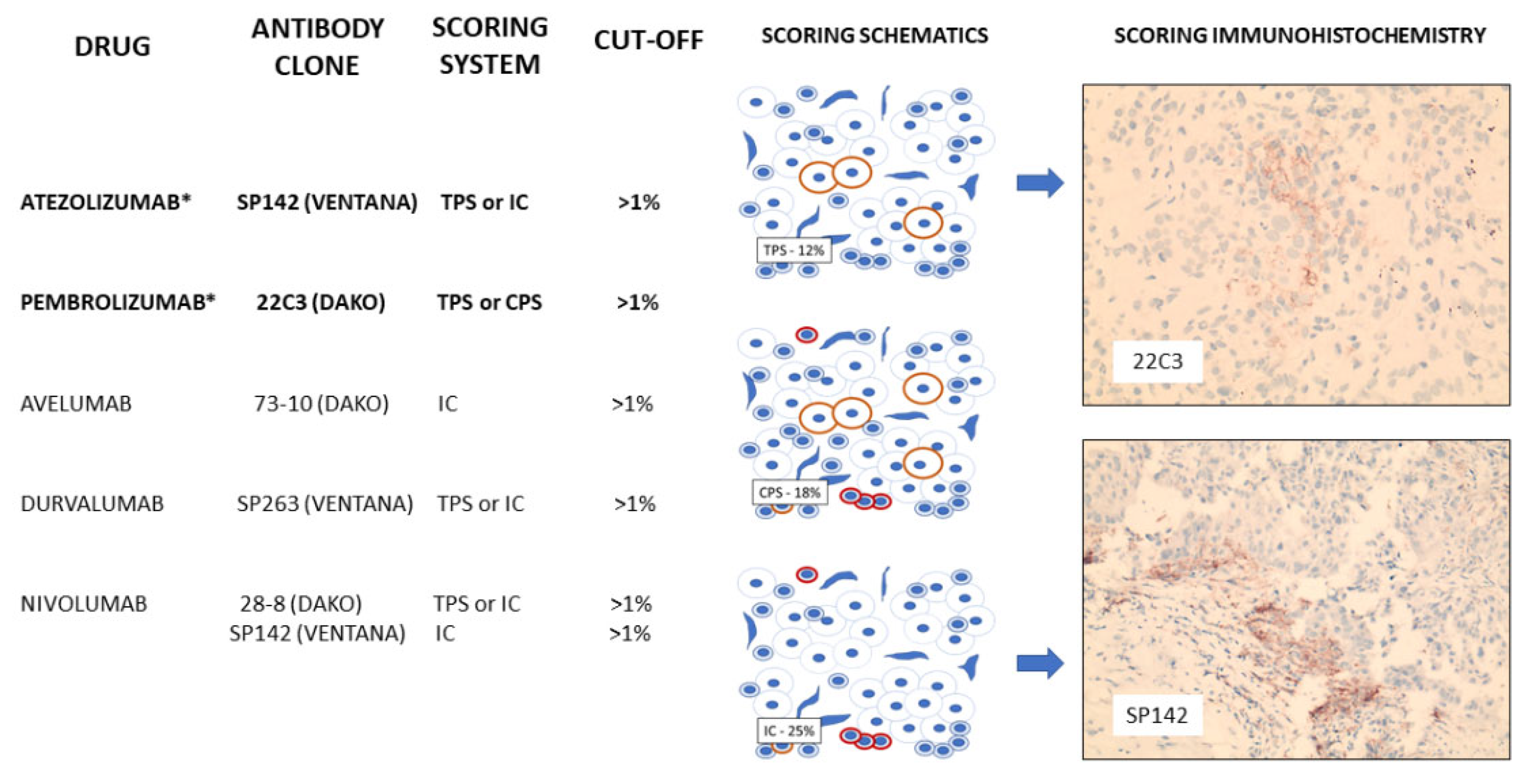
2.15. Programmed Cell Death Ligand-1 (PDL-1)
2.16. CXCL10/CXCR3
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Claus, E.B.; Risch, N.; Thompson, W.D. Genetic analysis of breast cancer in the cancer and steroid hormone study. Am. J. Hum. Genet. 1991, 48, 232–242. [Google Scholar] [PubMed]
- Newman, B.; Austin, M.A.; Lee, M.; King, M.C. Inheritance of human breast cancer: Evidence for autosomal dominant transmission in high-risk families. Proc. Natl. Acad. Sci. USA 1988, 85, 3044–3048. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.C.; Reis-Filho, J.S. Basal-like breast cancer and the BRCA1 phenotype. Oncogene 2006, 25, 5846–5853. [Google Scholar] [CrossRef]
- Deb, S.; Jene, N.; Fox, S.B. Genotypic and phenotypic analysis of familial male breast cancer shows under representation of the HER2 and basal subtypes in BRCA-associated carcinomas. BMC Cancer 2012, 12, 510. [Google Scholar] [CrossRef]
- Chen, S.; Parmigiani, G. Meta-analysis of BRCA1 and BRCA2 penetrance. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2007, 25, 1329–1333. [Google Scholar] [CrossRef]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.A.; Mooij, T.M.; Roos-Blom, M.J.; Jervis, S.; van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 2017, 317, 2402–2416. [Google Scholar] [CrossRef]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef]
- Jiang, X.; Li, X.; Li, W.; Bai, H.; Zhang, Z. PARP inhibitors in ovarian cancer: Sensitivity prediction and resistance mechanisms. J. Cell. Mol. Med. 2019, 23, 2303–2313. [Google Scholar] [CrossRef]
- Miglietta, F.; Cinquini, M.; Dieci, M.V.; Cortesi, L.; Criscitiello, C.; Montemurro, F.; Del Mastro, L.; Zambelli, A.; Biganzoli, L.; Levaggi, A.; et al. PARP-inhibitors for BRCA1/2-related advanced HER2-negative breast cancer: A meta-analysis and GRADE recommendations by the Italian Association of Medical Oncology. Breast 2022, 66, 293–304. [Google Scholar] [CrossRef]
- Geenen, J.J.J.; Linn, S.C.; Beijnen, J.H.; Schellens, J.H.M. PARP Inhibitors in the Treatment of Triple-Negative Breast Cancer. Clin. Pharmacokinet. 2018, 57, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Rottenberg, S.; Jaspers, J.E.; Kersbergen, A.; van der Burg, E.; Nygren, A.O.; Zander, S.A.; Derksen, P.W.; de Bruin, M.; Zevenhoven, J.; Lau, A.; et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc. Natl. Acad. Sci. USA 2008, 105, 17079–17084. [Google Scholar] [CrossRef] [PubMed]
- de Gooijer, M.C.; Buil, L.C.M.; Çitirikkaya, C.H.; Hermans, J.; Beijnen, J.H.; van Tellingen, O. ABCB1 Attenuates the Brain Penetration of the PARP Inhibitor AZD2461. Mol. Pharm. 2018, 15, 5236–5243. [Google Scholar] [CrossRef] [PubMed]
- Pettitt, S.J.; Krastev, D.B.; Brandsma, I.; Dréan, A.; Song, F.; Aleksandrov, R.; Harrell, M.I.; Menon, M.; Brough, R.; Campbell, J.; et al. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat. Commun. 2018, 9, 1849. [Google Scholar] [CrossRef]
- Gogola, E.; Duarte, A.A.; de Ruiter, J.R.; Wiegant, W.W.; Schmid, J.A.; de Bruijn, R.; James, D.I.; Guerrero Llobet, S.; Vis, D.J.; Annunziato, S.; et al. Selective Loss of PARG Restores PARylation and Counteracts PARP Inhibitor-Mediated Synthetic Lethality. Cancer Cell 2018, 33, 1078–1093. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Ceccaldi, R.; Shapiro, G.I.; D’Andrea, A.D. Homologous Recombination Deficiency: Exploiting the Fundamental Vulnerability of Ovarian Cancer. Cancer Discov. 2015, 5, 1137–1154. [Google Scholar] [CrossRef]
- Ray Chaudhuri, A.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 2016, 535, 382–387. [Google Scholar] [CrossRef]
- Rondinelli, B.; Gogola, E.; Yücel, H.; Duarte, A.A.; van de Ven, M.; van der Sluijs, R.; Konstantinopoulos, P.A.; Jonkers, J.; Ceccaldi, R.; Rottenberg, S.; et al. EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nat. Cell Biol. 2017, 19, 1371–1378. [Google Scholar] [CrossRef]
- Yazinski, S.A.; Comaills, V.; Buisson, R.; Genois, M.M.; Nguyen, H.D.; Ho, C.K.; Todorova Kwan, T.; Morris, R.; Lauffer, S.; Nussenzweig, A.; et al. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev. 2017, 31, 318–332. [Google Scholar] [CrossRef]
- Godin, S.K.; Sullivan, M.R.; Bernstein, K.A. Novel insights into RAD51 activity and regulation during homologous recombination and DNA replication. Biochem. Cell Biol. 2016, 94, 407–418. [Google Scholar] [CrossRef]
- Yun, M.H.; Hiom, K. CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature 2009, 459, 460–463. [Google Scholar] [CrossRef] [PubMed]
- Ning, J.; Wakimoto, H.; Peters, C.; Martuza, R.L.; Rabkin, S.D. Rad51 Degradation: Role in Oncolytic Virus-Poly(ADP-Ribose) Polymerase Inhibitor Combination Therapy in Glioblastoma. J. Natl. Cancer Inst. 2017, 109, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Noordermeer, S.M.; Adam, S.; Setiaputra, D.; Barazas, M.; Pettitt, S.J.; Ling, A.K.; Olivieri, M.; Álvarez-Quilón, A.; Moatti, N.; Zimmermann, M.; et al. The shieldin complex mediates 53BP1-dependent DNA repair. Nature 2018, 560, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Dev, H.; Chiang, T.W.; Lescale, C.; de Krijger, I.; Martin, A.G.; Pilger, D.; Coates, J.; Sczaniecka-Clift, M.; Wei, W.; Ostermaier, M.; et al. Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat. Cell Biol. 2018, 20, 954–965. [Google Scholar] [CrossRef] [PubMed]
- Jacot, W.; Thezenas, S.; Senal, R.; Viglianti, C.; Laberenne, A.C.; Lopez-Crapez, E.; Bibeau, F.; Bleuse, J.P.; Romieu, G.; Lamy, P.J. BRCA1 promoter hypermethylation, 53BP1 protein expression and PARP-1 activity as biomarkers of DNA repair deficit in breast cancer. BMC Cancer 2013, 13, 523. [Google Scholar] [CrossRef]
- Neboori, H.J.; Haffty, B.G.; Wu, H.; Yang, Q.; Aly, A.; Goyal, S.; Schiff, D.; Moran, M.S.; Golhar, R.; Chen, C.; et al. Low p53 binding protein 1 (53BP1) expression is associated with increased local recurrence in breast cancer patients treated with breast-conserving surgery and radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2012, 83, e677–e683. [Google Scholar] [CrossRef]
- Blum, J.L.; Laird, A.D.; Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Martin, M.; Roché, H.H.; Lee, K.H.; Goodwin, A.; et al. Determinants of Response to Talazoparib in Patients with HER2-Negative, Germline BRCA1/2-Mutated Breast Cancer. Clin. Cancer Res. 2022, 28, 1383–1390. [Google Scholar] [CrossRef]
- Castroviejo-Bermejo, M.; Cruz, C.; Llop-Guevara, A.; Gutiérrez-Enríquez, S.; Ducy, M.; Ibrahim, Y.H.; Gris-Oliver, A.; Pellegrino, B.; Bruna, A.; Guzmán, M.; et al. A RAD51 assay feasible in routine tumor samples calls PARP inhibitor response beyond BRCA mutation. EMBO Mol. Med. 2018, 10, e9172. [Google Scholar] [CrossRef]
- Mukhopadhyay, A.; Elattar, A.; Cerbinskaite, A.; Wilkinson, S.J.; Drew, Y.; Kyle, S.; Los, G.; Hostomsky, Z.; Edmondson, R.J.; Curtin, N.J. Development of a functional assay for homologous recombination status in primary cultures of epithelial ovarian tumor and correlation with sensitivity to poly(ADP-ribose) polymerase inhibitors. Clin. Cancer Res. 2010, 16, 2344–2351. [Google Scholar] [CrossRef]
- Naipal, K.A.; Verkaik, N.S.; Ameziane, N.; van Deurzen, C.H.; Ter Brugge, P.; Meijers, M.; Sieuwerts, A.M.; Martens, J.W.; O’Connor, M.J.; Vrieling, H.; et al. Functional ex vivo assay to select homologous recombination-deficient breast tumors for PARP inhibitor treatment. Clin. Cancer Res. 2014, 20, 4816–4826. [Google Scholar] [CrossRef]
- Cruz, C.; Castroviejo-Bermejo, M.; Gutiérrez-Enríquez, S.; Llop-Guevara, A.; Ibrahim, Y.H.; Gris-Oliver, A.; Bonache, S.; Morancho, B.; Bruna, A.; Rueda, O.M.; et al. RAD51 foci as a functional biomarker of homologous recombination repair and PARP inhibitor resistance in germline BRCA-mutated breast cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2018, 29, 1203–1210. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Xu, H.; Waddell, N.; Shield-Artin, K.; Haviv, I.; McKay, M.J.; Fox, S.B. Enhanced RAD21 cohesin expression confers poor prognosis in BRCA2 and BRCAX, but not BRCA1 familial breast cancers. Breast Cancer Res. 2012, 14, R69. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.A.; Ouchi, T. BRCA1 phosphorylation regulates caspase-3 activation in UV-induced apoptosis. Cancer Res. 2005, 65, 10657–10662. [Google Scholar] [CrossRef] [PubMed]
- López de Silanes, I.; Gorospe, M.; Taniguchi, H.; Abdelmohsen, K.; Srikantan, S.; Alaminos, M.; Berdasco, M.; Urdinguio, R.G.; Fraga, M.F.; Jacinto, F.V.; et al. The RNA-binding protein HuR regulates DNA methylation through stabilization of DNMT3b mRNA. Nucleic Acids Res. 2009, 37, 2658–2671. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Guo, Y.; Chu, H.; Guan, Y.; Bi, J.; Wang, B. Multiple functions of the RNA-binding protein HuR in cancer progression, treatment responses and prognosis. Int. J. Mol. Sci. 2013, 14, 10015–10041. [Google Scholar] [CrossRef]
- Heinonen, M.; Fagerholm, R.; Aaltonen, K.; Kilpivaara, O.; Aittomäki, K.; Blomqvist, C.; Heikkilä, P.; Haglund, C.; Nevanlinna, H.; Ristimäki, A. Prognostic role of HuR in hereditary breast cancer. Clin. Cancer Res. 2007, 13, 6959–6963. [Google Scholar] [CrossRef]
- Takada, M.; Nagai, S.; Haruta, M.; Sugino, R.P.; Tozuka, K.; Takei, H.; Ohkubo, F.; Inoue, K.; Kurosumi, M.; Miyazaki, M.; et al. BRCA1 alterations with additional defects in DNA damage response genes may confer chemoresistance to BRCA-like breast cancers treated with neoadjuvant chemotherapy. Genes Chromosomes Cancer 2017, 56, 405–420. [Google Scholar] [CrossRef]
- Paterson, M.C.; Dietrich, K.D.; Danyluk, J.; Paterson, A.H.; Lees, A.W.; Jamil, N.; Hanson, J.; Jenkins, H.; Krause, B.E.; McBlain, W.A.; et al. Correlation between c-erbB-2 amplification and risk of recurrent disease in node-negative breast cancer. Cancer Res. 1991, 51, 556–567. [Google Scholar]
- Ross, J.S.; Fletcher, J.A. The HER-2/neu oncogene in breast cancer: Prognostic factor, predictive factor, and target for therapy. Stem Cells 1998, 16, 413–428. [Google Scholar] [CrossRef]
- Incorvati, J.A.; Shah, S.; Mu, Y.; Lu, J. Targeted therapy for HER2 positive breast cancer. J. Hematol. Oncol. 2013, 6, 38. [Google Scholar] [CrossRef]
- Tomasello, G.; Gambini, D.; Petrelli, F.; Azzollini, J.; Arcanà, C.; Ghidini, M.; Peissel, B.; Manoukian, S.; Garrone, O. Characterization of the HER2 status in BRCA-mutated breast cancer: A single institutional series and systematic review with pooled analysis. ESMO Open 2022, 7, 100531. [Google Scholar] [CrossRef] [PubMed]
- Keeney, M.G.; Couch, F.J.; Visscher, D.W.; Lindor, N.M. Non-BRCA familial breast cancer: Review of reported pathology and molecular findings. Pathology 2017, 49, 363–370. [Google Scholar] [CrossRef] [PubMed]
- O’Shaughnessy, J.; Brezden-Masley, C.; Cazzaniga, M.; Dalvi, T.; Walker, G.; Bennett, J.; Ohsumi, S. Prevalence of germline BRCA mutations in HER2-negative metastatic breast cancer: Global results from the real-world, observational BREAKOUT study. Breast Cancer Res. 2020, 22, 114. [Google Scholar] [CrossRef] [PubMed]
- Curtit, E.; Benhamo, V.; Gruel, N.; Popova, T.; Manie, E.; Cottu, P.; Mariani, O.; Stoppa-Lyonnet, D.; Pivot, X.; Stern, M.H.; et al. First description of a sporadic breast cancer in a woman with BRCA1 germline mutation. Oncotarget 2015, 6, 35616–35624. [Google Scholar] [CrossRef] [PubMed]
- Viansone, A.; Pellegrino, B.; Omarini, C.; Pistelli, M.; Boggiani, D.; Sikokis, A.; Uliana, V.; Zanoni, D.; Tommasi, C.; Bortesi, B.; et al. Prognostic significance of germline BRCA mutations in patients with HER2-POSITIVE breast cancer. Breast 2022, 65, 145–150. [Google Scholar] [CrossRef]
- Conlon, N.T.; Kooijman, J.J.; van Gerwen, S.J.C.; Mulder, W.R.; Zaman, G.J.R.; Diala, I.; Eli, L.D.; Lalani, A.S.; Crown, J.; Collins, D.M. Comparative analysis of drug response and gene profiling of HER2-targeted tyrosine kinase inhibitors. Br. J. Cancer 2021, 124, 1249–1259. [Google Scholar] [CrossRef]
- Liu, W.; Shen, S.M.; Zhao, X.Y.; Chen, G.Q. Targeted genes and interacting proteins of hypoxia inducible factor-1. Int. J. Biochem. Mol. Biol. 2012, 3, 165–178. [Google Scholar]
- Wykoff, C.C.; Beasley, N.J.; Watson, P.H.; Turner, K.J.; Pastorek, J.; Sibtain, A.; Wilson, G.D.; Turley, H.; Talks, K.L.; Maxwell, P.H.; et al. Hypoxia-inducible expression of tumor-associated carbonic anhydrases. Cancer Res. 2000, 60, 7075–7083. [Google Scholar]
- Zhao, M.; Zhang, Y.; Zhang, H.; Wang, S.; Zhang, M.; Chen, X.; Wang, H.; Zeng, G.; Chen, X.; Liu, G.; et al. Hypoxia-induced cell stemness leads to drug resistance and poor prognosis in lung adenocarcinoma. Lung Cancer 2015, 87, 98–106. [Google Scholar] [CrossRef]
- Helmlinger, G.; Yuan, F.; Dellian, M.; Jain, R.K. Interstitial pH and pO2 gradients in solid tumors in vivo: High-resolution measurements reveal a lack of correlation. Nat. Med. 1997, 3, 177–182. [Google Scholar] [CrossRef]
- Bos, R.; van der Groep, P.; Greijer, A.E.; Shvarts, A.; Meijer, S.; Pinedo, H.M.; Semenza, G.L.; van Diest, P.J.; van der Wall, E. Levels of hypoxia-inducible factor-1alpha independently predict prognosis in patients with lymph node negative breast carcinoma. Cancer 2003, 97, 1573–1581. [Google Scholar] [CrossRef] [PubMed]
- Bos, R.; Zhong, H.; Hanrahan, C.F.; Mommers, E.C.; Semenza, G.L.; Pinedo, H.M.; Abeloff, M.D.; Simons, J.W.; van Diest, P.J.; van der Wall, E. Levels of hypoxia-inducible factor-1 alpha during breast carcinogenesis. J. Natl. Cancer Inst. 2001, 93, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Trastour, C.; Benizri, E.; Ettore, F.; Ramaioli, A.; Chamorey, E.; Pouysségur, J.; Berra, E. HIF-1alpha and CA IX staining in invasive breast carcinomas: Prognosis and treatment outcome. Int. J. Cancer J. Int. Cancer 2007, 120, 1451–1458. [Google Scholar] [CrossRef] [PubMed]
- Vleugel, M.M.; Greijer, A.E.; Shvarts, A.; van der Groep, P.; van Berkel, M.; Aarbodem, Y.; van Tinteren, H.; Harris, A.L.; van Diest, P.J.; van der Wall, E. Differential prognostic impact of hypoxia induced and diffuse HIF-1alpha expression in invasive breast cancer. J. Clin. Pathol. 2005, 58, 172–177. [Google Scholar] [CrossRef]
- Yan, M.; Rayoo, M.; Takano, E.A.; Fox, S.B. BRCA1 tumours correlate with a HIF-1alpha phenotype and have a poor prognosis through modulation of hydroxylase enzyme profile expression. Br. J. Cancer 2009, 101, 1168–1174. [Google Scholar] [CrossRef]
- van der Groep, P.; Bouter, A.; Menko, F.H.; van der Wall, E.; van Diest, P.J. High frequency of HIF-1alpha overexpression in BRCA1 related breast cancer. Breast Cancer Res. Treat. 2008, 111, 475–480. [Google Scholar] [CrossRef]
- Sharma, A.; Sinha, S.; Shrivastava, N. Therapeutic Targeting Hypoxia-Inducible Factor (HIF-1) in Cancer: Cutting Gordian Knot of Cancer Cell Metabolism. Front. Genet. 2022, 13, 849040. [Google Scholar] [CrossRef]
- Liu, Y.; Tamimi, R.M.; Collins, L.C.; Schnitt, S.J.; Gilmore, H.L.; Connolly, J.L.; Colditz, G.A. The association between vascular endothelial growth factor expression in invasive breast cancer and survival varies with intrinsic subtypes and use of adjuvant systemic therapy: Results from the Nurses’ Health Study. Breast Cancer Res. Treat. 2011, 129, 175–184. [Google Scholar] [CrossRef]
- Danza, K.; Pilato, B.; Lacalamita, R.; Addati, T.; Giotta, F.; Bruno, A.; Paradiso, A.; Tommasi, S. Angiogenetic axis angiopoietins/Tie2 and VEGF in familial breast cancer. Eur. J. Hum. Genet. 2013, 21, 824–830. [Google Scholar] [CrossRef]
- Severson, T.M.; Peeters, J.; Majewski, I.; Michaut, M.; Bosma, A.; Schouten, P.C.; Chin, S.F.; Pereira, B.; Goldgraben, M.A.; Bismeijer, T.; et al. BRCA1-like signature in triple negative breast cancer: Molecular and clinical characterization reveals subgroups with therapeutic potential. Mol. Oncol. 2015, 9, 1528–1538. [Google Scholar] [CrossRef]
- Kolyvas, E.A.; Caldas, C.; Kelly, K.; Ahmad, S.S. Androgen receptor function and targeted therapeutics across breast cancer subtypes. Breast Cancer Res. 2022, 24, 79. [Google Scholar] [CrossRef] [PubMed]
- Kono, M.; Fujii, T.; Lim, B.; Karuturi, M.S.; Tripathy, D.; Ueno, N.T. Androgen Receptor Function and Androgen Receptor-Targeted Therapies in Breast Cancer: A Review. JAMA Oncol. 2017, 3, 1266–1273. [Google Scholar] [CrossRef] [PubMed]
- Pristauz, G.; Petru, E.; Stacher, E.; Geigl, J.B.; Schwarzbraun, T.; Tsybrovskyy, O.; Winter, R.; Moinfar, F. Androgen receptor expression in breast cancer patients tested for BRCA1 and BRCA2 mutations. Histopathology 2010, 57, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Sukumar, J.; Gast, K.; Quiroga, D.; Lustberg, M.; Williams, N. Triple-negative breast cancer: Promising prognostic biomarkers currently in development. Expert Rev. Anticancer. Ther. 2021, 21, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Karanika, S.; Yang, G.; Wang, J.; Park, S.; Broom, B.M.; Manyam, G.C.; Wu, W.; Luo, Y.; Basourakos, S.; et al. Androgen receptor inhibitor-induced “BRCAness” and PARP inhibition are synthetically lethal for castration-resistant prostate cancer. Sci. Signal. 2017, 10, eaam7479. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Jang, M.H.; Kim, E.J.; Kim, H.J.; Lee, H.J.; Kim, Y.J.; Kim, J.H.; Kang, E.; Kim, S.W.; Kim, I.A.; et al. High EGFR gene copy number predicts poor outcome in triple-negative breast cancer. Mod. Pathol. 2014, 27, 1212–1222. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Conchas, G.A.; Rodriguez-Romo, L.; Hernandez-Barajas, D.; Gonzalez-Guerrero, J.F.; Rodriguez-Fernandez, I.A.; Verdines-Perez, A.; Templeton, A.J.; Ocana, A.; Seruga, B.; Tannock, I.F.; et al. Epidermal growth factor receptor overexpression and outcomes in early breast cancer: A systematic review and a meta-analysis. Cancer Treat. Rev. 2018, 62, 1–8. [Google Scholar] [CrossRef]
- Abdelrahman, A.E.; Rashed, H.E.; Abdelgawad, M.; Abdelhamid, M.I. Prognostic impact of EGFR and cytokeratin 5/6 immunohistochemical expression in triple-negative breast cancer. Ann. Diagn. Pathol. 2017, 28, 43–53. [Google Scholar] [CrossRef]
- Danzinger, S.; Tan, Y.Y.; Rudas, M.; Kastner, M.T.; Weingartshofer, S.; Muhr, D.; Singer, C.F. Differential Claudin 3 and EGFR Expression Predicts BRCA1 Mutation in Triple-Negative Breast Cancer. Cancer Investig. 2018, 36, 378–388. [Google Scholar] [CrossRef]
- Li, L.; Wang, H.; Yang, E.S.; Arteaga, C.L.; Xia, F. Erlotinib attenuates homologous recombinational repair of chromosomal breaks in human breast cancer cells. Cancer Res. 2008, 68, 9141–9146. [Google Scholar] [CrossRef]
- Nowsheen, S.; Cooper, T.; Stanley, J.A.; Yang, E.S. Synthetic lethal interactions between EGFR and PARP inhibition in human triple negative breast cancer cells. PLoS ONE 2012, 7, e46614. [Google Scholar] [CrossRef]
- Morales, S.; Monzo, M.; Navarro, A. Epigenetic regulation mechanisms of microRNA expression. Biomol. Concepts 2017, 8, 203–212. [Google Scholar] [CrossRef]
- Nassar, F.J.; Chamandi, G.; Tfaily, M.A.; Zgheib, N.K.; Nasr, R. Peripheral Blood-Based Biopsy for Breast Cancer Risk Prediction and Early Detection. Front. Med. 2020, 7, 28. [Google Scholar] [CrossRef]
- Setti, G.; Pezzi, M.E.; Viani, M.V.; Pertinhez, T.A.; Cassi, D.; Magnoni, C.; Bellini, P.; Musolino, A.; Vescovi, P.; Meleti, M. Salivary MicroRNA for Diagnosis of Cancer and Systemic Diseases: A Systematic Review. Int. J. Mol. Sci. 2020, 21, 907. [Google Scholar] [CrossRef] [PubMed]
- Gasparri, M.L.; Casorelli, A.; Bardhi, E.; Besharat, A.R.; Savone, D.; Ruscito, I.; Farooqi, A.A.; Papadia, A.; Mueller, M.D.; Ferretti, E.; et al. Beyond circulating microRNA biomarkers: Urinary microRNAs in ovarian and breast cancer. Tumour Biol. 2017, 39, 1010428317695525. [Google Scholar] [CrossRef] [PubMed]
- Shen, J. Evaluation of environmental and personal susceptibility characteristics that modify genetic risks. Methods Mol. Biol. 2009, 471, 163–177. [Google Scholar] [CrossRef] [PubMed]
- Abdul, Q.A.; Yu, B.P.; Chung, H.Y.; Jung, H.A.; Choi, J.S. Epigenetic modifications of gene expression by lifestyle and environment. Arch. Pharmacal Res. 2017, 40, 1219–1237. [Google Scholar] [CrossRef]
- Murria Estal, R.; Palanca Suela, S.; de Juan Jiménez, I.; Egoavil Rojas, C.; García-Casado, Z.; Juan Fita, M.J.; Sánchez Heras, A.B.; Segura Huerta, A.; Chirivella González, I.; Sánchez-Izquierdo, D.; et al. MicroRNA signatures in hereditary breast cancer. Breast Cancer Res. Treat. 2013, 142, 19–30. [Google Scholar] [CrossRef]
- Yan, M.; Shield-Artin, K.; Byrne, D.; Deb, S.; Waddell, N.; Haviv, I.; Fox, S.B. Comparative microRNA profiling of sporadic and BRCA1 associated basal-like breast cancers. BMC Cancer 2015, 15, 506. [Google Scholar] [CrossRef]
- Tanic, M.; Yanowski, K.; Gómez-López, G.; Rodriguez-Pinilla, M.S.; Marquez-Rodas, I.; Osorio, A.; Pisano, D.G.; Martinez-Delgado, B.; Benítez, J. MicroRNA expression signatures for the prediction of BRCA1/2 mutation-associated hereditary breast cancer in paraffin-embedded formalin-fixed breast tumors. Int. J. Cancer J. Int. Cancer 2015, 136, 593–602. [Google Scholar] [CrossRef]
- Pessôa-Pereira, D.; Evangelista, A.F.; Causin, R.L.; da Costa Vieira, R.A.; Abrahão-Machado, L.F.; Santana, I.V.V.; da Silva, V.D.; de Souza, K.C.B.; de Oliveira-Silva, R.J.; Fernandes, G.C.; et al. miRNA expression profiling of hereditary breast tumors from BRCA1- and BRCA2-germline mutation carriers in Brazil. BMC Cancer 2020, 20, 143. [Google Scholar] [CrossRef] [PubMed]
- Block, I.; Burton, M.; Sørensen, K.P.; Andersen, L.; Larsen, M.J.; Bak, M.; Cold, S.; Thomassen, M.; Tan, Q.; Kruse, T.A. Association of miR-548c-5p, miR-7-5p, miR-210-3p, miR-128-3p with recurrence in systemically untreated breast cancer. Oncotarget 2018, 9, 9030–9042. [Google Scholar] [CrossRef]
- Cao, Z.G.; Li, J.J.; Yao, L.; Huang, Y.N.; Liu, Y.R.; Hu, X.; Song, C.G.; Shao, Z.M. High expression of microRNA-454 is associated with poor prognosis in triple-negative breast cancer. Oncotarget 2016, 7, 64900–64909. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.; Katsaros, D.; Lu, L.; Preti, M.; Durando, A.; Arisio, R.; Mu, L.; Yu, H. High miR-21 expression in breast cancer associated with poor disease-free survival in early stage disease and high TGF-beta1. Breast Cancer Res. Treat. 2009, 117, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Toyama, T.; Kondo, N.; Endo, Y.; Sugiura, H.; Yoshimoto, N.; Iwasa, M.; Takahashi, S.; Fujii, Y.; Yamashita, H. High expression of microRNA-210 is an independent factor indicating a poor prognosis in Japanese triple-negative breast cancer patients. Jpn. J. Clin. Oncol. 2012, 42, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Zhao, B.; Ren, X.; Wu, S.; Liu, M.; Wang, Z.; Liu, W. MiR-27a-3p Targeting GSK3β Promotes Triple-Negative Breast Cancer Proliferation and Migration Through Wnt/β-Catenin Pathway. Cancer Manag. Res. 2020, 12, 6241–6249. [Google Scholar] [CrossRef]
- Grimaldi, A.M.; Nuzzo, S.; Condorelli, G.; Salvatore, M.; Incoronato, M. Prognostic and Clinicopathological Significance of MiR-155 in Breast Cancer: A Systematic Review. Int. J. Mol. Sci. 2020, 21, 5834. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, A.; Bao, P.P.; Lin, L.; Wang, Y.; Wu, H.; Shu, X.O.; Liu, A.; Cai, Q. MicroRNA-374b inhibits breast cancer progression through regulating CCND1 and TGFA genes. Carcinogenesis 2021, 42, 528–536. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, Z.; Li, S.; Dong, L.; Li, Y.; Mao, Y.; Liang, Y.; Tao, Y.; Ma, J. Inhibition of miR-214 attenuates the migration and invasion of triple-negative breast cancer cells. Mol. Med. Rep. 2019, 19, 4035–4042. [Google Scholar] [CrossRef]
- Garcia, A.I.; Buisson, M.; Bertrand, P.; Rimokh, R.; Rouleau, E.; Lopez, B.S.; Lidereau, R.; Mikaélian, I.; Mazoyer, S. Down-regulation of BRCA1 expression by miR-146a and miR-146b-5p in triple negative sporadic breast cancers. EMBO Mol. Med. 2011, 3, 279–290. [Google Scholar] [CrossRef]
- Moskwa, P.; Buffa, F.M.; Pan, Y.; Panchakshari, R.; Gottipati, P.; Muschel, R.J.; Beech, J.; Kulshrestha, R.; Abdelmohsen, K.; Weinstock, D.M.; et al. miR-182-mediated downregulation of BRCA1 impacts DNA repair and sensitivity to PARP inhibitors. Mol. Cell 2011, 41, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Meghani, K.; Fuchs, W.; Detappe, A.; Drané, P.; Gogola, E.; Rottenberg, S.; Jonkers, J.; Matulonis, U.; Swisher, E.M.; Konstantinopoulos, P.A.; et al. Multifaceted Impact of MicroRNA 493-5p on Genome-Stabilizing Pathways Induces Platinum and PARP Inhibitor Resistance in BRCA2-Mutated Carcinomas. Cell Rep. 2018, 23, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Tang, L.; Xu, Y.; Xu, J.; Zhang, W.; Xie, H.; Wang, S.; Guan, X. PARP inhibitor increases chemosensitivity by upregulating miR-664b-5p in BRCA1-mutated triple-negative breast cancer. Sci. Rep. 2017, 7, 42319. [Google Scholar] [CrossRef] [PubMed]
- Strumidło, A.; Skiba, S.; Scott, R.J.; Lubiński, J. The potential role of miRNAs in therapy of breast and ovarian cancers associated with BRCA1 mutation. Hered. Cancer Clin. Pract. 2017, 15, 15. [Google Scholar] [CrossRef] [PubMed]
- Sempere, L.F.; Azmi, A.S.; Moore, A. microRNA-based diagnostic and therapeutic applications in cancer medicine. RNA 2021, 12, e1662. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, S.; Hughes, E.; Wagner, S.; Tshiaba, P.; Rosenthal, E.; Roa, B.B.; Kurian, A.W.; Domchek, S.M.; Garber, J.; Lancaster, J.; et al. Association of a Polygenic Risk Score With Breast Cancer Among Women Carriers of High- and Moderate-Risk Breast Cancer Genes. JAMA Netw. Open 2020, 3, e208501. [Google Scholar] [CrossRef]
- Zeng, C.; Zhang, J. A narrative review of five multigenetic assays in breast cancer. Transl. Cancer Res. 2022, 11, 897–907. [Google Scholar] [CrossRef]
- Blanter, J.; Zimmerman, B.; Tharakan, S.; Ru, M.; Cascetta, K.; Tiersten, A. BRCA Mutation Association with Recurrence Score and Discordance in a Large Oncotype Database. Oncology 2020, 98, 248–251. [Google Scholar] [CrossRef]
- Layman, R.M.; Lin, H.; Gutierrez Barrera, A.M.; Karuturi, M.S.; Yam, C.; Arun, B.K. Clinical outcomes and Oncotype DX Breast Recurrence Score® in early-stage BRCA-associated hormone receptor-positive breast cancer. Cancer Med. 2022, 11, 1474–1483. [Google Scholar] [CrossRef]
- Halpern, N.; Sonnenblick, A.; Uziely, B.; Divinsky, L.; Goldberg, Y.; Hamburger, T.; Peretz, T.; Kadouri, L. Oncotype Dx recurrence score among BRCA1/2 germline mutation carriers with hormone receptors positive breast cancer. Int. J. Cancer J. Int. Cancer 2017, 140, 2145–2149. [Google Scholar] [CrossRef]
- Lewin, R.; Sulkes, A.; Shochat, T.; Tsoref, D.; Rizel, S.; Liebermann, N.; Hendler, D.; Neiman, V.; Ben-Aharon, I.; Friedman, E.; et al. Oncotype-DX recurrence score distribution in breast cancer patients with BRCA1/2 mutations. Breast Cancer Res. Treat. 2016, 157, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Vollebergh, M.A.; Lips, E.H.; Nederlof, P.M.; Wessels, L.F.A.; Schmidt, M.K.; van Beers, E.H.; Cornelissen, S.; Holtkamp, M.; Froklage, F.E.; de Vries, E.G.E.; et al. An aCGH classifier derived from BRCA1-mutated breast cancer and benefit of high-dose platinum-based chemotherapy in HER2-negative breast cancer patients. Ann. Oncol. 2011, 22, 1561–1570. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Degasperi, A.; Zou, X.; Amarante, T.D.; Martinez-Martinez, A.; Koh, G.C.C.; Dias, J.M.L.; Heskin, L.; Chmelova, L.; Rinaldi, G.; Wang, V.Y.W.; et al. Substitution mutational signatures in whole-genome-sequenced cancers in the UK population. Science 2022, 376, abl9283. [Google Scholar] [CrossRef] [PubMed]
- Gulhan, D.C.; Lee, J.J.; Melloni, G.E.M.; Cortes-Ciriano, I.; Park, P.J. Detecting the mutational signature of homologous recombination deficiency in clinical samples. Nat. Genet. 2019, 51, 912–919. [Google Scholar] [CrossRef]
- Frampton, G.M.; Fichtenholtz, A.; Otto, G.A.; Wang, K.; Downing, S.R.; He, J.; Schnall-Levin, M.; White, J.; Sanford, E.M.; An, P.; et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat. Biotechnol. 2013, 31, 1023–1031. [Google Scholar] [CrossRef]
- Telli, M.L.; Timms, K.M.; Reid, J.; Hennessy, B.; Mills, G.B.; Jensen, K.C.; Szallasi, Z.; Barry, W.T.; Winer, E.P.; Tung, N.M.; et al. Homologous Recombination Deficiency (HRD) Score Predicts Response to Platinum-Containing Neoadjuvant Chemotherapy in Patients with Triple-Negative Breast Cancer. Clin. Cancer Res. 2016, 22, 3764–3773. [Google Scholar] [CrossRef]
- Myriad_Genetics. myChoice HRD Technical Specifications Effective Date: June 2017. Available online: https://myriad-web.s3.amazonaws.com/myChoice/downloads/myChoiceHRDTechSpecs.pdf (accessed on 6 February 2023).
- Buisson, A.; Saintigny, P.; Pujade-Lauraine, E.; Montoto-Grillot, C.; Vacirca, D.; Barberis, M.; Colombo, N.; Harle, A.; Gilson, P.; Roma, C.; et al. A deep learning solution for detection of homologous recombination deficiency in ovarian cancer using low pass whole-genome sequencing: Evaluation of the analytical performance. J. Clin. Oncol. 2022, 40, e17599. [Google Scholar] [CrossRef]
- Davies, H.; Glodzik, D.; Morganella, S.; Yates, L.R.; Staaf, J.; Zou, X.; Ramakrishna, M.; Martin, S.; Boyault, S.; Sieuwerts, A.M.; et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat. Med. 2017, 23, 517–525. [Google Scholar] [CrossRef]
- Chen, B.; Zhang, G.; Li, X.; Ren, C.; Wang, Y.; Li, K.; Mok, H.; Cao, L.; Wen, L.; Jia, M.; et al. Comparison of BRCA versus non-BRCA germline mutations and associated somatic mutation profiles in patients with unselected breast cancer. Aging 2020, 12, 3140–3155. [Google Scholar] [CrossRef] [PubMed]
- Ruangapirom, L.; Sutivijit, N.; Teerapakpinyo, C.; Mutirangura, A.; Doungkamchan, C. Identification of Shared Neoantigens in BRCA1-Related Breast Cancer. Vaccines 2022, 10, 1597. [Google Scholar] [CrossRef]
- De, P.; Sun, Y.; Carlson, J.H.; Friedman, L.S.; Leyland-Jones, B.R.; Dey, N. Doubling down on the PI3K-AKT-mTOR pathway enhances the antitumor efficacy of PARP inhibitor in triple negative breast cancer model beyond BRCA-ness. Neoplasia 2014, 16, 43–72. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Li, C.; Zhang, Y.; Wang, M.; Jiang, N.; Xiang, L.; Li, T.; Roberts, T.M.; Zhao, J.J.; Cheng, H.; et al. Combined inhibition of PI3K and PARP is effective in the treatment of ovarian cancer cells with wild-type PIK3CA genes. Gynecol. Oncol. 2016, 142, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wang, M.; Jiang, N.; Zhang, Y.; Bian, X.; Wang, X.; Roberts, T.M.; Zhao, J.J.; Liu, P.; Cheng, H. Effective use of PI3K inhibitor BKM120 and PARP inhibitor Olaparib to treat PIK3CA mutant ovarian cancer. Oncotarget 2016, 7, 13153–13166. [Google Scholar] [CrossRef] [PubMed]
- Aktas, B.Y.; Guner, G.; Guven, D.C.; Arslan, C.; Dizdar, O. Exploiting DNA repair defects in breast cancer: From chemotherapy to immunotherapy. Expert Rev. Anticancer. Ther. 2019, 19, 589–601. [Google Scholar] [CrossRef]
- Chabanon, R.M.; Rouanne, M.; Lord, C.J.; Soria, J.C.; Pasero, P.; Postel-Vinay, S. Targeting the DNA damage response in immuno-oncology: Developments and opportunities. Nat. Rev. Cancer 2021, 21, 701–717. [Google Scholar] [CrossRef]
- Nolan, E.; Savas, P.; Policheni, A.N.; Darcy, P.K.; Vaillant, F.; Mintoff, C.P.; Dushyanthen, S.; Mansour, M.; Pang, J.B.; Fox, S.B.; et al. Combined immune checkpoint blockade as a therapeutic strategy for BRCA1-mutated breast cancer. Sci. Transl. Med. 2017, 9, eaal4922. [Google Scholar] [CrossRef]
- van Wilpe, S.; Tolmeijer, S.H.; Koornstra, R.H.T.; de Vries, I.J.M.; Gerritsen, W.R.; Ligtenberg, M.; Mehra, N. Homologous Recombination Repair Deficiency and Implications for Tumor Immunogenicity. Cancers 2021, 13, 2249. [Google Scholar] [CrossRef]
- Solinas, C.; Marcoux, D.; Garaud, S.; Vitória, J.R.; Van den Eynden, G.; de Wind, A.; De Silva, P.; Boisson, A.; Craciun, L.; Larsimont, D.; et al. BRCA gene mutations do not shape the extent and organization of tumor infiltrating lymphocytes in triple negative breast cancer. Cancer Lett. 2019, 450, 88–97. [Google Scholar] [CrossRef]
- Grandal, B.; Evrevin, C.; Laas, E.; Jardin, I.; Rozette, S.; Laot, L.; Dumas, E.; Coussy, F.; Pierga, J.Y.; Brain, E.; et al. Impact of BRCA Mutation Status on Tumor Infiltrating Lymphocytes (TILs), Response to Treatment, and Prognosis in Breast Cancer Patients Treated with Neoadjuvant Chemotherapy. Cancers 2020, 12, 3681. [Google Scholar] [CrossRef]
- Sønderstrup, I.M.H.; Jensen, M.B.; Ejlertsen, B.; Eriksen, J.O.; Gerdes, A.M.; Kruse, T.A.; Larsen, M.J.; Thomassen, M.; Laenkholm, A.V. Evaluation of tumor-infiltrating lymphocytes and association with prognosis in BRCA-mutated breast cancer. Acta Oncol. 2019, 58, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, N.; Hviid, T.V.F.; Nielsen, L.B.; Sønderstrup, I.M.H.; Eriksen, J.O.; Ejlertsen, B.; Gerdes, A.M.; Kruse, T.A.; Thomassen, M.; Jensen, M.B.; et al. Tumour-infiltrating CD4-, CD8- and FOXP3-positive immune cells as predictive markers of mortality in BRCA1- and BRCA2-associated breast cancer. Br. J. Cancer 2021, 125, 1388–1398. [Google Scholar] [CrossRef] [PubMed]
- Ali, H.R.; Provenzano, E.; Dawson, S.J.; Blows, F.M.; Liu, B.; Shah, M.; Earl, H.M.; Poole, C.J.; Hiller, L.; Dunn, J.A.; et al. Association between CD8+ T-cell infiltration and breast cancer survival in 12,439 patients. Ann. Oncol. 2014, 25, 1536–1543. [Google Scholar] [CrossRef] [PubMed]
- Bottai, G.; Raschioni, C.; Losurdo, A.; Di Tommaso, L.; Tinterri, C.; Torrisi, R.; Reis-Filho, J.S.; Roncalli, M.; Sotiriou, C.; Santoro, A.; et al. An immune stratification reveals a subset of PD-1/LAG-3 double-positive triple-negative breast cancers. Breast Cancer Res. 2016, 18, 121. [Google Scholar] [CrossRef]
- Oshi, M.; Asaoka, M.; Tokumaru, Y.; Angarita, F.A.; Yan, L.; Matsuyama, R.; Zsiros, E.; Ishikawa, T.; Endo, I.; Takabe, K. Abundance of Regulatory T Cell (Treg) as a Predictive Biomarker for Neoadjuvant Chemotherapy in Triple-Negative Breast Cancer. Cancers 2020, 12, 3038. [Google Scholar] [CrossRef]
- Kos, K.; de Visser, K.E. The Multifaceted Role of Regulatory T Cells in Breast Cancer. Annu. Rev. Cancer Biol. 2021, 5, 291–310. [Google Scholar] [CrossRef]
- Sobral-Leite, M.; Van de Vijver, K.; Michaut, M.; van der Linden, R.; Hooijer, G.K.J.; Horlings, H.M.; Severson, T.M.; Mulligan, A.M.; Weerasooriya, N.; Sanders, J.; et al. Assessment of PD-L1 expression across breast cancer molecular subtypes, in relation to mutation rate, BRCA1-like status, tumor-infiltrating immune cells and survival. Oncoimmunology 2018, 7, e1509820. [Google Scholar] [CrossRef]
- Jenzer, M.; Keß, P.; Nientiedt, C.; Endris, V.; Kippenberger, M.; Leichsenring, J.; Stögbauer, F.; Haimes, J.; Mishkin, S.; Kudlow, B.; et al. The BRCA2 mutation status shapes the immune phenotype of prostate cancer. Cancer Immunol. Immunother. 2019, 68, 1621–1633. [Google Scholar] [CrossRef]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar]
- Patsoukis, N.; Brown, J.; Petkova, V.; Liu, F.; Li, L.; Boussiotis, V.A. Selective effects of PD-1 on Akt and Ras pathways regulate molecular components of the cell cycle and inhibit T cell proliferation. Sci. Signal. 2012, 5, ra46. [Google Scholar] [CrossRef]
- Sheppard, K.A.; Fitz, L.J.; Lee, J.M.; Benander, C.; George, J.A.; Wooters, J.; Qiu, Y.; Jussif, J.M.; Carter, L.L.; Wood, C.R.; et al. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. FEBS Lett. 2004, 574, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Yuan, P.; Wu, H.; Chen, J.; Fu, J.; Li, P.; Lu, J.; Wei, W. Dendritic cells with an increased PD-L1 by TGF-β induce T cell anergy for the cytotoxicity of hepatocellular carcinoma cells. Int. Immunopharmacol. 2014, 20, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Sun, H.; Zhao, S.; Wang, Y.; Pu, H.; Wang, Y.; Zhang, Q. Expression of PD-L1 and prognosis in breast cancer: A meta-analysis. Oncotarget 2017, 8, 31347–31354. [Google Scholar] [CrossRef] [PubMed]
- Rozenblit, M.; Huang, R.; Danziger, N.; Hegde, P.; Alexander, B.; Ramkissoon, S.; Blenman, K.; Ross, J.S.; Rimm, D.L.; Pusztai, L. Comparison of PD-L1 protein expression between primary tumors and metastatic lesions in triple negative breast cancers. J. Immunother. Cancer 2020, 8, e001558. [Google Scholar] [CrossRef]
- Pang, J.B.; Castles, B.; Byrne, D.J.; Button, P.; Hendry, S.; Lakhani, S.R.; Sivasubramaniam, V.; Cooper, W.A.; Armes, J.; Millar, E.K.A.; et al. SP142 PD-L1 Scoring Shows High Interobserver and Intraobserver Agreement in Triple-negative Breast Carcinoma But Overall Low Percentage Agreement with Other PD-L1 Clones SP263 and 22C3. Am. J. Surg. Pathol. 2021, 45, 1108–1117. [Google Scholar] [CrossRef]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Miles, D.; Gligorov, J.; André, F.; Cameron, D.; Schneeweiss, A.; Barrios, C.; Xu, B.; Wardley, A.; Kaen, D.; Andrade, L.; et al. Primary results from IMpassion131, a double-blind, placebo-controlled, randomised phase III trial of first-line paclitaxel with or without atezolizumab for unresectable locally advanced/metastatic triple-negative breast cancer. Ann. Oncol. 2021, 32, 994–1004. [Google Scholar] [CrossRef]
- Roche., N.r. Roche Provides Update on Tecentriq US Indication for PD-L1-Positive, Metastatic Triple-Negative Breast Cancer. 27 August 2021. Available online: https://www.globenewswire.com/news-release/2021/08/27/2287799/0/en/Roche-provides-update-on-Tecentriq-US-indication-for-PD-L1-positive-metastatic-triple-negative-breast-cancer.html (accessed on 6 February 2023).
- Franzoi, M.A.; de Azambuja, E. Atezolizumab in metastatic triple-negative breast cancer: IMpassion130 and 131 trials - how to explain different results? ESMO Open 2020, 5, e001112. [Google Scholar] [CrossRef]
- Cortes, J.; Cescon, D.W.; Rugo, H.S.; Nowecki, Z.; Im, S.A.; Yusof, M.M.; Gallardo, C.; Lipatov, O.; Barrios, C.H.; Holgado, E.; et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): A randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet 2020, 396, 1817–1828. [Google Scholar] [CrossRef]
- Emens, L.A.; Molinero, L.; Loi, S.; Rugo, H.S.; Schneeweiss, A.; Diéras, V.; Iwata, H.; Barrios, C.H.; Nechaeva, M.; Nguyen-Duc, A.; et al. Atezolizumab and nab-Paclitaxel in Advanced Triple-Negative Breast Cancer: Biomarker Evaluation of the IMpassion130 Study. J. Natl. Cancer Inst. 2021, 113, 1005–1016. [Google Scholar] [CrossRef]
- Chen, Q.; Sun, L.; Chen, Z.J. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 2016, 17, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Pantelidou, C.; Sonzogni, O.; De Oliveria Taveira, M.; Mehta, A.K.; Kothari, A.; Wang, D.; Visal, T.; Li, M.K.; Pinto, J.; Castrillon, J.A.; et al. PARP Inhibitor Efficacy Depends on CD8(+) T-cell Recruitment via Intratumoral STING Pathway Activation in BRCA-Deficient Models of Triple-Negative Breast Cancer. Cancer Discov. 2019, 9, 722–737. [Google Scholar] [CrossRef] [PubMed]
- Yum, S.; Li, M.; Fang, Y.; Chen, Z.J. TBK1 recruitment to STING activates both IRF3 and NF-κB that mediate immune defense against tumors and viral infections. Proc. Natl. Acad. Sci. USA 2021, 118, e2100225118. [Google Scholar] [CrossRef]
- Liu, H.; Li, H.; Zhang, J.; Meng, Q.; Ma, L. Correlation of TBK1, AR, and other serum cancer-related biomarkers in breast cancer patients: An observational study. Medicine 2022, 101, e29996. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Guo, S.; Stiles, J.K. The emerging role of CXCL10 in cancer (Review). Oncol. Lett. 2011, 2, 583–589. [Google Scholar] [CrossRef]
- Mulligan, A.M.; Raitman, I.; Feeley, L.; Pinnaduwage, D.; Nguyen, L.T.; O’Malley, F.P.; Ohashi, P.S.; Andrulis, I.L. Tumoral lymphocytic infiltration and expression of the chemokine CXCL10 in breast cancers from the Ontario Familial Breast Cancer Registry. Clin. Cancer Res. 2013, 19, 336–346. [Google Scholar] [CrossRef]
- Isaac, D.; Karapetyan, L.; Tamkus, D. Association of Germline PALB2 Mutation and Response to Platinum-Based Chemotherapy in Metastatic Breast Cancer: A Case Series. JCO Precis. Oncol. 2018, 2, 1–5. [Google Scholar] [CrossRef]
- Vikas, P.; Borcherding, N.; Chennamadhavuni, A.; Garje, R. Therapeutic Potential of Combining PARP Inhibitor and Immunotherapy in Solid Tumors. Front. Oncol. 2020, 10, 570. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deb, S.; Chakrabarti, A.; Fox, S.B. Prognostic and Predictive Biomarkers in Familial Breast Cancer. Cancers 2023, 15, 1346. https://doi.org/10.3390/cancers15041346
Deb S, Chakrabarti A, Fox SB. Prognostic and Predictive Biomarkers in Familial Breast Cancer. Cancers. 2023; 15(4):1346. https://doi.org/10.3390/cancers15041346
Chicago/Turabian StyleDeb, Siddhartha, Anannya Chakrabarti, and Stephen B. Fox. 2023. "Prognostic and Predictive Biomarkers in Familial Breast Cancer" Cancers 15, no. 4: 1346. https://doi.org/10.3390/cancers15041346
APA StyleDeb, S., Chakrabarti, A., & Fox, S. B. (2023). Prognostic and Predictive Biomarkers in Familial Breast Cancer. Cancers, 15(4), 1346. https://doi.org/10.3390/cancers15041346