
Systems Biology Approaches for the Improvement of Oncolytic Virus-Based Immunotherapies

 , , , , ,
, , , , ,  and
and 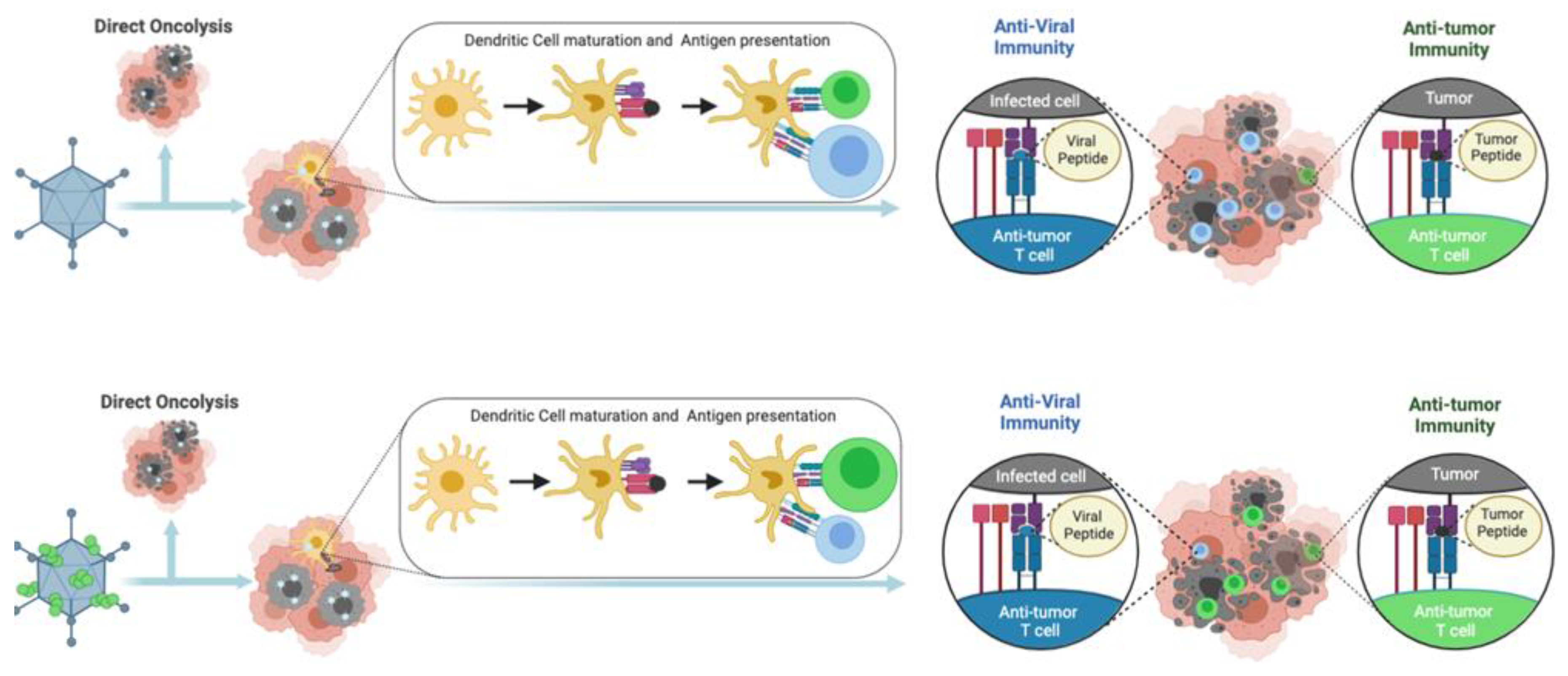{kind=link}
{kind=link}
{kind=link}
{kind=link}
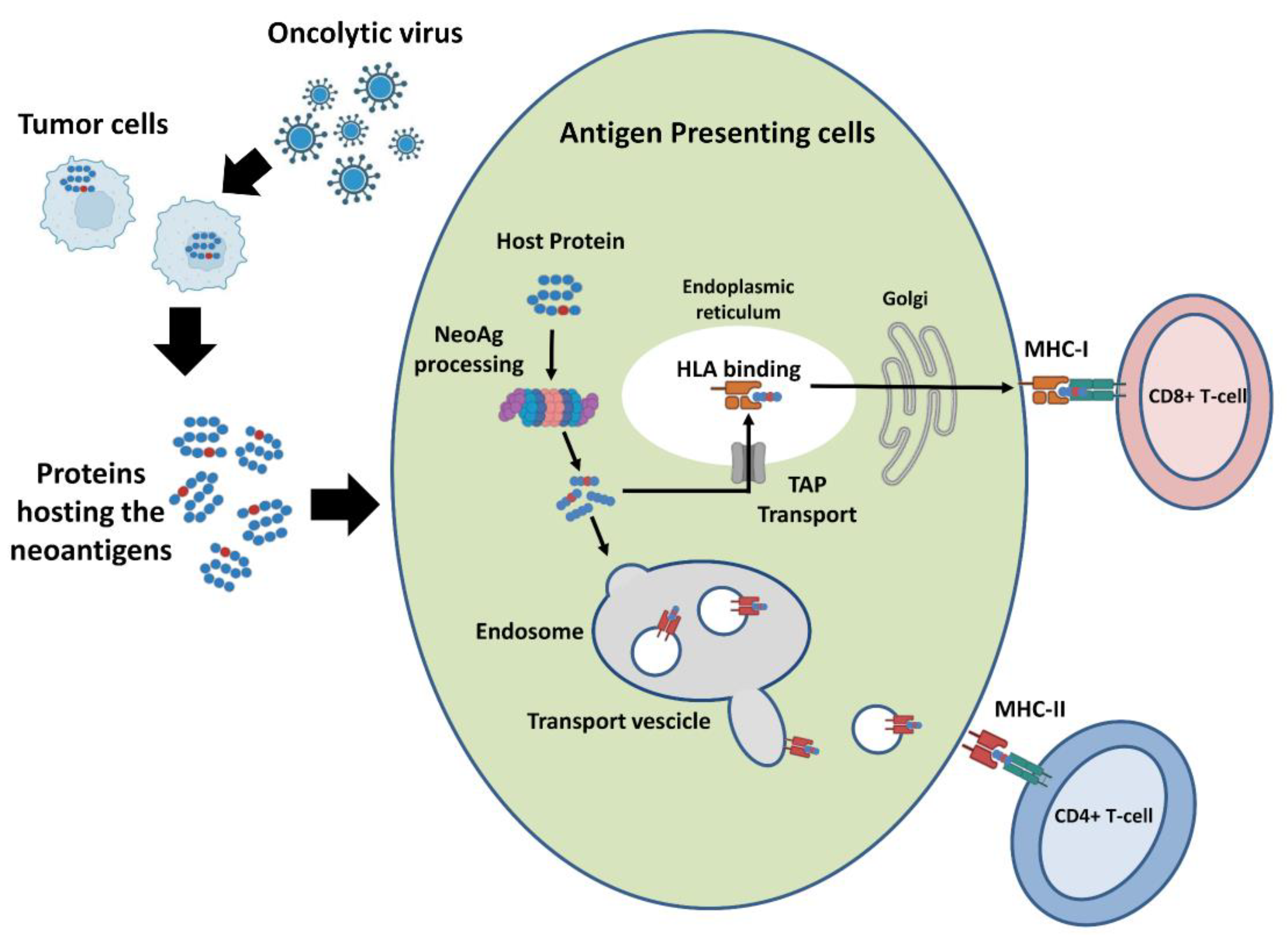{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
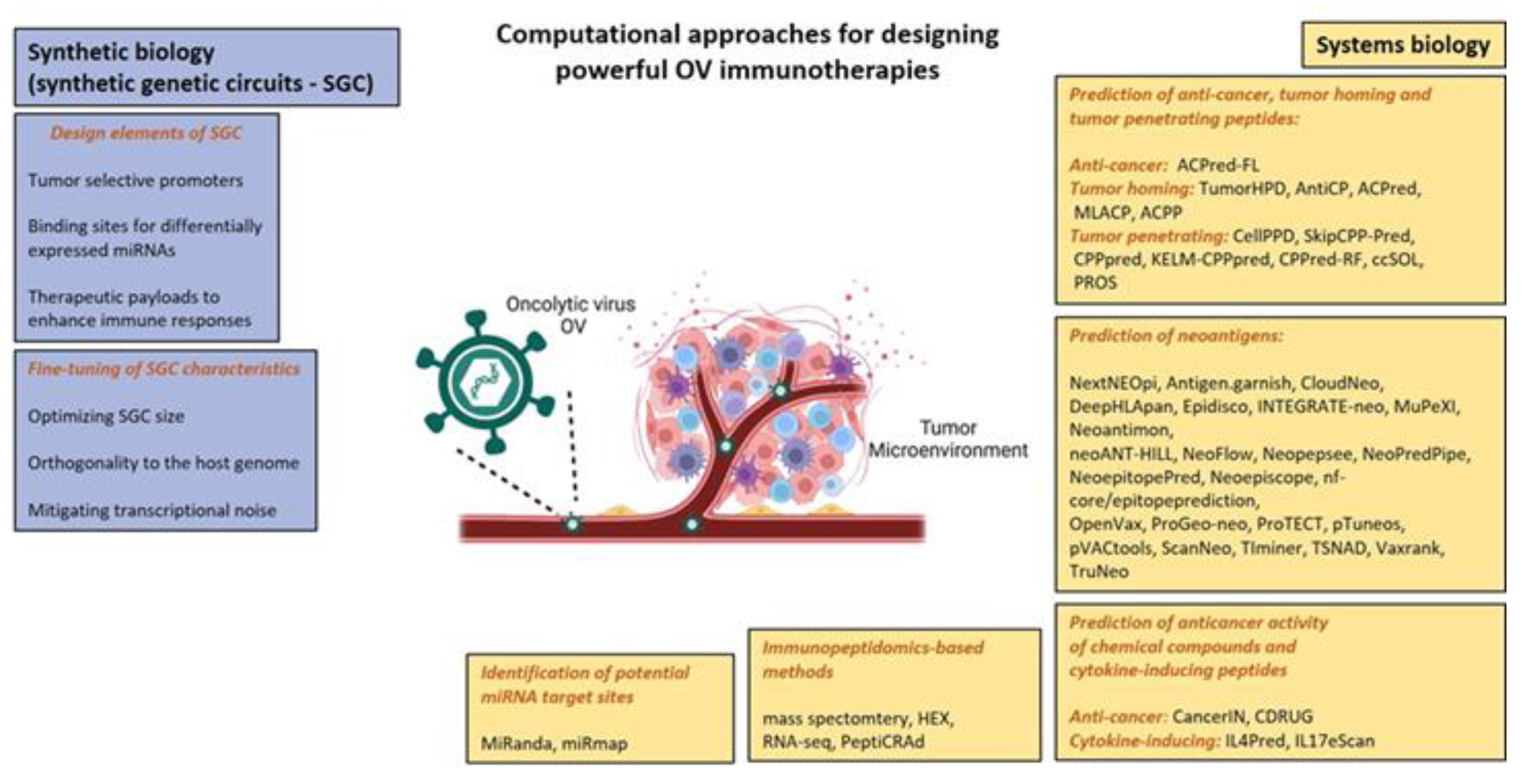
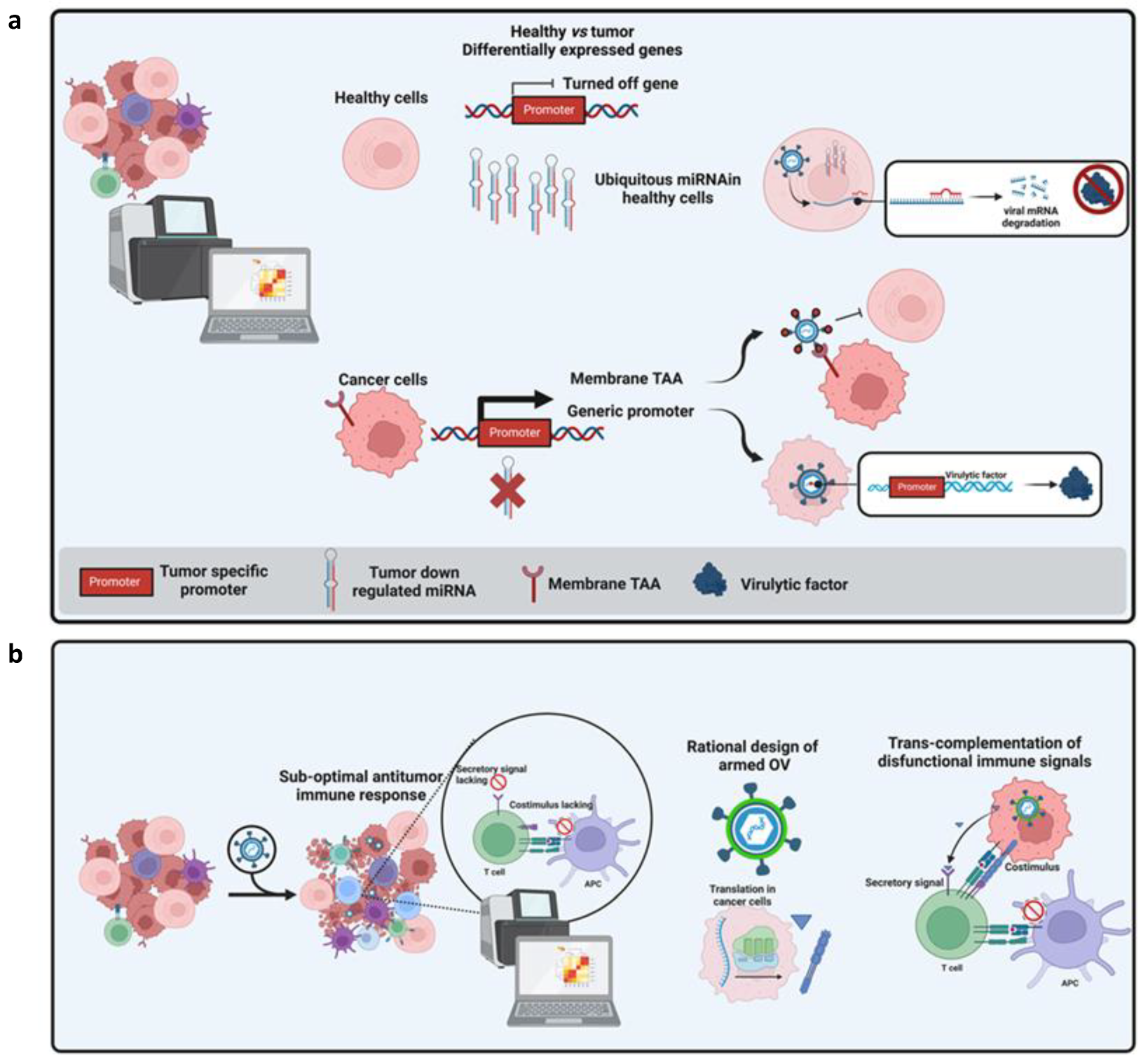
Synthetic Biology as a Tool to Optimize OV Design
2. Applications of System Biology in Target Discovery to Design New OV-Based Cancer Vaccines
2.1. Ligandome Analysis for Therapeutic Cancer Approaches
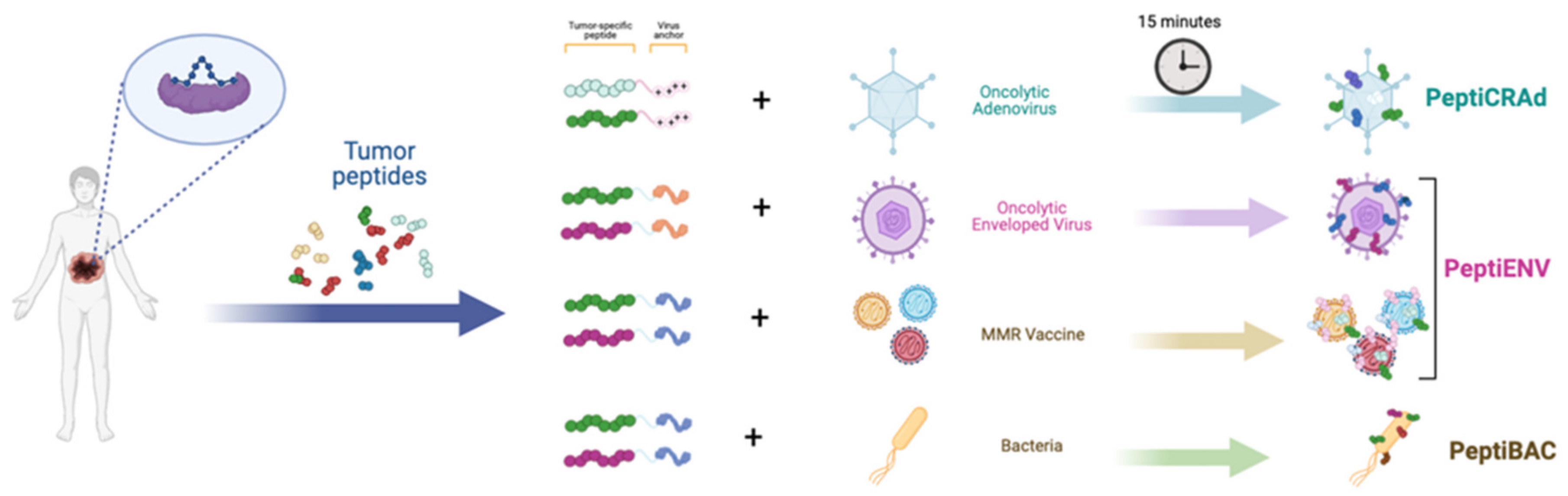
2.2. Systems Biology Approaches for the Improvement of OV-Based Immunotherapies
2.3. The Right One at the Right Time: Exploiting System Biology to Implement Safety and Efficacy of OVs
2.4. Other Bioinformatic Tools to Find Out Immunogenic Peptides/Neoantigens to Insert in Vectors
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liu, M.; Guo, F. Recent Updates on Cancer Immunotherapy. Precis. Clin. Med. 2018, 1, pby011. [Google Scholar] [CrossRef] [PubMed]
- Klebanoff, C.A.; Rosenberg, S.A.; Restifo, N.P. Prospects for Gene-Engineered T Cell Immunotherapy for Solid Cancers. Nat. Med. 2016, 22, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Soria, J.-C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive Correlates of Response to the Anti-PD-L1 Antibody MPDL3280A in Cancer Patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Sasso, E.; D’Avino, C.; Passariello, M.; D’Alise, A.M.; Siciliano, D.; Esposito, M.L.; Froechlich, G.; Cortese, R.; Scarselli, E.; Zambrano, N.; et al. Massive Parallel Screening of Phage Libraries for the Generation of Repertoires of Human Immunomodulatory Monoclonal Antibodies. Mabs 2018, 10, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Tripodi, L.; Vitale, M.; Cerullo, V.; Pastore, L. Oncolytic Adenoviruses for Cancer Therapy. Int. J. Mol. Sci. 2021, 22, 2517. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.I.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 2017, 170, 1109–1119. [Google Scholar] [CrossRef] [PubMed]
- Bommareddy, P.K.; Patel, A.; Hossain, S.; Kaufman, H.L. Talimogene Laherparepvec (T-VEC) and Other Oncolytic Viruses for the Treatment of Melanoma. Am. J. Clin. Dermatol. 2017, 18, 1–15. [Google Scholar] [CrossRef]
- Puzanov, I.; Chesney, J.; Collichio, F.; Singh, P.; Milhem, M.; Glaspy, J.; Hamid, O.; Ross, M.; Friedlander, P.; Garbe, C.; et al. Talimogene laherparepvec (T-VEC) in combination with ipilimumab (ipi) versus ipi alone for advanced melanoma: 4-year interim analysis of a randomized, open-label, phase 2 trial. J. Immunother. Cancer 2020, 8 (Suppl. 3), A1–A559. [Google Scholar]
- Carr, M.J.; Sun, J.; DePalo, D.; Rothermel, L.D.; Song, Y.; Straker, R.J.; Baecher, K.; Louie, R.J.; Stahlie, E.H.A.; Wright, G.P.; et al. Talimogene Laherparepvec (T-VEC) for the Treatment of Advanced Locoregional Melanoma After Failure of Immunotherapy: An International Multi-Institutional Experience. Ann. Surg. Oncol. 2022, 29, 791–801. [Google Scholar] [CrossRef]
- Chesney, J.A.; Ribas, A.; Long, G.V.; Kirkwood, J.M.; Dummer, R.; Puzanov, I.; Hoeller, C.; Gajewski, T.F.; Gutzmer, R.; Rutkowski, P.; et al. Randomized, Double-Blind, Placebo-Controlled, Global Phase III Trial of Talimogene Laherparepvec Combined With Pembrolizumab for Advanced Melanoma. J. Clin. Oncol. 2023, 41, 528–540. [Google Scholar] [CrossRef]
- Lin, Y.; Zheng, C. A Tug of War: DNA-Sensing Antiviral Innate Immunity and Herpes Simplex Virus Type I Infection. Front. Microbiol. 2019, 10, 2627. [Google Scholar] [CrossRef]
- Ye, G.; Liu, H.; Zhou, Q.; Liu, X.; Huang, L.; Weng, C. A Tug of War: Pseudorabies Virus and Host Antiviral Innate Immunity. Viruses 2022, 14, 547. [Google Scholar] [CrossRef]
- Macedo, N.; Miller, D.M.; Haq, R.; Kaufman, H.L. Clinical Landscape of Oncolytic Virus Research in 2020. J. Immunother. Cancer 2020, 8, e001486. [Google Scholar] [CrossRef] [PubMed]
- Vitale, M.; Scialò, F.; Passariello, M.; Leggiero, E.; D’Agostino, A.; Tripodi, L.; Gentile, L.; Bianco, A.; Castaldo, G.; Cerullo, V.; et al. Oncolytic Adenoviral Vector-Mediated Expression of an Anti-PD-L1-ScFv Improves Anti-Tumoral Efficacy in a Melanoma Mouse Model. Front. Oncol. 2022, 12, 902190. [Google Scholar] [CrossRef] [PubMed]
- Tripodi, L.; Passariello, M.; D’Argenio, V.; Leggiero, E.; Vitale, M.; Colicchio, R.; Salvatore, P.; Cerullo, V.; Pastore, C.D.L.L. Evaluation of the Antiproliferative Effect of Bifidobacterium Longum BB-536 in Solid Tumor Cell Lines, Co-Cultured with Murine Splenocytes. Biochim. Clin. 2021, 45, 242–247. [Google Scholar] [CrossRef]
- Huang, H.; Liu, Y.; Liao, W.; Cao, Y.; Liu, Q.; Guo, Y.; Lu, Y.; Xie, Z. Oncolytic Adenovirus Programmed by Synthetic Gene Circuit for Cancer Immunotherapy. Nat. Commun. 2019, 10, 4801. [Google Scholar] [CrossRef]
- Serrano, L. Synthetic Biology: Promises and Challenges. Mol. Syst. Biol. 2007, 3, 158. [Google Scholar] [CrossRef]
- Monie, D.D.; Bhandarkar, A.R.; Parney, I.F.; Correia, C.; Sarkaria, J.N.; Vile, R.G.; Li, H. Synthetic and Systems Biology Principles in the Design of Programmable Oncolytic Virus Immunotherapies for Glioblastoma. Neurosurg. Focus 2021, 50, E10. [Google Scholar] [CrossRef]
- Leventhal, D.S.; Sokolovska, A.; Li, N.; Plescia, C.; Kolodziej, S.A.; Gallant, C.W.; Christmas, R.; Gao, J.-R.; James, M.J.; Abin-Fuentes, A.; et al. Immunotherapy with Engineered Bacteria by Targeting the STING Pathway for Anti-Tumor Immunity. Nat. Commun. 2020, 11, 2739. [Google Scholar] [CrossRef]
- Swift, S.L.; Stojdl, D.F. Big Data Offers Novel Insights for Oncolytic Virus Immunotherapy. Viruses 2016, 8, 45. [Google Scholar] [CrossRef]
- Ideker, T.; Galitski, T.; Hood, L. A new approach to decoding life: Systems Biology. Annu. Rev. Genom. Hum. G 2001, 2, 343–372. [Google Scholar] [CrossRef] [PubMed]
- GuhaThakurta, D.; Sheikh, N.A.; Meagher, T.C.; Letarte, S.; Trager, J.B. Applications of Systems Biology in Cancer Immunotherapy: From Target Discovery to Biomarkers of Clinical Outcome. Expert. Rev. Clin. Phar. 2013, 6, 387–401. [Google Scholar] [CrossRef]
- Feola, S.; Chiaro, J.; Martins, B.; Cerullo, V. Uncovering the Tumor Antigen Landscape: What to Know about the Discovery Process. Cancers 2020, 12, 1660. [Google Scholar] [CrossRef] [PubMed]
- Freudenmann, L.K.; Marcu, A.; Stevanović, S. Mapping the Tumour Human Leukocyte Antigen (HLA) Ligandome by Mass Spectrometry. Immunology 2018, 154, 331–345. [Google Scholar] [CrossRef] [PubMed]
- Kovalchik, K.A.; Wessling, L.; Saab, F.; Ma, Q.; Despault, J.; Kubiniok, P.; Hamelin, D.; Faridi, P.; Li, C.; Purcell, A.; et al. Immunopeptidomics for Dummies: Detailed Experimental Protocols and Rapid, User-Friendly Visualization of MHC I and II Ligand Datasets with MhcVizPipe. biorXiv 2020. [Google Scholar] [CrossRef]
- Sharma, P.; Wagner, K.; Wolchok, J.D.; Allison, J.P. Novel Cancer Immunotherapy Agents with Survival Benefit: Recent Successes and next Steps. Nat. Rev. Cancer 2011, 11, 805–812. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. Immune Checkpoint Targeting in Cancer Therapy: Toward Combination Strategies with Curative Potential. Cell 2015, 161, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The Blockade of Immune Checkpoints in Cancer Immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Bonaventura, P.; Shekarian, T.; Alcazer, V.; Valladeau-Guilemond, J.; Valsesia-Wittmann, S.; Amigorena, S.; Caux, C.; Depil, S. Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front. Immunol. 2019, 10, 168. [Google Scholar] [CrossRef]
- Kowalewski, D.J.; Schuster, H.; Backert, L.; Berlin, C.; Kahn, S.; Kanz, L.; Salih, H.R.; Rammensee, H.-G.; Stevanovic, S.; Stickel, J.S. HLA Ligandome Analysis Identifies the Underlying Specificities of Spontaneous Antileukemia Immune Responses in Chronic Lymphocytic Leukemia (CLL). Proc. Natl. Acad. Sci. USA 2015, 112, E166–E175. [Google Scholar] [CrossRef]
- Löffler, M.W.; Kowalewski, D.J.; Backert, L.; Bernhardt, J.; Adam, P.; Schuster, H.; Dengler, F.; Backes, D.; Kopp, H.-G.; Beckert, S.; et al. Mapping the HLA Ligandome of Colorectal Cancer Reveals an Imprint of Malignant Cell Transformation. Cancer Res. 2018, 78, 4627–4641. [Google Scholar] [CrossRef] [PubMed]
- Bassani-Sternberg, M.; Bräunlein, E.; Klar, R.; Engleitner, T.; Sinitcyn, P.; Audehm, S.; Straub, M.; Weber, J.; Slotta-Huspenina, J.; Specht, K.; et al. Direct Identification of Clinically Relevant Neoepitopes Presented on Native Human Melanoma Tissue by Mass Spectrometry. Nat. Commun. 2016, 7, 13404. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.P.; Kim, Y.; Clements, D.R.; Konda, P.; Schuster, H.; Kowalewski, D.J.; Paulo, J.A.; Cohen, A.M.; Stevanovic, S.; Gygi, S.P.; et al. Therapy-Induced MHC I Ligands Shape Neo-Antitumor CD8 T Cell Responses during Oncolytic Virus-Based Cancer Immunotherapy. J. Proteome Res. 2019, 18, 2666–2675. [Google Scholar] [CrossRef]
- Marin, I.; Boix, O.; Garcia-Garijo, A.; Sirois, I.; Caballe, A.; Zarzuela, E.; Ruano, I.; Attolini, C.S.-O.; Prats, N.; Lopez-Dominguez, J.A.; et al. Cellular Senescence Is Immunogenic and Promotes Anti-Tumor Immunity. Cancer Discov. 2022, 2022, CD-0523. [Google Scholar] [CrossRef]
- Chong, C.; Müller, M.; Pak, H.; Harnett, D.; Huber, F.; Grun, D.; Leleu, M.; Auger, A.; Arnaud, M.; Stevenson, B.J.; et al. Integrated Proteogenomic Deep Sequencing and Analytics Accurately Identify Non-Canonical Peptides in Tumor Immunopeptidomes. Nat. Commun. 2020, 11, 1293. [Google Scholar] [CrossRef]
- Laumont, C.M.; Vincent, K.; Hesnard, L.; Audemard, É.; Bonneil, É.; Laverdure, J.-P.; Gendron, P.; Courcelles, M.; Hardy, M.-P.; Côté, C.; et al. Noncoding Regions Are the Main Source of Targetable Tumor-Specific Antigens. Sci. Transl. Med. 2018, 10, eaau5516. [Google Scholar] [CrossRef]
- Peltonen, K.; Feola, S.; Umer, H.M.; Chiaro, J.; Mermelekas, G.; Ylösmäki, E.; Pesonen, S.; Branca, R.M.M.; Lehtiö, J.; Cerullo, V. Therapeutic Cancer Vaccination with Immunopeptidomics-Discovered Antigens Confers Protective Antitumor Efficacy. Cancers 2021, 13, 3408. [Google Scholar] [CrossRef]
- Chiaro, J.; Antignani, G.; Feola, S.; Feodoroff, M.; Martins, B.; Cojoc, H.; Ferrari, V.; Ciampi, D.; Ilonen, I.; Räsänen, J.; et al. Development of Mesothelioma-Specific Oncolytic Vaccine Exploiting Immunopeptidomic Analysis of Murine and Human Tumors. 08 November 2022, PREPRINT (Version 1) available at Research Square. Available online: https://www.researchsquare.com/article/rs-2238403/v1 (accessed on 20 November 2022).
- Capasso, C.; Hirvinen, M.; Garofalo, M.; Romaniuk, D.; Kuryk, L.; Sarvela, T.; Vitale, A.; Antopolsky, M.; Magarkar, A.; Viitala, T.; et al. Oncolytic Adenoviruses Coated with MHC-I Tumor Epitopes Increase the Antitumor Immunity and Efficacy against Melanoma. Oncoimmunology 2015, 5, e1105429. [Google Scholar] [CrossRef]
- Kuryk, L.; Haavisto, E.; Garofalo, M.; Capasso, C.; Hirvinen, M.; Pesonen, S.; Ranki, T.; Vassilev, L.; Cerullo, V. Synergistic Anti-Tumor Efficacy of Immunogenic Adenovirus ONCOS-102 (Ad5/3-D24-GM-CSF) and Standard of Care Chemotherapy in Preclinical Mesothelioma Model. Int. J. Cancer 2016, 139, 1883–1893. [Google Scholar] [CrossRef] [PubMed]
- Capasso, C.; Magarkar, A.; Cervera-Carascon, V.; Fusciello, M.; Feola, S.; Muller, M.; Garofalo, M.; Kuryk, L.; Tähtinen, S.; Pastore, L.; et al. A Novel in Silico Framework to Improve MHC-I Epitopes and Break the Tolerance to Melanoma. OncoImmunology 2017, 6, e1319028. [Google Scholar] [CrossRef] [PubMed]
- Feola, S.; Capasso, C.; Fusciello, M.; Martins, B.; Tähtinen, S.; Medeot, M.; Carpi, S.; Frascaro, F.; Ylosmäki, E.; Peltonen, K.; et al. Oncolytic Vaccines Increase the Response to PD-L1 Blockade in Immunogenic and Poorly Immunogenic Tumors. OncoImmunology 2018, 7, e1457596. [Google Scholar] [CrossRef]
- Feola, S.; Chiaro, J.; Martins, B.; Russo, S.; Fusciello, M.; Ylösmäki, E.; Bonini, C.; Ruggiero, E.; Hamdan, F.; Feodoroff, M.; et al. A Novel Immunopeptidomic-Based Pipeline for the Generation of Personalized Oncolytic Cancer Vaccines. eLife 2022, 11, e71156. [Google Scholar] [CrossRef]
- Ylösmäki, E.; Malorzo, C.; Capasso, C.; Honkasalo, O.; Fusciello, M.; Martins, B.; Ylösmäki, L.; Louna, A.; Feola, S.; Paavilainen, H.; et al. Personalized Cancer Vaccine Platform for Clinically Relevant Oncolytic Enveloped Viruses. Mol. Ther. 2018, 26, 2315–2325. [Google Scholar] [CrossRef] [PubMed]
- Ylösmäki, E.; Fusciello, M.; Martins, B.; Feola, S.; Hamdan, F.; Chiaro, J.; Ylösmäki, L.; Vaughan, M.J.; Viitala, T.; Kulkarni, P.S.; et al. Novel Personalized Cancer Vaccine Platform Based on Bacillus Calmette-Guèrin. J. Immunother. Cancer 2021, 9, e002707. [Google Scholar] [CrossRef] [PubMed]
- Fusciello, M.; Ylösmäki, E.; Feola, S.; Uoti, A.; Martins, B.; Aalto, K.; Hamdan, F.; Chiaro, J.; Russo, S.; Viitala, T.; et al. A Novel Cancer Vaccine for Melanoma Based on an Approved Vaccine against Measles, Mumps, and Rubella. Mol. Ther.-Oncolytics 2022, 25, 137–145. [Google Scholar] [CrossRef]
- Jensen, S.M.; Potts, G.K.; Ready, D.B.; Patterson, M.J. Specific MHC-I Peptides Are Induced Using PROTACs. Front. Immunol. 2018, 9, 2697. [Google Scholar] [CrossRef] [PubMed]
- Feola, S.; Haapala, M.; Peltonen, K.; Capasso, C.; Martins, B.; Antignani, G.; Federico, A.; Pietiäinen, V.; Chiaro, J.; Feodoroff, M.; et al. PeptiCHIP: A Microfluidic Platform for Tumor Antigen Landscape Identification. ACS Nano 2021, 15, 15992–16010. [Google Scholar] [CrossRef]
- Alberts, P.; Tilgase, A.; Rasa, A.; Bandere, K.; Venskus, D. The Advent of Oncolytic Virotherapy in Oncology: The Rigvir® Story. Eur. J. Pharmacol. 2018, 837, 117–126. [Google Scholar] [CrossRef]
- Liang, M. Oncorine, the World First Oncolytic Virus Medicine and Its Update in China. Curr. Cancer Drug Tar. 2018, 18, 171–176. [Google Scholar] [CrossRef]
- Andtbacka, R.H.I.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef]
- Mucaj, V.; Lee, S.S.; Skuli, N.; Giannoukos, D.N.; Qiu, B.; Eisinger-Mathason, T.S.K.; Nakazawa, M.S.; Shay, J.E.S.; Gopal, P.P.; Venneti, S.; et al. MicroRNA-124 Expression Counteracts pro-Survival Stress Responses in Glioblastoma. Oncogene 2015, 34, 2204–2214. [Google Scholar] [CrossRef] [PubMed]
- Brophy, J.A.N.; Voigt, C.A. Principles of Genetic Circuit Design. Nat. Methods 2014, 11, 508–520. [Google Scholar] [CrossRef]
- Gardner, T.S.; Cantor, C.R.; Collins, J.J. Construction of a Genetic Toggle Switch in Escherichia Coli. Nature 2000, 403, 339–342. [Google Scholar] [CrossRef] [PubMed]
- Danino, T.; Mondragón-Palomino, O.; Tsimring, L.; Hasty, J. A Synchronized Quorum of Genetic Clocks. Nature 2010, 463, 326–330. [Google Scholar] [CrossRef]
- Daniel, R.; Rubens, J.R.; Sarpeshkar, R.; Lu, T.K. Synthetic Analog Computation in Living Cells. Nature 2013, 497, 619–623. [Google Scholar] [CrossRef]
- Naoum, G.E.; Zhu, Z.B.; Buchsbaum, D.J.; Curiel, D.T.; Arafat, W.O. Survivin a Radiogenetic Promoter for Glioblastoma Viral Gene Therapy Independently from CArG Motifs. Clin. Transl. Med. 2017, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Ali, N.; Zhong, L.; Shi, J. MicroRNAs as Biomarkers for Human Glioblastoma: Progress and Potential. Acta Pharmacol. Sin. 2018, 39, 1405–1413. [Google Scholar] [CrossRef]
- Mazzacurati, L.; Marzulli, M.; Reinhart, B.; Miyagawa, Y.; Uchida, H.; Goins, W.F.; Li, A.; Kaur, B.; Caligiuri, M.; Cripe, T.; et al. Use of MiRNA Response Sequences to Block Off-Target Replication and Increase the Safety of an Unattenuated, Glioblastoma-Targeted Oncolytic HSV. Mol. Ther. 2015, 23, 99–107. [Google Scholar] [CrossRef]
- Crommentuijn, M.H.; Kantar, R.; Noske, D.P.; Vandertop, W.P.; Badr, C.E.; Würdinger, T.; Maguire, C.A.; Tannous, B.A. Systemically Administered AAV9-STRAIL Combats Invasive Glioblastoma in a Patient-Derived Orthotopic Xenograft Model. Mol. Ther.-Oncolytics 2016, 3, 16017. [Google Scholar] [CrossRef]
- Xie, Z.; Wroblewska, L.; Prochazka, L.; Weiss, R.; Benenson, Y. Multi-Input RNAi-Based Logic Circuit for Identification of Specific Cancer Cells. Science vol 333 2011. [CrossRef] [PubMed]
- Schlabach, M.R.; Hu, J.K.; Li, M.; Elledge, S.J. Synthetic Design of Strong Promoters. Proc. Natl. Acad. Sci. USA 2010, 107, 2538–2543. [Google Scholar] [CrossRef] [PubMed]
- Eldar, A.; Elowitz, M.B. Functional Roles for Noise in Genetic Circuits. Nature 2010, 467, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Marqus, S.; Pirogova, E.; Piva, T.J. Evaluation of the Use of Therapeutic Peptides for Cancer Treatment. J. Biomed. Sci. 2017, 24, 21. [Google Scholar] [CrossRef] [PubMed]
- Lathwal, A.; Kumar, R.; Raghava, G.P.S. Computer-Aided Designing of Oncolytic Viruses for Overcoming Translational Challenges of Cancer Immunotherapy. Drug. Discov. Today 2020, 25, 1198–1205. [Google Scholar] [CrossRef] [PubMed]
- Groisberg, R.; Maymani, H.; Subbiah, V. Immunotherapy and Next-Generation Sequencing Guided Therapy for Precision Oncology: What Have We Learnt and What Does the Future Hold? Expert. Rev. Precis. Med. Drug Dev. 2018, 3, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Kang, X.; Chen, K.S.; Jehng, T.; Jones, L.; Chen, J.; Huang, X.F.; Chen, S.-Y. An Engineered Oncolytic Virus Expressing PD-L1 Inhibitors Activates Tumor Neoantigen-Specific T Cell Responses. Nat. Commun. 2020, 11, 1395. [Google Scholar] [CrossRef]
- Russell, S.J.; Barber, G.N. Oncolytic Viruses as Antigen-Agnostic Cancer Vaccines. Cancer Cell 2018, 33, 599–605. [Google Scholar] [CrossRef]
- Fotakis, G.; Trajanoski, Z.; Rieder, D. Computational Cancer Neoantigen Prediction: Current Status and Recent Advances. Immuno-Oncol. Technol. 2021, 12, 100052. [Google Scholar] [CrossRef] [PubMed]
- Wells, D.K.; van Buuren, M.M.; Dang, K.K.; Hubbard-Lucey, V.M.; Sheehan, K.C.F.; Campbell, K.M.; Lamb, A.; Ward, J.P.; Sidney, J.; Blazquez, A.B.; et al. Key Parameters of Tumor Epitope Immunogenicity Revealed Through a Consortium Approach Improve Neoantigen Prediction. Cell 2020, 183, 818–834. [Google Scholar] [CrossRef]
- Ghorani, E.; Rosenthal, R.; McGranahan, N.; Reading, J.L.; Lynch, M.; Peggs, K.S.; Swanton, C.; Quezada, S.A. Differential Binding Affinity of Mutated Peptides for MHC Class I Is a Predictor of Survival in Advanced Lung Cancer and Melanoma. Ann. Oncol. 2018, 29, 271–279. [Google Scholar] [CrossRef]
- Łuksza, M.; Riaz, N.; Makarov, V.; Balachandran, V.P.; Hellmann, M.D.; Solovyov, A.; Rizvi, N.A.; Merghoub, T.; Levine, A.J.; Chan, T.A.; et al. A Neoantigen Fitness Model Predicts Tumour Response to Checkpoint Blockade Immunotherapy. Nature 2017, 551, 517–520. [Google Scholar] [CrossRef] [PubMed]
- Goncharova, E.P.; Ruzhenkova, J.S.; Petrov, I.S.; Shchelkunov, S.N.; Zenkova, M.A. Oncolytic Virus Efficiency Inhibited Growth of Tumour Cells with Multiple Drug Resistant Phenotype in Vivo and in Vitro. J. Transl. Med. 2016, 14, 241. [Google Scholar] [CrossRef] [PubMed]
- Stopfer, L.E.; D’Souza, A.D.; White, F.M. 1,2,3, MHC: A Review of Mass Spectrometry-Based Immunopeptidomics Methods for Relative and Absolute Quantification of PMHCs. Immuno-Oncol. Technol. 2021, 11, 100042. [Google Scholar] [CrossRef]
- Chiaro, J.; Kasanen, H.H.E.; Whalley, T.; Capasso, C.; Grönholm, M.; Feola, S.; Peltonen, K.; Hamdan, F.; Hernberg, M.; Mäkelä, S.; et al. Viral Molecular Mimicry Influences the Antitumor Immune Response in Murine and Human Melanoma. Cancer Immunol. Res. 2021, 9, 981–993. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Kuncheria, L.; Roulstone, V.; Kyula, J.N.; Mansfield, D.; Bommareddy, P.K.; Smith, H.; Kaufman, H.L.; Harrington, K.J.; Coffin, R.S. Development of a New Fusion-Enhanced Oncolytic Immunotherapy Platform Based on Herpes Simplex Virus Type 1. J. Immunother. Cancer 2019, 7, 214. [Google Scholar] [CrossRef] [PubMed]
- Naumenko, V.A.; Stepanenko, A.A.; Lipatova, A.V.; Vishnevskiy, D.A. Infection of Non-Cancer Cells: A Barrier or Support for Oncolytic Virotherapy? Mol. Ther.-Oncolytics 2022, 24, 663–682. [Google Scholar] [CrossRef]
- Menotti, L.; Avitabile, E.; Gatta, V.; Malatesta, P.; Petrovic, B.; Campadelli-Fiume, G. HSV as A Platform for the Generation of Retargeted, Armed, and Reporter-Expressing Oncolytic Viruses. Viruses 2018, 10, 352. [Google Scholar] [CrossRef]
- Froechlich, G.; Gentile, C.; Infante, L.; Caiazza, C.; Pagano, P.; Scatigna, S.; Cotugno, G.; D’Alise, A.M.; Lahm, A.; Scarselli, E.; et al. Generation of a Novel Mesothelin-Targeted Oncolytic Herpes Virus and Implemented Strategies for Manufacturing. Int. J. Mol. Sci. 2021, 22, 477. [Google Scholar] [CrossRef]
- Sharabi, O.; Greenshpan, Y.; Ofir, N.; Ottolenghi, A.; Levi, T.; Olender, L.; Adler-Agmon, Z.; Porgador, A.; Gazit, R. High Throughput Screen for the Improvement of Inducible Promoters for Tumor Microenvironment Cues. Sci. Rep. 2022, 12, 7169. [Google Scholar] [CrossRef] [PubMed]
- Mullen, J.T.; Kasuya, H.; Yoon, S.S.; Carroll, N.M.; Pawlik, T.M.; Chandrasekhar, S.; Nakamura, H.; Donahue, J.M.; Tanabe, K.K. Regulation of Herpes Simplex Virus 1 Replication Using Tumor-Associated Promoters. Ann. Surg. 2002, 236, 502–513. [Google Scholar] [CrossRef]
- Kambara, H.; Okano, H.; Chiocca, E.A.; Saeki, Y. An Oncolytic HSV-1 Mutant Expressing ICP34.5 under Control of a Nestin Promoter Increases Survival of Animals Even When Symptomatic from a Brain Tumor. Cancer Res. 2005, 65, 2832–2839. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Liu, X.; Jiang, B.; Wei, S.; Xiang, B.; Liao, R.; Wang, Q.; He, X. A Genome-Wide Investigation of Effects of Aberrant DNA Methylation on the Usage of Alternative Promoters in Hepatocellular Carcinoma. Front. Oncol. 2022, 11, 780266. [Google Scholar] [CrossRef]
- Everts, B.; Poel, H.G. van der Replication-Selective Oncolytic Viruses in the Treatment of Cancer. Cancer Gene Ther. 2005, 12, 141–161. [Google Scholar] [CrossRef] [PubMed]
- Chiocca, E.A.; Nakashima, H.; Kasai, K.; Fernandez, S.A.; Oglesbee, M. Preclinical Toxicology of RQNestin34.5v.2: An Oncolytic Herpes Virus with Transcriptional Regulation of the ICP34.5 Neurovirulence Gene. Mol. Ther.-Methods Clin. Dev. 2020, 17, 871–893. [Google Scholar] [CrossRef]
- Sasso, E.; Froechlich, G.; Cotugno, G.; D’Alise, A.M.; Gentile, C.; Bignone, V.; Lucia, M.D.; Petrovic, B.; Campadelli-Fiume, G.; Scarselli, E.; et al. Replicative Conditioning of Herpes Simplex Type 1 Virus by Survivin Promoter, Combined to ERBB2 Retargeting, Improves Tumour Cell-Restricted Oncolysis. Sci. Rep. 2020, 10, 4307. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R.C.; Farh, K.K.-H.; Burge, C.B.; Bartel, D.P. Most Mammalian MRNAs Are Conserved Targets of MicroRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef]
- José, A.; Sobrevals, L.; Camacho-Sánchez, J.M.; Huch, M.; Andreu, N.; Ayuso, E.; Navarro, P.; Alemany, R.; Fillat, C. Intraductal Delivery of Adenoviruses Targets Pancreatic Tumors in Transgenic Ela-Myc Mice and Orthotopic Xenografts. Oncotarget 2013, 4, 94–105. [Google Scholar] [CrossRef] [PubMed]
- John, B.; Enright, A.J.; Aravin, A.; Tuschl, T.; Sander, C.; Marks, D.S. Human MicroRNA Targets. PLoS Biol. 2004, 2, e363. [Google Scholar] [CrossRef]
- Vejnar, C.E.; Zdobnov, E.M. MiRmap: Comprehensive Prediction of MicroRNA Target Repression Strength. Nucleic Acids Res. 2012, 40, 11673–11683. [Google Scholar] [CrossRef]
- Li, X.; Su, Y.; Sun, B.; Ji, W.; Peng, Z.; Xu, Y.; Wu, M.; Su, C. An Artificially Designed Interfering LncRNA Expressed by Oncolytic Adenovirus Competitively Consumes OncomiRs to Exert Antitumor Efficacy in Hepatocellular Carcinoma. Mol. Cancer Ther. 2016, 15, 1436–1451. [Google Scholar] [CrossRef]
- Sasso, E.; Latino, D.; Froechlich, G.; Succoio, M.; Passariello, M.; Lorenzo, C.D.; Nicosia, A.; Zambrano, N. A Long Non-Coding SINEUP RNA Boosts Semi-Stable Production of Fully Human Monoclonal Antibodies in HEK293E Cells. Mabs 2018, 10, 730–737. [Google Scholar] [CrossRef]
- De Graaf, J.F.; de Vor, L.; Fouchier, R.A.M.; Hoogen, B.G. van den Armed Oncolytic Viruses: A Kick-Start for Anti-Tumor Immunity. ytokine Growth Factor Rev. 2018, 41, 28–39. [Google Scholar] [CrossRef]
- Sasso, E.; D’Alise, A.M.; Zambrano, N.; Scarselli, E.; Folgori, A.; Nicosia, A. New Viral Vectors for Infectious Diseases and Cancer. Semin. Immunol. 2020, 50, 101430. [Google Scholar] [CrossRef]
- Kumar, A.; Khani, A.T.; Ortiz, A.S.; Swaminathan, S. GM-CSF: A Double-Edged Sword in Cancer Immunotherapy. Front. Immunol. 2022, 13, 901277. [Google Scholar] [CrossRef] [PubMed]
- Lucia, M.D.; Cotugno, G.; Bignone, V.; Garzia, I.; Nocchi, L.; Langone, F.; Petrovic, B.; Sasso, E.; Pepe, S.; Froechlich, G.; et al. Retargeted and Multi-Cytokine-Armed Herpes Virus Is a Potent Cancer Endovaccine for Local and Systemic Anti-Tumor Treatment. Mol. Ther.-Oncolytics 2020, 19, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Quixabeira, D.C.A.; Zafar, S.; Santos, J.M.; Cervera-Carrascon, V.; Havunen, R.; Kudling, T.V.; Basnet, S.; Anttila, M.; Kanerva, A.; Hemminki, A. Oncolytic Adenovirus Coding for a Variant Interleukin 2 (VIL-2) Cytokine Re-Programs the Tumor Microenvironment and Confers Enhanced Tumor Control. Front. Immunol. 2021, 12, 674400. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Dai, E.; Liu, Z.; Ma, C.; Guo, Z.S.; Bartlett, D.L. In Situ Therapeutic Cancer Vaccination with an Oncolytic Virus Expressing Membrane-Tethered IL-2. Mol. Ther.-Oncolytics 2020, 17, 350–360. [Google Scholar] [CrossRef]
- Liu, L.; Li, H.; Xu, Q.; Wu, Y.; Chen, D.; Yu, F. Antitumor Activity of Recombinant Oncolytic Vaccinia Virus with Human IL2. Open Med. 2022, 17, 1084–1091. [Google Scholar] [CrossRef] [PubMed]
- Froechlich, G.; Caiazza, C.; Gentile, C.; D’Alise, A.M.; Lucia, M.D.; Langone, F.; Leoni, G.; Cotugno, G.; Scisciola, V.; Nicosia, A.; et al. Integrity of the Antiviral STING-Mediated DNA Sensing in Tumor Cells Is Required to Sustain the Immunotherapeutic Efficacy of Herpes Simplex Oncolytic Virus. Cancers 2020, 12, 3407. [Google Scholar] [CrossRef] [PubMed]
- Zamarin, D.; Holmgaard, R.B.; Ricca, J.; Plitt, T.; Palese, P.; Sharma, P.; Merghoub, T.; Wolchok, J.D.; Allison, J.P. Intratumoral Modulation of the Inducible Co-Stimulator ICOS by Recombinant Oncolytic Virus Promotes Systemic Anti-Tumour Immunity. Nat. Commun. 2017, 8, 14340. [Google Scholar] [CrossRef]
- Rivadeneira, D.B.; DePeaux, K.; Wang, Y.; Kulkarni, A.; Tabib, T.; Menk, A.V.; Sampath, P.; Lafyatis, R.; Ferris, R.L.; Sarkar, S.N.; et al. Oncolytic Viruses Engineered to Enforce Leptin Expression Reprogram Tumor-Infiltrating T Cell Metabolism and Promote Tumor Clearance. Immunity 2019, 51, 548–560. [Google Scholar] [CrossRef] [PubMed]
- Gentile, C.; Finizio, A.; Froechlich, G.; D’Alise, A.M.; Cotugno, G.; Amiranda, S.; Nicosia, A.; Scarselli, E.; Zambrano, N.; Sasso, E. Generation of a Retargeted Oncolytic Herpes Virus Encoding Adenosine Deaminase for Tumor Adenosine Clearance. Int. J. Mol. Sci. 2021, 22, 13521. [Google Scholar] [CrossRef] [PubMed]
- Allard, B.; Allard, D.; Buisseret, L.; Stagg, J. The Adenosine Pathway in Immuno-Oncology. Nat. Rev. Clin. Oncol. 2020, 17, 611–629. [Google Scholar] [CrossRef] [PubMed]
- Ramelyte, E.; Tastanova, A.; Balázs, Z.; Ignatova, D.; Turko, P.; Menzel, U.; Guenova, E.; Beisel, C.; Krauthammer, M.; Levesque, M.P.; et al. Oncolytic Virotherapy-Mediated Anti-Tumor Response: A Single-Cell Perspective. Cancer Cell 2021, 39, 394–406. [Google Scholar] [CrossRef]
- Lang, F.; Schrörs, B.; Löwer, M.; Türeci, Ö.; Sahin, U. Identification of Neoantigens for Individualized Therapeutic Cancer Vaccines. Nat. Rev. Drug Discov. 2022, 21, 261–282. [Google Scholar] [CrossRef]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An Immunogenic Personal Neoantigen Vaccine for Patients with Melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.-P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA Mutanome Vaccines Mobilize Poly-Specific Therapeutic Immunity against Cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef]
- Gupta, R.G.; Li, F.; Roszik, J.; Lizée, G. Exploiting Tumor Neoantigens to Target Cancer Evolution: Current Challenges and Promising Therapeutic ApproachesExploiting Tumor Neoantigens to Target Cancer Evolution. Cancer Discov. 2021, 11, 1024–1039. [Google Scholar] [CrossRef]
- Mark, Y.; Alexander, H.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef]
- Morgan, R.A.; Chinnasamy, N.; Abate-Daga, D.; Gros, A.; Robbins, P.F.; Zheng, Z.; Dudley, M.E.; Feldman, S.A.; Yang, J.C.; Sherry, R.M.; et al. Cancer Regression and Neurological Toxicity Following Anti-MAGE-A3 TCR Gene Therapy. J. Immunother. 2013, 36, 133–151. [Google Scholar] [CrossRef]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational Landscape and Significance across 12 Major Cancer Types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.K.; Riley, T.P.; Baker, S.C.B.; Borrman, T.; Weng, Z.; Baker, B.M. Emerging Concepts in TCR Specificity: Rationalizing and (Maybe) Predicting Outcomes. J. Immunol. 2017, 199, 2203–2213. [Google Scholar] [CrossRef] [PubMed]
- Hundal, J.; Carreno, B.M.; Petti, A.A.; Linette, G.P.; Griffith, O.L.; Mardis, E.R.; Griffith, M. PVAC-Seq: A Genome-Guided in Silico Approach to Identifying Tumor Neoantigens. Genome Med. 2016, 8, 11. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, M.; Fenoy, E.; Harndahl, M.; Kristensen, A.B.; Nielsen, I.K.; Nielsen, M.; Buus, S. Pan-Specific Prediction of Peptide–MHC Class I Complex Stability, a Correlate of T Cell Immunogenicity. J. Immunol. 2016, 197, 1517–1524. [Google Scholar] [CrossRef]
- Tappeiner, E.; Finotello, F.; Charoentong, P.; Mayer, C.; Rieder, D.; Trajanoski, Z. TIminer: NGS Data Mining Pipeline for Cancer Immunology and Immunotherapy. Bioinformatics 2017, 33, 3140–3141. [Google Scholar] [CrossRef]
- Li, Y.; Wang, G.; Tan, X.; Ouyang, J.; Zhang, M.; Song, X.; Liu, Q.; Leng, Q.; Chen, L.; Xie, L. ProGeo-Neo: A Customized Proteogenomic Workflow for Neoantigen Prediction and Selection. BMC Med. Genom. 2020, 13, 52. [Google Scholar] [CrossRef]
- Bjerregaard, A.-M.; Nielsen, M.; Hadrup, S.R.; Szallasi, Z.; Eklund, A.C. MuPeXI: Prediction of Neo-Epitopes from Tumor Sequencing Data. Cancer Immunol. Immunother. 2017, 66, 1123–1130. [Google Scholar] [CrossRef]
- Bulik-Sullivan, B.; Busby, J.; Palmer, C.D.; Davis, M.J.; Murphy, T.; Clark, A.; Busby, M.; Duke, F.; Yang, A.; Young, L.; et al. Deep Learning Using Tumor HLA Peptide Mass Spectrometry Datasets Improves Neoantigen Identification. Nat. Biotechnol. 2019, 37, 55–63. [Google Scholar] [CrossRef]
- Rubinsteyn, A.; Hodes, I.; Kodysh, J.; Hammerbacher, J. Vaxrank: A Computational Tool for Designing Personalized Cancer Vaccines. Biorxiv 2018, 142919. [Google Scholar] [CrossRef]
- Leoni, G.; D’Alise, A.M.; Tucci, F.G.; Micarelli, E.; Garzia, I.; Lucia, M.D.; Langone, F.; Nocchi, L.; Cotugno, G.; Bartolomeo, R.; et al. VENUS, a Novel Selection Approach to Improve the Accuracy of Neoantigens’ Prediction. Nato Adv. Sci. Inst. Se. 2021, 9, 880. [Google Scholar] [CrossRef]
- Duan, F.; Duitama, J.; Seesi, S.A.; Ayres, C.M.; Corcelli, S.A.; Pawashe, A.P.; Blanchard, T.; McMahon, D.; Sidney, J.; Sette, A.; et al. Genomic and Bioinformatic Profiling of Mutational Neoepitopes Reveals New Rules to Predict Anticancer Immunogenicity. J. Exp. Med. 2014, 211, 2231–2248. [Google Scholar] [CrossRef] [PubMed]
- Wooldridge, L.; Ekeruche-Makinde, J.; van den Berg, H.A.; Skowera, A.; Miles, J.J.; Tan, M.P.; Dolton, G.; Clement, M.; Llewellyn-Lacey, S.; Price, D.A.; et al. A Single Autoimmune T Cell Receptor Recognizes More Than a Million Different Peptides. J. Biol. Chem. 2012, 287, 1168–1177. [Google Scholar] [CrossRef] [PubMed]
- Łuksza, M.; Sethna, Z.M.; Rojas, L.A.; Lihm, J.; Bravi, B.; Elhanati, Y.; Soares, K.; Amisaki, M.; Dobrin, A.; Hoyos, D.; et al. Neoantigen Quality Predicts Immunoediting in Survivors of Pancreatic Cancer. Nature 2022, 606, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Gfeller, D.; Guillaume, P.; Michaux, J.; Pak, H.-S.; Daniel, R.T.; Racle, J.; Coukos, G.; Bassani-Sternberg, M. The Length Distribution and Multiple Specificity of Naturally Presented HLA-I Ligands. J. Immunol. 2018, 201, 3705–3716. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tripodi, L.; Sasso, E.; Feola, S.; Coluccino, L.; Vitale, M.; Leoni, G.; Szomolay, B.; Pastore, L.; Cerullo, V. Systems Biology Approaches for the Improvement of Oncolytic Virus-Based Immunotherapies. Cancers 2023, 15, 1297. https://doi.org/10.3390/cancers15041297
Tripodi L, Sasso E, Feola S, Coluccino L, Vitale M, Leoni G, Szomolay B, Pastore L, Cerullo V. Systems Biology Approaches for the Improvement of Oncolytic Virus-Based Immunotherapies. Cancers. 2023; 15(4):1297. https://doi.org/10.3390/cancers15041297
Chicago/Turabian StyleTripodi, Lorella, Emanuele Sasso, Sara Feola, Ludovica Coluccino, Maria Vitale, Guido Leoni, Barbara Szomolay, Lucio Pastore, and Vincenzo Cerullo. 2023. "Systems Biology Approaches for the Improvement of Oncolytic Virus-Based Immunotherapies" Cancers 15, no. 4: 1297. https://doi.org/10.3390/cancers15041297
APA StyleTripodi, L., Sasso, E., Feola, S., Coluccino, L., Vitale, M., Leoni, G., Szomolay, B., Pastore, L., & Cerullo, V. (2023). Systems Biology Approaches for the Improvement of Oncolytic Virus-Based Immunotherapies. Cancers, 15(4), 1297. https://doi.org/10.3390/cancers15041297